Phys · neurological
Guillain-Barre Syndrome
Also known as GBS · acute inflammatory demyelinating polyradiculoneuropathy · AIDP · Landry ascending paralysis · Miller Fisher syndrome · AMAN · AMSAN · Bickerstaff brainstem encephalitis
Consultant-physician-depth guide to Guillain-Barre syndrome — immune-mediated demyelination of peripheral nerves via molecular mimicry and anti-ganglioside antibodies, ascending weakness with areflexia, CSF albuminocytologic dissociation and nerve conduction studies, IVIG versus plasma exchange, respiratory and autonomic monitoring, and the CIDP boundary — for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Guillain-Barre Syndrome

The answer first
Guillain-Barre syndrome (GBS) is an acute, immune-mediated polyradiculoneuropathy. The clinical signature is progressive, symmetric, ascending weakness with areflexia that reaches its nadir within 4 weeks. It is the most common cause of acute generalized paralysis since polio was eradicated, and it is a medical emergency: around one in four patients needs mechanical ventilation, and autonomic failure causes a substantial share of deaths. [1]
Three facts organise everything else: [1]
- It is treatable. Both intravenous immunoglobulin (IVIG) and plasma exchange speed recovery if given within 2-4 weeks of onset. Corticosteroids do not work and may worsen the disease.
- The threat to life is respiratory and autonomic, not the weakness itself. Bedside lung function monitoring (FVC, MIP, MEP), not the strength of the grip, decides who goes to ICU and who is intubated.
- The diagnosis is clinical and supported by CSF and nerve conduction studies. A normal CSF in the first week does not exclude GBS — albuminocytologic dissociation appears after 1-2 weeks. [1]
The organising principle on the ward is: diagnose early, monitor lung function relentlessly, treat with IVIG or plasma exchange (not both), never give steroids, and watch the clock for the 8-week boundary that converts the diagnosis to CIDP. [1]
Classification and the spectrum
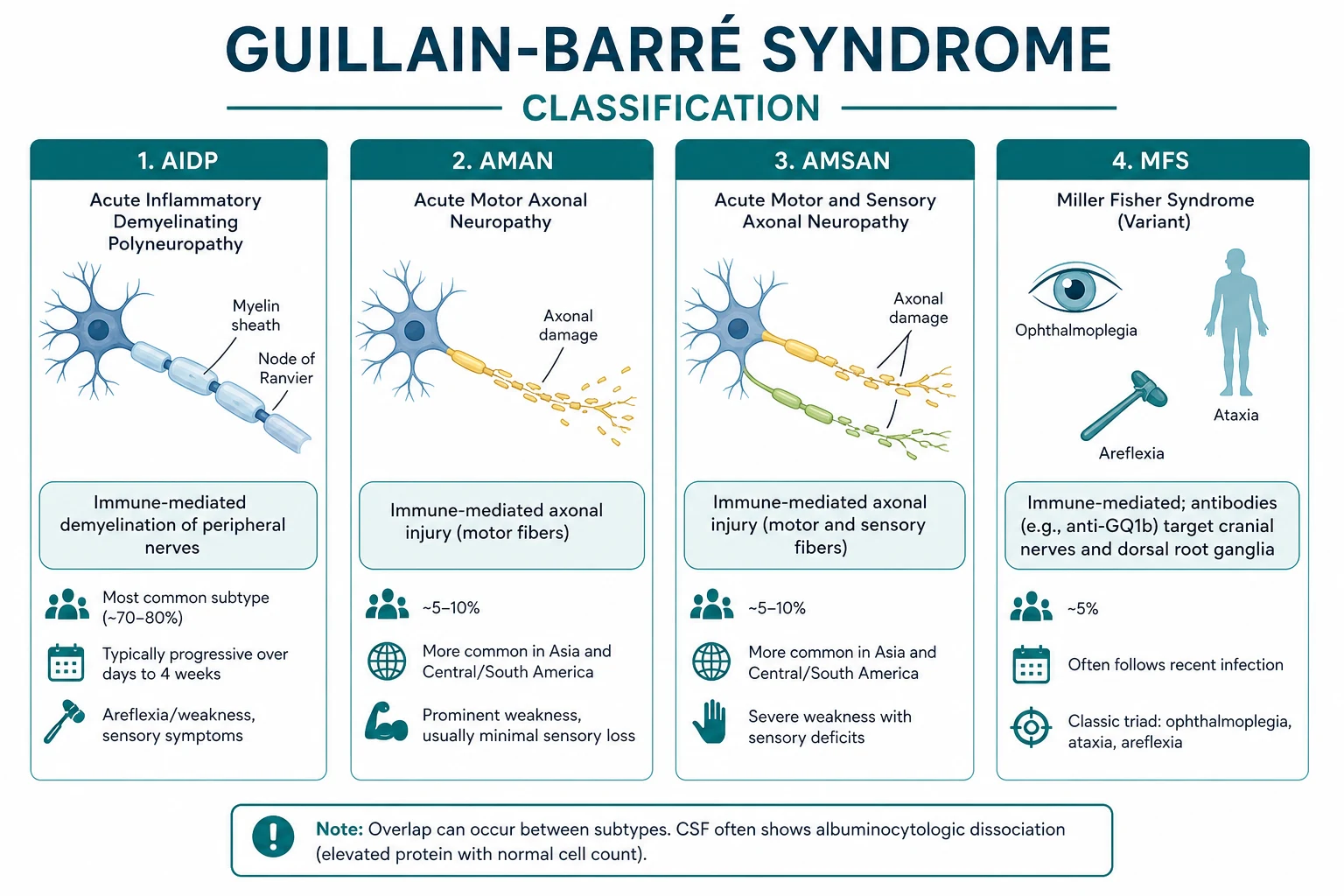

GBS is a syndrome with several subtypes defined by the site of immune attack and the electrophysiology. The variant determines the antibody, the trigger, and sometimes the prognosis. [1]
The major variants
| Variant | Frequency | Mechanism | Typical antibody | Notes |
|---|---|---|---|---|
| AIDP (acute inflammatory demyelinating polyradiculoneuropathy) | around 85-90% in Western countries | Demyelination | Variable | Most common; the prototype |
| AMAN (acute motor axonal neuropathy) | Common in Asia, Latin America | Axonal motor | Anti-GM1, anti-GD1a | Pure motor; post-Campylobacter; faster nadir |
| AMSAN (acute motor-sensory axonal neuropathy) | Less common | Axonal motor and sensory | Anti-GM1 | Severe; slow, incomplete recovery |
| Miller Fisher syndrome | around 5% | Cranial nerve and muscle spindle | Anti-GQ1b | Ophthalmoplegia, ataxia, areflexia |
| Bickerstaff brainstem encephalitis | Rare | Brainstem overlap | Anti-GQ1b | Miller-Fisher plus altered consciousness (coma) |
| Pharyngeal-cervical-brachial variant | Rare | Upper-body predominant | Anti-GT1a | Isolated bulbar and arm weakness |
DWE high-yield: The most tested association is anti-GQ1b with Miller Fisher syndrome (ophthalmoplegia, ataxia, areflexia). The second is anti-GM1 with AMAN after Campylobacter jejuni. Know both. The ataxia in Miller Fisher is sensory (proprioceptive, from muscle spindle involvement), not cerebellar. [1]
The GBS disability scale
This 0-6 scale (the Hughes scale) is the universal outcome measure and the decision tool for treatment. [1]
| Grade | Function |
|---|---|
| 0 | Healthy |
| 1 | Minor symptoms, able to run |
| 2 | Able to walk 10 m without assistance |
| 3 | Able to walk 10 m with assistance |
| 4 | Chairbound/bedbound |
| 5 | Requiring assisted ventilation for some of the day |
| 6 | Dead |
Treatment threshold is grade 3 or worse (unable to walk unaided), or any sign of respiratory, bulbar, or autonomic decline. [1]
Pathophysiology
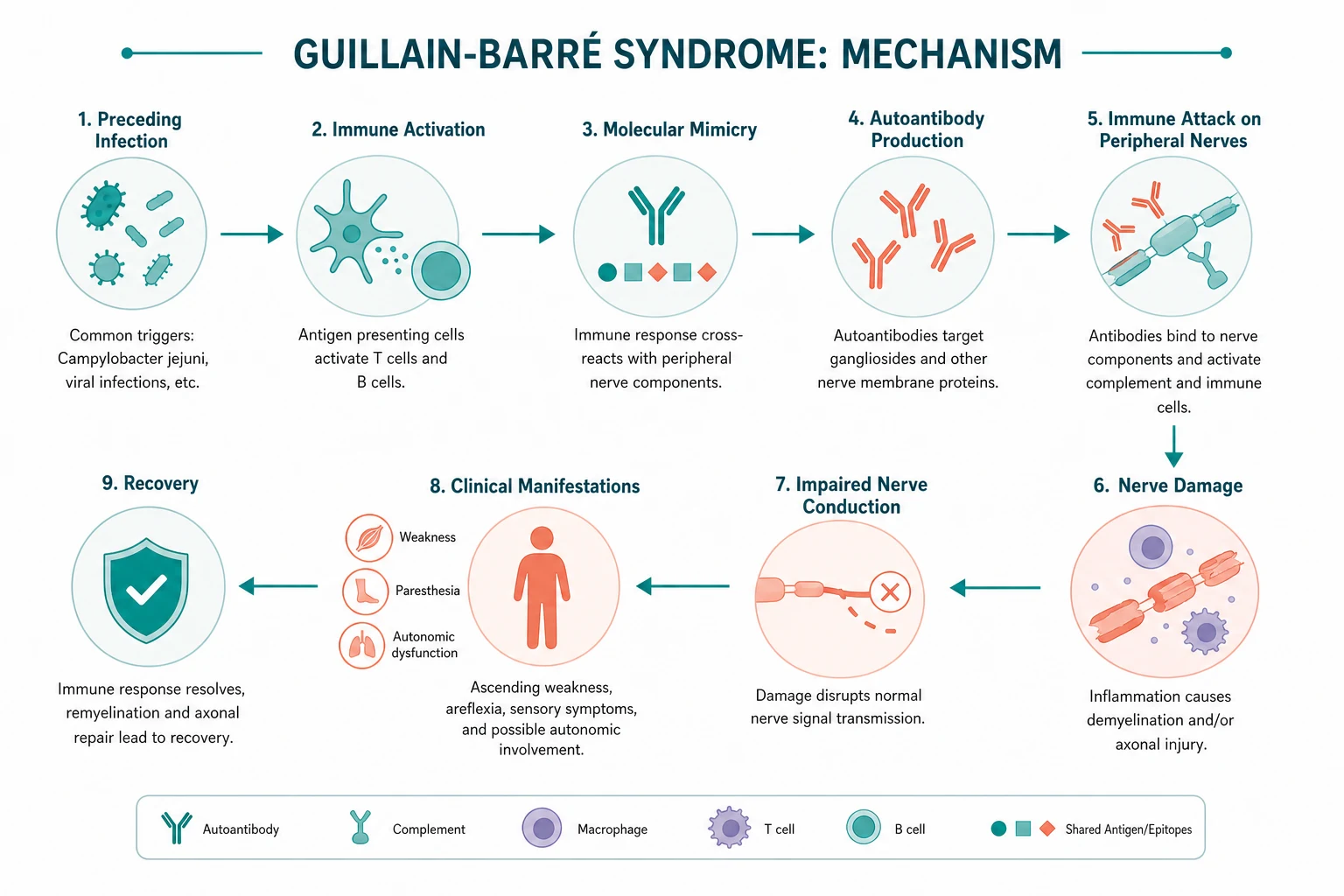
Molecular mimicry — the mechanism that links the trigger to the nerve
GBS is a post-infectious, immune-mediated disease. About two-thirds of patients report an infection in the preceding 1-3 weeks. The mechanism is molecular mimicry: the infecting organism expresses surface molecules that resemble gangliosides on human peripheral nerves. The resulting antibody response cross-reacts with nerve tissue. [1]
The best-characterised example is Campylobacter jejuni. Its lipo-oligosaccharide coat carries ganglioside-like structures (GM1, GD1a, GT1a). Antibodies raised against the bacterium bind to gangliosides at the nodes of Ranvier and on myelin. This was proven experimentally: immunising rabbits with C. jejuni LPS from GBS-associated strains induces anti-GM1 and anti-GQ1b antibodies and a flaccid paralysis resembling the human disease (Ang 2001, PMID 11254608). [1]
| Trigger | Approximate frequency | Typical antibody | Variant association |
|---|---|---|---|
| Campylobacter jejuni (gastroenteritis) | around 30% | Anti-GM1, anti-GD1a | AMAN, AMSAN, AIDP |
| Cytomegalovirus (CMV) | around 10% | Anti-GM2 | AIDP, prominent sensory and cranial nerve |
| Epstein-Barr virus | around 5% | Variable | AIDP |
| Mycoplasma pneumoniae | less common | Anti-GalNAc-GD1a | Often in children |
| Haemophilus influenzae | less common | Variable | AIDP |
| Zika virus | Outbreak-related | Variable | AIDP (French Polynesia, Latin America) |
| Influenza | less common | Variable | AIDP |
| Surgery, trauma, immunisation | around 5% | Variable | AIDP |
DCE viva trap: When asked "is GBS caused by vaccines?", the honest answer is: the influenza vaccine carries a tiny excess risk (around one extra case per million doses), far smaller than the risk of GBS after influenza itself. The 1976 swine-flu vaccine is the historical outlier. Modern vaccines are safe. The Cao-Lormeau 2016 case-control study (PMID 26948433) established Zika as a robust outbreak trigger. [1]
Demyelinating versus axonal — why electrophysiology matters
In AIDP, antibodies and complement activate macrophages that strip myelin from the Schwann-cell unit, particularly at the nerve roots and proximal segments. Demyelination causes conduction block and slowing — the signal cannot traverse the demyelinated segment. Because the axon is intact, remyelination can restore function, which is why AIDP generally recovers well. [1]
In AMAN/AMSAN, the attack is at the node of Ranvier: macrophages insert between the myelin and the axon and there is direct axonal injury (Wallerian-like degeneration). Conduction velocities are relatively preserved, but compound muscle action potential (CMAP) amplitudes fall because axons are lost. Axonal variants have a less predictable and often slower recovery, because recovery depends on axonal regrowth (around 1 mm/day, or about 1 inch/month). [1]
The antibody target explains the regional pattern of injury. GQ1b is densely expressed on oculomotor, trochlear, and abducens nerves and on muscle spindles — hence ophthalmoplegia and areflexia (loss of the stretch reflex) in Miller Fisher syndrome (Chiba 1993, PMID 8413947; Kusunoki 2008, PMID 17976386). [1]
DWE high-yield: The reflex loss in GBS is not a sensory problem alone — it is the loss of the afferent limb (muscle spindle Ia afferent) due to ganglioside-targeted attack. This is why areflexia can be disproportionate to the weakness. [1]
Clinical features
The classic presentation — ascending weakness
The patient develops bilateral, symmetric weakness that begins in the legs and ascends to the arms, trunk, and cranial nerves over days. The weakness is flaccid with absent or depressed reflexes. Key features: [1]
- Time course — progression over days to a maximum of 4 weeks to nadir (the defining criterion). Median time to nadir is around 9-12 days.
- Symmetry — usually striking, though minor asymmetry occurs.
- Ascending pattern — legs first, then arms. Cranial nerves and respiration may follow.
- Areflexia — the reflexes disappear early, often before weakness is severe. Preserved reflexes in a weak limb should prompt reconsideration of the diagnosis.
- Sensory involvement — less prominent than motor, but paraesthesia and numbness in the hands and feet are common early features. Sensory loss is usually minor.
- Pain — up to two-thirds have pain (back, thigh, calf, paraesthesia), which may precede weakness and mislead toward mechanical back pain or radiculopathy. [1]
Cranial nerve involvement
- Bilateral facial (VII) palsy — occurs in around half of severe cases. Bilateral lower motor neuron facial weakness is a red flag for GBS and should never be dismissed as bilateral Bell palsy.
- Bulbar weakness — dysphagia, dysarthria, weak cough — signals imminent aspiration and ventilatory risk.
- Ophthalmoplegia — in Miller Fisher syndrome and in severe AIDP. [1]
Autonomic dysfunction — a leading killer
Autonomic involvement is common and may be life-threatening. It is driven by involvement of the autonomic fibres and is most dangerous in ventilated patients: [1]
- Cardiac arrhythmia — bradycardia, asystole, atrial and ventricular arrhythmias. Sudden death from dysautonomia is a recognised cause of mortality.
- Blood pressure swings — severe labile hypertension and hypotension, sometimes orthostatic.
- Ileus, urinary retention, sweating disturbances, SIADH. [1]
DCE trap: Every GBS inpatient needs continuous cardiac monitoring until stable. Avoid drugs that destabilise the autonomic system; use short-acting vasoactive agents. A sudden bradycardic arrest in a ventilated GBS patient is usually dysautonomia, not a primary cardiac event. [1]
Miller Fisher syndrome — the triad to know
The Miller Fisher triad is ophthalmoplegia, ataxia, and areflexia. The ataxia is sensory (cerebellar testing — finger-to-nose, heel-to-shin — is normal; the problem is proprioceptive input from muscle spindles). Anti-GQ1b antibody is present in over 90% of cases and is essentially diagnostic. Miller Fisher overlaps on a spectrum with Bickerstaff brainstem encephalitis, which adds altered consciousness (drowsiness, coma) and sometimes extensor plantar responses. [1]
What to find on examination
| Domain | Typical finding |
|---|---|
| Tone | Flaccid, reduced |
| Power | Symmetric weakness, legs worse than arms, distal often worse early |
| Reflexes | Areflexia or hyporeflexia (early, disproportionate) |
| Plantars | Down (flexor) — UMN signs are absent |
| Sensation | Minor glove-and-stocking sensory loss; marked proprioceptive loss in Miller Fisher |
| Coordination | Limited by weakness; ataxia is sensory in Miller Fisher |
| Cranial nerves | Bilateral facial palsy, bulbar weakness, ophthalmoplegia in variants |
| Autonomic | Labile BP, arrhythmia, ileus, urinary retention |
Differential diagnosis
Rapidly progressive ascending weakness with areflexia has a focused differential. The dangerous mimics are the spinal cord and electrolyte disorders. [1]
| Diagnosis | Distinguishing features |
|---|---|
| Transverse myelitis / acute cord lesion | A clear sensory level, sphincter (bladder/bowel) involvement, UMN signs below the lesion (hyperreflexia, extensor plantar). MRI of the spine is urgent. |
| Spinal cord compression | Back pain, UMN signs below lesion, often asymmetric. Imaging is an emergency. |
| Hypokalaemia / hypokalaemic periodic paralysis | Check potassium urgently. Treatable. Periodic paralysis is episodic and recurrent. |
| Tick paralysis | Ascending paralysis with areflexia; a tick is attached. Removal reverses it. |
| Botulism | Descending paralysis, cranial nerve onset, dilated pupils, anticholinergic features, food/wound/IV-drug history. |
| Diphtheritic neuropathy | Pharyngitis then palatal paralysis weeks later; bull neck; unimmunised. |
| Acute porphyria | Abdominal pain, psychiatric features, motor neuropathy; check urine porphobilinogen. |
| Critical illness polyneuropathy/myopathy | Arises in the context of severe critical illness and multi-organ failure, not at presentation. |
| Myasthenia gravis | Fatigable weakness, fluctuating, ocular onset, reflexes preserved, worsens through the day. |
| West Nile / polio | Asymmetric weakness, fever, meningitic features; polio is rare in immunised populations. |
DWE high-yield: The single most tested discriminator is the reflex pattern and the presence of a sensory level / sphincter involvement. Areflexia with a downgoing plantar and no sensory level points to GBS; hyperreflexia with extensor plantars and a sensory level points to a cord lesion and demands urgent MRI. Never attribute bilateral facial palsy to two separate Bell palsies. [1]
Investigations

Cerebrospinal fluid — albuminocytologic dissociation
The CSF hallmark is albuminocytologic dissociation: a raised protein with a normal cell count. This reflects inflammation of the nerve roots (where the blood-nerve barrier is permeable) with sparing of the meninges. [1]
- Protein — elevated, often 0.55-2.0 g/L. But it is frequently normal in the first week. A normal early CSF does not exclude GBS.
- White cells — normal (under 5 cells/microL). A cell count above 50 cells/microL should prompt a search for an alternative — HIV-seroconversion GBS, leptospirosis, malignant infiltration, or infection.
- Timing — CSF protein rises over 1-2 weeks. If the first LP is normal and suspicion persists, repeat after 1-2 weeks. [1]
DWE trap: The two classic CSF errors are (1) excluding GBS because the first-week CSF is normal, and (2) attributing a high white-cell CSF to "atypical GBS" without testing HIV. Always send an HIV test in any GBS with CSF pleocytosis. [1]
Nerve conduction studies (NCS)
NCS confirm the diagnosis, define the variant (demyelinating versus axonal), and inform prognosis. Early studies may be normal; the abnormalities evolve. [1]
| Parameter | AIDP (demyelinating) | AMAN (axonal) |
|---|---|---|
| Distal motor latency | Prolonged | Normal or mildly prolonged |
| Conduction velocity | Slowed (below 70% lower limit) | Normal or near-normal |
| Conduction block / temporal dispersion | Present (key finding) | Absent |
| F-waves | Prolonged or absent (early sign — root involvement) | May be absent |
| CMAP amplitude | May be reduced | Markedly reduced (axon loss) |
| Sensory action potentials | Often reduced | Normal in pure AMAN |
Prolonged or absent F-waves are an early sign because the nerve roots are involved early. Conduction block (the signal fails to traverse a segment) is the electrophysiological signature of demyelination. [1]
Anti-ganglioside antibodies
These support specific variants but are not required for acute management: [1]
- Anti-GQ1b — Miller Fisher syndrome (over 90% sensitive), and a marker of ophthalmoplegia in any GBS variant.
- Anti-GM1, anti-GD1a — AMAN, post-Campylobacter.
- Anti-GalNAc-GD1a — pharyngeal-cervical-brachial variant. [1]
MRI
MRI is mainly used to exclude cord compression and transverse myelitis. Post-contrast MRI of the nerve roots may show enhancement of the cauda equina and nerve roots, which supports the diagnosis when NCS is equivocal. The brain is typically normal. [1]
Bedside respiratory and autonomic monitoring
This is the single most important inpatient monitoring. Measure at least every 4-6 hours, more often if deteriorating: [1]
- Forced vital capacity (FVC) — the key parameter.
- Maximum inspiratory pressure (MIP, or negative inspiratory force NIF) and maximum expiratory pressure (MEP) — more sensitive than FVC for early diaphragm failure.
- Single breath count (count aloud in one breath) — a practical surrogate.
- Bulbar assessment — cough, swallow, gag. [1]
Brighton Collaboration criteria
For diagnostic certainty in research and surveillance, the Brighton Collaboration case definition assigns levels 1-3 based on the combination of bilateral and progressive weakness, areflexia, monophasic course, CSF findings, and NCS. The Fokke 2014 validation (PMID 24163275) confirmed that most clinically diagnosed GBS meet level 1 or 2. [1]
Acute management


The three pillars
- Disease-modifying therapy — IVIG or plasma exchange (equally effective; not combined).
- Supportive care — respiratory, autonomic, DVT, nutrition, pain, skin, psychological.
- No corticosteroids — they do not work and may be harmful. [1]
Disease-modifying therapy
| Therapy | Regimen | Onset of effect | Role |
|---|---|---|---|
| IVIG | 0.4 g/kg/day for 5 days (total 2 g/kg) | Within 2 weeks | First-line in most centres — easier, no central access, fewer haemodynamic risks |
| Plasma exchange | 5 sessions over 1-2 weeks, total 250-300 mL/kg | Within 2 weeks | Equivalent; preferred if IVIG contraindicated (IgA deficiency, prior severe reaction) |
Both speed recovery by around 40-50% compared with supportive care alone. The evidence base: [1]
- Plasma exchange — the 1985 GBS Study Group RCT (PMID 4022342) first showed plasmapheresis reduced time to recover walking versus supportive care.
- IVIG — the 1992 Dutch trial (PMID 1552913) showed IVIG was at least as effective as plasma exchange.
- Equivalence — the 1997 Plasma Exchange/Sandoglobulin Trial (PMID 9014908, 383 patients) showed PE, IVIG, and the combination were equivalent; combination added no benefit. This is why we give one, not both. [1]
Indication: any patient who cannot walk unaided (grade 3 or worse), or who has respiratory decline, bulbar weakness, or significant autonomic dysfunction, within 4 weeks of onset. Mild, ambulatory patients (grade 2 or better) who are stable may be observed without treatment, but most physicians treat if there is any progression. [1]
DWE high-yield trap: The most common treatment error is giving both IVIG and plasma exchange, or giving corticosteroids. The evidence is unambiguous: combination is not better (PS GBS Trial Group 1997), and steroids alone or added to IVIG are ineffective (Hughes Cochrane 2016, PMID 27775812; van Koningsveld 2004, PMID 14738791). The methylprednisolone-plus-IVIG trial showed only a non-significant trend toward benefit and is not enough to change practice. [1]
Why steroids fail — and the CIDP contrast
Corticosteroids are ineffective in GBS and may slow recovery. The mechanism is debated (steroids may impair the macrophage-mediated clearance that follows the antibody attack). This is the key contrast with chronic inflammatory demyelinating polyneuropathy (CIDP), in which corticosteroids are first-line. The difference is sometimes summarised: CIDP is steroid-responsive; GBS is not. If you are tempted to give steroids, re-check the diagnosis and the time course. [1]
IVIG practical points
- Dose — 0.4 g/kg/day for 5 days. Total 2 g/kg.
- Adverse effects — thrombosis (stroke, MI, DVT), acute kidney injury (especially with sucrose-stabilised products and pre-existing renal impairment), aseptic meningitis, haemolytic anaemia (non-O blood group), transfusion reactions. Check IgA level if there is a history of anaphylaxis to blood products (IgA deficiency).
- Precautions — slow the infusion in older patients, those with cardiac or renal disease, and those with hyperviscosity. Ensure hydration. Thromboprophylaxis. [1]
Plasma exchange practical points
- Regimen — 5 exchanges, usually every other day, 1-1.5 plasma volumes per session, replacement with albumin or fresh-frozen plasma.
- Access — large-bore central or peripheral access.
- Contraindications — haemodynamic instability, severe sepsis, bleeding diathesis, difficult vascular access.
- Adverse effects — hypotension, bleeding (from citrate and clotting-factor removal), line infection, hypocalcaemia. [1]
Respiratory care and intubation thresholds
Around 20-30% of patients require mechanical ventilation. Intubate electively before respiratory arrest, never after desaturation (desaturation is a late, pre-terminal sign). [1]
| Parameter | Threshold for ICU review | Threshold favouring intubation |
|---|---|---|
| FVC | Below 30 mL/kg, or falling greater than 30% | Below 20 mL/kg |
| MEP | Below 50 cmH2O | Below 40 cmH2O |
| Bulbar / cough | Weak cough, poor swallow | Bulbar weakness with aspiration risk |
| Other | Rapidly progressive course | Autonomic instability, single breath count below 20 |
DCE viva answer pattern: "I would intubate this patient electively. Her FVC has fallen from 30 to 19 mL/kg, she has a weak cough and bulbar weakness, and she is tiring. Waiting for desaturation is dangerous — desaturation is a late sign in young, fit patients, and by then the arrest is imminent. I would transfer her to ICU, ventilate, and continue IVIG and supportive care." [1]
The Walgaard 2010 model (PMID 20517939) identified predictors of respiratory insufficiency: short interval from onset to admission, low FVC and MIP, weak neck flexion, and bulbar weakness. Use these to triage early ICU involvement. [1]
Autonomic monitoring
- Continuous ECG until stable — treat bradycardia (atropine, pacing if severe) and tachyarrhythmia.
- Blood pressure — use short-acting agents for both hypertension and hypotension. Avoid sudden position changes. A labile BP often resolves as the disease plateaus.
- Urinary catheter if retention; treat ileus and SIADH if present. [1]
The supportive care bundle
| Domain | Intervention |
|---|---|
| DVT prophylaxis | LMWH (e.g. enoxaparin 40 mg SC daily, dose-adjusted in renal impairment) plus intermittent pneumatic compression — risk of DVT/PE is around 30% without prophylaxis |
| Pain | Gabapentin or pregabalin first-line for neuropathic pain; amitriptyline, carbamazepine; opioids for severe pain; avoid pure opioid monotherapy |
| Skin / pressure care | Regular turning, pressure-relieving mattress |
| Physiotherapy | Passive range of motion to prevent contractures; early mobilisation as strength returns |
| Tracheostomy | Consider if ventilation beyond 2-3 weeks is anticipated; aids weaning and pulmonary toilet |
| Psychological | Screen for depression, anxiety, PTSD; the awake, paralysed, ventilated patient is at high risk |
Treatment-related fluctuations and A-CIDP
Around 5-10% of patients treated with IVIG have a treatment-related fluctuation (TRF) — improvement followed by recurrence within 2 months. Treat with a second course of IVIG. If weakness progresses or relapses beyond 8 weeks, re-diagnose as acute-onset CIDP (A-CIDP) — management shifts to corticosteroids, repeat IVIG, and long-term immunotherapy. The 8-week clock is a defining boundary. [1]
Prognosis
| Outcome | Frequency |
|---|---|
| Full or near-full recovery | around 80% |
| Mortality | around 5% (3-10% across series) |
| Unable to walk unaided at 6 months | around 20% |
| Persistent severe disability | around 5-10% |
| Persistent pain | up to 50% |
Favourable prognostic factors: younger age, AIDP variant, antecedent upper-respiratory (rather than diarrhoeal/Campylobacter) illness, mild nadir, early treatment, and rapid improvement after IVIG. [1]
Adverse prognostic factors (the EGRIS/EGOS inputs): older age, diarrhoeal/Campylobacter antecedent, axonal variant (AMAN/AMSAN), severe nadir (ventilated, grade 4-5), weak neck and bulbar muscles, low CMAP amplitudes on NCS, and a high Erasmus GBS Outcome Score (EGOS, PMID 17537676). [1]
The EGOS at 2 weeks uses age, antecedent diarrhoea, and GBS disability score to predict the probability of independent walking at 6 months. A modified EGOS (mEGOS) incorporates the Medical Research Council sum score. Use it to set realistic expectations and to flag the patients who will need prolonged rehabilitation. [1]
DCE long-case trap: When presenting a recovering GBS patient, state the variant, the nadir disability grade, whether ventilation was required, the current grade, and the EGOS-predicted 6-month outcome. Examiners want to see that you understand prognosis is determined at nadir, not at presentation. [1]
CIDP — the chronic boundary
Chronic inflammatory demyelinating polyneuropathy (CIDP) is the chronic cousin of GBS. The defining difference is time course: progression continues beyond 8 weeks (GBS reaches nadir within 4 weeks). CIDP may follow a relapsing-remitting or progressive course. [1]
The critical management contrast: [1]
| Feature | GBS | CIDP |
|---|---|---|
| Time to nadir | Under 4 weeks | Over 8 weeks |
| Course | Monophasic | Chronic, relapsing or progressive |
| Corticosteroids | Ineffective | Effective (first-line) |
| IVIG | Effective | Effective |
| Plasma exchange | Effective | Effective |
DWE high-yield: The most tested contrast is the steroid response: CIDP responds to steroids, GBS does not. A patient who is given steroids and improves may have had CIDP from the start. The diagnosis is made on time course, not on steroid response after the fact. [1]
DCE long-case approach
Opening statement (SASPOP)
"Mr Davies is a 54-year-old mechanic who presents with four days of progressive weakness beginning in his feet and ascending to his legs and hands, with paraesthesia in his fingers and toes, and now difficulty standing. Two weeks ago he had a week of diarrhoea, diagnosed as Campylobacter jejuni enteritis by his GP. He is otherwise well, takes no regular medications, and is a non-smoker. [1]
On examination he is afebrile, alert, and oriented. His FVC is 28 mL/kg and falling, MIP is -45 cmH2O. He has symmetric flaccid weakness, power 3/5 in the legs and 4/5 in the arms, with absent reflexes throughout and downgoing plantars. Sensation is reduced to pinprick in a glove-and-stocking distribution. Cranial nerves are intact except for mild bilateral facial weakness. [1]
CSF shows protein 1.2 g/L with 3 white cells. Nerve conduction studies show prolonged distal latencies, conduction block, and absent F-waves consistent with AIDP. [1]
His main problems are:
- Acute Guillain-Barre syndrome, AIDP variant, post-Campylobacter, currently GBS disability grade 4 — disease-modifying therapy indicated
- Respiratory function declining — high risk of ventilatory failure, requires ICU-level monitoring
- Autonomic risk — requires continuous cardiac monitoring
- Pain and immobility — VTE prophylaxis, analgesia, physiotherapy
- Psychological impact and prognosis communication" [1]
Integrated management plan
Present in three phases: [1]
- Immediate (hours): Confirm diagnosis (CSF, NCS). Transfer to a monitored bed (HDU/ICU if FVC below 30 mL/kg or falling). Start bedside respiratory monitoring every 4 hours. Continuous ECG. Establish IV access. Check IgA and renal function before IVIG. Start IVIG 0.4 g/kg/day for 5 days. Do not give steroids. Insert a urinary catheter if retention. Start DVT prophylaxis.
- Subacute (days to weeks): Continue respiratory and cardiac monitoring; intubate electively at the thresholds above. Continue supportive care — early enteral nutrition, pain management, physiotherapy, pressure care. Watch for treatment-related fluctuation (re-treat with IVIG) and for progression beyond 8 weeks (re-diagnose as A-CIDP). If ventilation is prolonged, plan tracheostomy at 2-3 weeks.
- Recovery and rehabilitation: As strength returns, mobilise with physiotherapy. Address persistent pain, fatigue, and psychological sequelae. Set functional goals. Plan outpatient follow-up to assess residual disability and return to work and driving. [1]
DCE examiner probing questions you must anticipate:
- "Why IVIG and not plasma exchange?" — Both are equivalent; IVIG is easier, avoids central access and haemodynamic instability, and is preferred in most ANZ and UK centres. Choose plasma exchange if IVIG is contraindicated.
- "Will steroids help?" — No. Steroids are ineffective in GBS and may be harmful. The van Koningsveld 2004 trial and the Cochrane review are definitive.
- "When will you intubate?" — Electively at FVC below 20 mL/kg, MIP more negative than -30 cmH2O, MEP below 40 cmH2O, or with bulbar weakness and aspiration risk. Not after desaturation.
- "What is his prognosis?" — Around 80% recover well; he has adverse features (Campylobacter antecedent, grade 4 at nadir). I would quote him a guarded-but-hopeful outlook with a realistic chance of independent walking by 6 months, and intensive rehabilitation to maximise recovery. [1]
DCE short-case approach: neurological examination in suspected GBS
Instruction: "Examine this patient's neurological system. They were admitted three days ago with progressive leg weakness." [1]
Systematic routine
- Observe from the end of the bed: posture, work of breathing (use of accessory muscles, paradoxical abdominal movement suggesting diaphragm weakness), ability to speak in full sentences.
- Bedside respiratory: FVC, single breath count, cough — state you would do this first in a real exam.
- Cranial nerves: facial power (bilateral LMN facial palsy), eye movements (ophthalmoplegia in Miller Fisher), bulbar (cough, gag, swallow), tongue and palate symmetry.
- Motor: tone (flaccid), power (symmetric, legs worse than arms, distal worse early), and reflexes (the key finding — areflexia or hyporeflexia).
- Sensory: pinprick, light touch, joint position sense, vibration — minor glove-and-stocking loss; marked proprioceptive loss in Miller Fisher.
- Coordination: limited by weakness; test for sensory ataxia (positive Romberg) in Miller Fisher.
- Gait: if able — stamping, steppage gait. [1]
Presentation template
"I examined Mr Davies's neurological system. He is alert and cooperative but is using accessory muscles to breathe and speaks in short phrases. [1]
On general inspection there is no facial asymmetry at rest. Cranial nerve examination reveals mild bilateral lower motor neuron facial weakness with reduced eye closure and a symmetrical smile. Eye movements are full. The cough is weak. [1]
In the upper limbs, tone is reduced. Power is 4 out of 5 in the arms proximally and distally, symmetric. In the lower limbs, tone is reduced. Power is 2 out of 5 in the legs, symmetric, with the feet and toes most affected. Reflexes are absent in all four limbs. The plantar responses are downgoing. [1]
Sensation is reduced to pinprick and light touch in a glove-and-stocking distribution. Joint position sense is impaired at the toes. Coordination cannot be tested in the legs due to weakness but is intact in the arms. [1]
In summary, these findings — symmetric flaccid weakness with areflexia and a downgoing plantar, plus bilateral facial palsy and declining respiratory function — are consistent with acute Guillain-Barre syndrome. I would immediately assess his ventilatory capacity with FVC and inspiratory pressures and arrange CSF examination and nerve conduction studies." [1]
Discussion questions
Q: "Why are the plantars downgoing in a patient this weak?" [1]
"Because GBS is a lower motor neuron lesion — the pathology is in the peripheral nerve and nerve root, below the anterior horn cell. Upper motor neuron signs (spasticity, hyperreflexia, extensor plantars) require a lesion of the corticospinal tract. The presence of downgoing plantars in a profoundly weak patient with areflexia is itself a clue that the lesion is in the peripheral nerve, not the spinal cord." [1]
Q: "How would you exclude a spinal cord lesion?" [1]
"The presence of a sensory level, sphincter disturbance (urinary retention can occur in both, but a clear bowel/bladder level is cord-suggestive), and upper motor neuron signs would point to a cord lesion. If any of these were present, or if the weakness were asymmetric or predominantly proximal with sparing of the face, I would arrange urgent MRI of the spine. In a classical symmetric ascending presentation with areflexia and downgoing plantars, MRI is mainly confirmatory or to exclude mimics." [1]
Key DWE MCQ patterns
- The treatment choice: a patient who cannot walk unaided within 4 weeks — give IVIG or plasma exchange, not both, not steroids.
- The CSF pattern: high protein, normal white cells, appearing after 1-2 weeks. A normal first-week CSF does not exclude GBS.
- The variant-antibody match: Miller Fisher and anti-GQ1b; AMAN and anti-GM1 (post-Campylobacter).
- The Miller Fisher triad: ophthalmoplegia, ataxia, areflexia — ataxia is sensory.
- The steroid trap: steroids are ineffective in GBS but effective in CIDP.
- The intubation threshold: FVC below 20 mL/kg or MIP more negative than -30 cmH2O.
- The trigger: Campylobacter jejuni is the most common identifiable antecedent infection.
- The 8-week boundary: progression beyond 8 weeks is CIDP, not GBS. [1]
References
[1] Willison, Jacobs, van Doorn — Guillain-Barre syndrome. The Lancet 2016 comprehensive review of pathogenesis, clinical spectrum, and treatment. [2] Leonhard et al. 2019 — Diagnosis and management of GBS in ten steps; the practical bedside algorithm. [3] van den Berg et al. 2014 — GBS pathogenesis, diagnosis, treatment and prognosis; the Nat Rev Neurol reference. [4] GBS Study Group 1985 — Plasmapheresis RCT (245 patients); established plasma exchange over supportive care. [5] van der Meche and Schmitz 1992 — IVIG at least as effective as plasma exchange (150 patients); the IVIG landmark. [6] PS GBS Trial Group 1997 — PE, IVIG, and combined (383 patients); equivalence; combination not better. [7] van Koningsveld et al. 2004 — Methylprednisolone added to IVIG showed only a non-significant trend; steroids not added routinely. [8] Hughes Cochrane 2016 — Corticosteroids not effective for GBS (and may slow recovery). [9] van Koningsveld et al. 2007 — The Erasmus GBS Outcome Score (EGOS) prognostic model. [10] Walgaard et al. 2010 — Prediction of respiratory insufficiency in GBS (EGRIS inputs). [11] Fokke et al. 2014 — Diagnosis of GBS and validation of the Brighton Collaboration criteria. [12] Chiba et al. 1993 — Anti-GQ1b IgG associated with ophthalmoplegia in Miller Fisher syndrome and GBS. [13] Kusunoki and Kaida 2008 — Antibodies against gangliosides and ganglioside complexes in GBS. [14] Ang, Jacobs et al. 2001 — Campylobacter jejuni LPS from GBS/MFS strains induce anti-GM1 and anti-GQ1b antibodies in rabbits (molecular mimicry proof). [15] Cao-Lormeau et al. 2016 — GBS outbreak associated with Zika virus infection in French Polynesia.
European Academy of Neurology / Peripheral Nerve Society Guideline on GBS (2021); AAN/AANEM Practice Parameter on Immunotherapy for GBS (2012); NICE Guideline NG127 (2021); Therapeutic Guidelines (eTG) Neurology. [1]
References
- [1]Willison HJ, Jacobs BC, van Doorn PA Guillain-Barré syndrome Lancet, 2016.PMID 26948435
- [2]Leonhard SE, Mandarakas MR, Gondim FAA, et al. Diagnosis and management of Guillain-Barré syndrome in ten steps Nat Rev Neurol, 2019.PMID 31541214
- [3]van den Berg B, Walgaard C, Drenthen J, et al. Guillain-Barré syndrome: pathogenesis, diagnosis, treatment and prognosis Nat Rev Neurol, 2014.PMID 25023340
- [4]The Guillain-Barre Syndrome Study Group Plasmapheresis and acute Guillain-Barré syndrome. The Guillain-Barré syndrome Study Group Neurology, 1985.PMID 4022342
- [5]van der Meche FGA, Schmitz PIM A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. Dutch Guillain-Barré Study Group N Engl J Med, 1992.PMID 1552913
- [6]Plasma Exchange/Sandoglobulin Guillain-Barre Syndrome Trial Group Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barré syndrome. Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group Lancet, 1997.PMID 9014908
- [7]van Koningsveld R, Schmitz PIM, van der Meche FGA, et al. Effect of methylprednisolone when added to standard treatment with intravenous immunoglobulin for Guillain-Barré syndrome: randomised trial Lancet, 2004.PMID 14738791
- [8]Hughes RA, Brassington R, Gunn AA, van Doorn PA Corticosteroids for Guillain-Barré syndrome Cochrane Database Syst Rev, 2016.PMID 27775812
- [9]van Koningsveld R, Steyerberg EW, Hughes RA, et al. A clinical prognostic scoring system for Guillain-Barré syndrome Lancet Neurol, 2007.PMID 17537676
- [10]Walgaard C, Lingsma HF, Ruts L, et al. Prediction of respiratory insufficiency in Guillain-Barré syndrome Ann Neurol, 2010.PMID 20517939
- [11]Fokke C, van den Berg B, Drenthen J, et al. Diagnosis of Guillain-Barré syndrome and validation of Brighton criteria Brain, 2014.PMID 24163275
- [12]Chiba A, Kusunoki S, Obata H, et al. Serum anti-GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain-Barré syndrome: clinical and immunohistochemical studies Neurology, 1993.PMID 8413947
- [13]Kusunoki S, Kaida K, Ueda M Antibodies against gangliosides and ganglioside complexes in Guillain-Barré syndrome: new aspects of research Biochim Biophys Acta, 2008.PMID 17976386
- [14]Ang CW, De Klerk MA, Endtz HP, Jacobs BC, et al. Guillain-Barré syndrome- and Miller Fisher syndrome-associated Campylobacter jejuni lipopolysaccharides induce anti-GM1 and anti-GQ1b Antibodies in rabbits Infect Immun, 2001.PMID 11254608
- [15]Cao-Lormeau VM, Blake A, Mons S, et al. Guillain-Barré Syndrome outbreak associated with Zika virus infection in French Polynesia: a case-control study Lancet, 2016.PMID 26948433