Phys · neurological
Motor Neuron Disease
Also known as motor neuron disease · MND · amyotrophic lateral sclerosis · ALS · Lou Gehrig disease · upper and lower motor neuron signs · primary lateral sclerosis · PLS · progressive muscular atrophy · PMA · progressive bulbar palsy · pseudobulbar affect · C9orf72 repeat expansion · SOD1 mutation
Consultant-physician-depth guide to motor neuron disease (amyotrophic lateral sclerosis): degeneration of both upper and lower motor neurons, glutamate excitotoxicity and C9orf72/SOD1 genetics, the combined UMN-and-LMN-in-the-same-region diagnostic hallmark, El Escorial / Awaji / Gold Coast criteria, exclusion of mimics (cervical myelopathy, multifocal motor neuropathy, Kennedy disease), riluzole and edaravone, and multidisciplinary care with non-invasive ventilation, PEG and advance care planning for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Motor Neuron Disease

The answer first
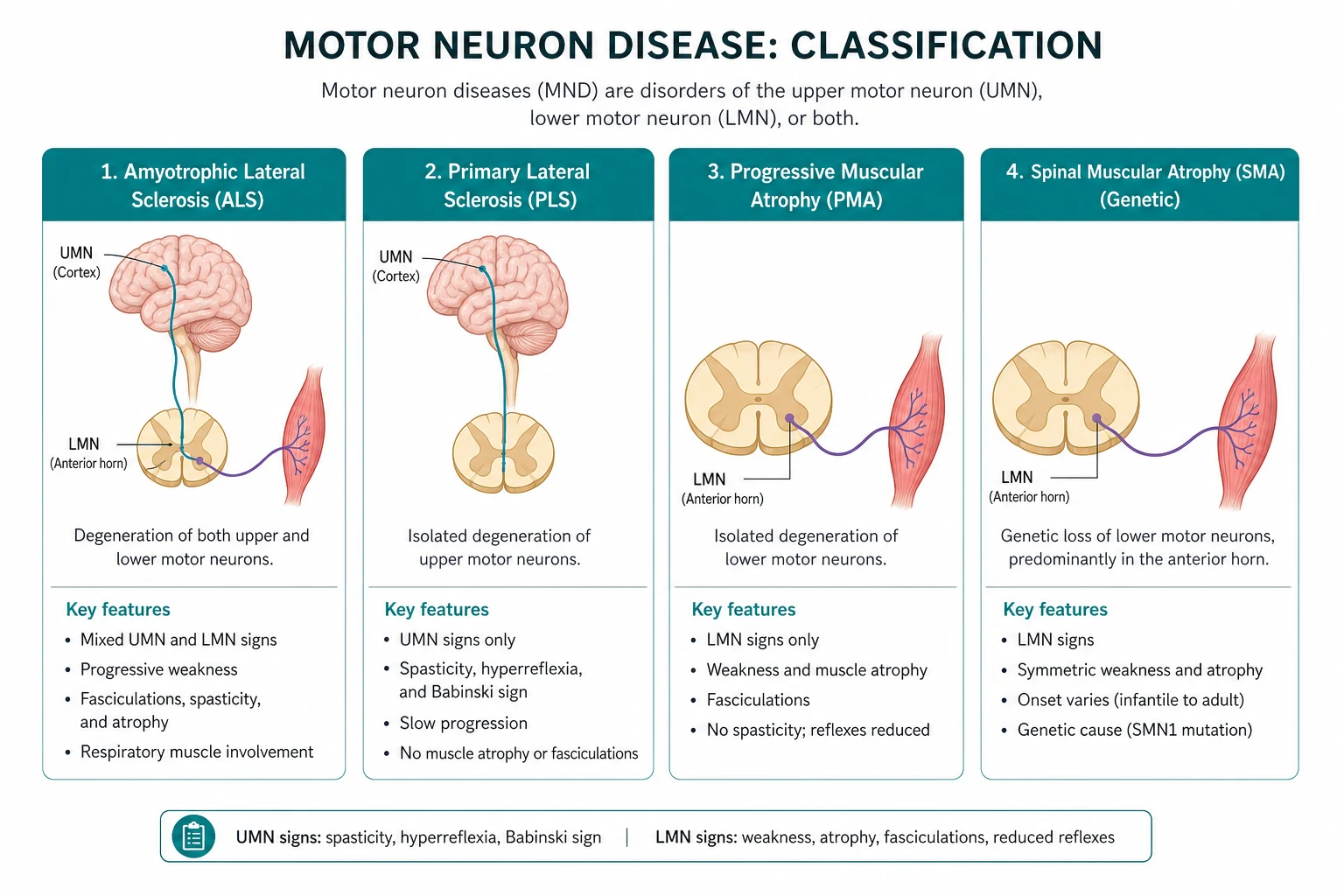
Motor neuron disease (MND) is a progressive, fatal neurodegenerative disorder of motor neurons in which both upper motor neurons (UMN) and lower motor neurons (LMN) degenerate, producing a combination of UMN and LMN signs. Amyotrophic lateral sclerosis (ALS) is the commonest form and the two terms are used interchangeably in practice. The diagnostic signature is UMN and LMN signs coexisting in the same body region (for example, a spastic, hyperreflexic arm that is also wasted and fasciculating). No other common neurological disease does this [9][10].
The diagnosis is clinical, supported by electromyography that confirms widespread LMN denervation and by exclusion of the mimics. The three questions that drive every decision are: is this truly MND (combined UMN and LMN signs in multiple regions, not a mimic)?; which body systems are involved now (bulbar, respiratory, nutritional, cognitive)?; and what is the respiratory status — because respiratory failure is the usual cause of death? [1]
The clinical decision rules a registrar must own at viva: [1]
- Combined UMN and LMN signs in the same region is the hallmark. A wasted, fasciculating tongue with a brisk jaw jerk is bulbar ALS. A spastic leg with an extensor plantar response that is also wasted and areflexic is paradoxical — that paradox is MND. The mistake is to attribute all signs to one process (a cervical myelopathy) when two levels of the neuraxis are involved [2].
- MND is a clinical diagnosis, made by excluding mimics. There is no confirmatory blood test or scan. The job is to demonstrate combined UMN/LMN involvement in multiple regions, confirm active denervation with EMG, and actively exclude cervical myelopathy, multifocal motor neuropathy with conduction block (anti-GM1, treatable), Kennedy disease (X-linked, androgen receptor), B12 deficiency and lead toxicity [8].
- Respiratory failure is the cause of death, and non-invasive ventilation is the single most powerful survival intervention. Median survival without respiratory support is 3 to 5 years; NIV extends survival by a median of 7 to 13 months and improves quality of life [7]. A sniff nasal inspiratory pressure below 40 cmH2O, or an FVC below 50 per cent predicted (or below 1.5 L), is the trigger to discuss and offer NIV.
- Multidisciplinary care and riluzole both extend survival. Attendance at a specialist MND clinic independently prolongs survival [14], and riluzole 50 mg twice daily adds a median of about 3 months [5]. These are the two disease-modifying interventions with robust evidence; everything else is symptom control.
The organising principle is multidisciplinary, symptom-led, prognosis-aware care: make the diagnosis accurately and kindly, exclude the treatable mimics, start riluzole early, surveil respiratory and nutritional function, intervene with NIV and PEG at the right time, and begin advance care planning early. [1]
Pathophysiology — why both motor neurons die
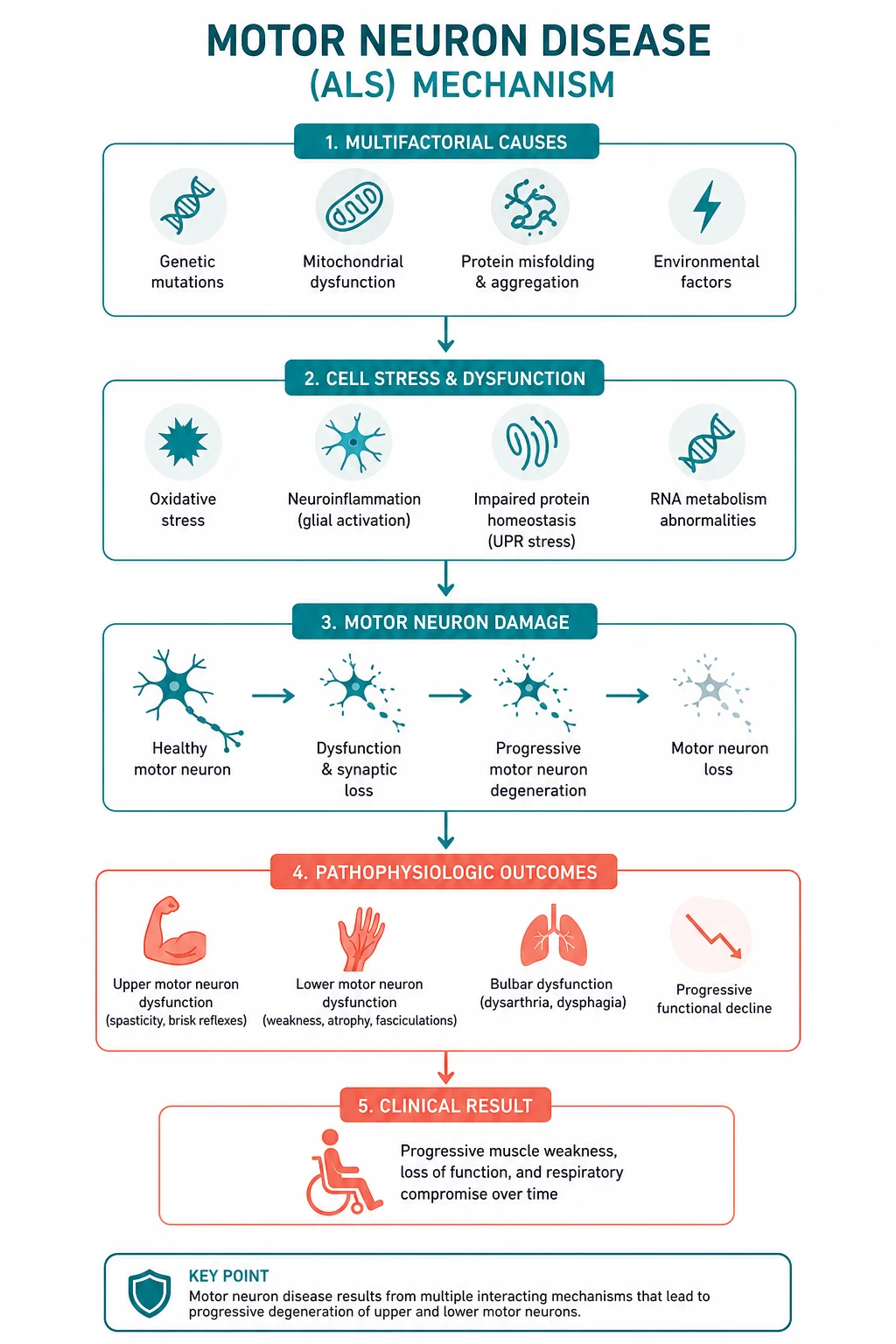

MND is characterised by progressive degeneration of the cortical motor neurons (the large pyramidal Betz cells of the motor cortex and their corticospinal axons — the UMN) and the anterior horn cells of the spinal cord and brainstem motor nuclei (the LMN). The region of onset determines the phenotype: limb cortex and anterior horn give limb-onset ALS; brainstem nuclei (nucleus ambiguus, hypoglossal nucleus) give bulbar-onset disease [9].
Four mechanisms are proposed, and most pathologists regard them as converging on a final common pathway of motor neuron death [10]:
- Glutamate excitotoxicity. Glutamate is the principal excitatory neurotransmitter at the corticomotoneuronal synapse. Astrocytic re-uptake via the EAAT2 transporter is impaired in ALS, leaving excess glutamate in the synaptic cleft. Sustained activation of neuronal AMPA and NMDA receptors drives calcium influx, and motor neurons are unusually vulnerable because they express low levels of calcium-binding proteins and lack the GluR2 subunit that confers calcium impermeability. This is why riluzole, which inhibits glutamate release, works — modestly but reproducibly [5].
- Oxidative stress. About 20 per cent of familial ALS is caused by mutations in SOD1, the gene for copper/zinc superoxide dismutase [12]. Mutant SOD1 is toxic through a gain-of-function (protein misfolding and aggregation) rather than loss of dismutase activity. Free radical damage to lipids, proteins and DNA accumulates. This is the rationale for edaravone, a free-radical scavenger [6].
- Protein aggregation. The pathological hallmark of ALS is the TDP-43-positive inclusion in the cytoplasm of degenerating motor neurons. TDP-43 (transactive response DNA-binding protein 43 kDa) normally lives in the nucleus and regulates RNA; in ALS it mislocalises to the cytoplasm, aggregates, and forms the ubiquitinated inclusions seen in about 97 per cent of cases. Mutations in the TARDBP gene (encoding TDP-43) and FUS (fused in sarcoma) cause rare familial ALS through the same RNA-handling failure.
- Neuroinflammation. Activated microglia and astrocytes surround degenerating motor neurons and release pro-inflammatory cytokines (TNF-alpha, IL-1). The role of inflammation is partly toxic and partly protective; it is a target for therapy but not yet a successful one.
Genetics — C9orf72 is the commonest cause
About 10 per cent of ALS is familial (a first- or second-degree relative affected), and about 70 per cent of familial cases have an identifiable mutation. The four principal genes [11][12]:
| Gene | Inheritance | Frequency | Key phenotype and teaching point |
|---|---|---|---|
| C9orf72 | Autosomal dominant | 25 to 40 per cent of familial ALS; 5 to 10 per cent of sporadic ALS — the commonest single cause | GGGGCC hexanucleotide repeat expansion in intron 1. Causes ALS, frontotemporal dementia, or both. Strongly associated with behavioural-variant FTD, psychosis, and a family history of dementia. The genetic bridge between ALS and FTD |
| SOD1 | Autosomal dominant (some recessive) | 20 per cent of familial ALS | First ALS gene identified (1993). Relatively pure motor syndrome, slow cognition sparing. The target of tofersen (antisense oligonucleotide) |
| TARDBP | Autosomal dominant | 4 to 5 per cent of familial ALS | Encodes TDP-43 — the aggregated protein found in nearly all ALS. Confirms TDP-43 proteinopathy as the core pathology |
| FUS | Autosomal dominant (some recessive) | 4 to 5 per cent of familial ALS | Juvenile-onset ALS, rapidly progressive. FUS is another RNA-binding protein that aggregates in the cytoplasm |
DWE high-yield: The single most testable genetic fact is that C9orf72 is the commonest genetic cause of ALS (and of the ALS-FTD overlap syndrome). SOD1 was the first discovered and is the target of the new antisense oligonucleotide tofersen, but it is no longer the commonest. The TDP-43 inclusion, not the SOD1 mutation, is the pathological hallmark in 97 per cent of cases [11][12].
Clinical spectrum — the signature is combined UMN and LMN signs
The cardinal clinical principle is that UMN and LMN signs coexist. Learn to read them in the same body region, because that coexistence is the diagnostic trigger [2][9].
| Upper motor neuron signs | Lower motor neuron signs |
|---|---|
| Spasticity (velocity-dependent tone increase) | Wasting (atrophy) |
| Hyperreflexia (brisk reflexes, including jaw jerk) | Fasciculations |
| Extensor plantar response (Babinski) | Weakness |
| Clonus | Hyporeflexia or areflexia in a wasted muscle |
| Slowed rapid alternating movements | Muscle cramps |
| Pseudobulbar affect (involuntary laughter or crying) | Hypophonic, nasal speech from palatal weakness |
| Emotional lability, frontal release signs | Tongue wasting and fasciculations |
The clinical paradox that defines ALS: a limb can be spastic (UMN) and wasted (LMN) at once. A wasted hand with brisk finger jerks, or a spastic leg with an extensor plantar that is also weak and cramping, is MND until proven otherwise. Sensation, eye movements, sphincter function and cognition (in the pure motor form) are characteristically spared — sensory or sphincter involvement should prompt a search for an alternative diagnosis [9].
Site of onset and the four body regions
The diagnostic criteria divide the body into four regions: bulbar, cervical, thoracic, lumbar. Onset determines the phenotype: [1]
- Limb-onset ALS (about 70 per cent). The commonest form. Usually begins asymmetrically — a clumsy hand, foot drop, or a dragging leg. A wasted first dorsal interosseous with a brisk finger jerk is the classic examination finding.
- Bulbar-onset ALS (about 25 per cent). Slurred speech (dysarthria) and swallowing difficulty (dysphagia) predominate. Older women are over-represented. Prognosis is worse than limb-onset because respiratory failure arrives sooner and aspiration is a constant threat. A wasted, fasciculating tongue with a brisk jaw jerk is pathognomonic.
- Respiratory-onset ALS (about 5 per cent). Diaphragmatic weakness as the first symptom — presenting as dyspnoea, orthopnoea, or morning headache. Often misdiagnosed as cardiac or pulmonary disease. The most rapidly progressive phenotype.
- Weight loss and cramps may precede weakness by months. [1]
Bulbar involvement
Bulbar disease is feared because it threatens airway, nutrition and communication [8]:
- Dysarthria — a spastic (strained, effortful) or flaccid (nasal, breathy) speech, or a mixed dysarthria. Spastic dysarthria reflects UMN pseudobulbar palsy; flaccid reflects LMN brainstem involvement.
- Dysphagia — first for liquids (nasal regurgitation, coughing), then solids. Leads to weight loss, dehydration and aspiration pneumonia.
- Sialorrhoea (drooling) — not from excess saliva but from impaired swallowing of normal saliva production. Distressing and socially isolating; the target of anticholinergic and botulinum-toxin therapy.
- Pseudobulbar affect — involuntary, emotionally incongruent episodes of laughing or crying, present in up to half of patients. Distinct from depression and treatable with dextromethorphan-quinidine [13].
- Jaw jerk and gag reflex are brisk in pseudobulbar (UMN) disease.
Respiratory involvement — the cause of death
Diaphragmatic and intercostal muscle weakness produces a restrictive ventilatory defect and type 2 respiratory failure. Death is usually from respiratory failure, aspiration pneumonia, or a combination [7].
Symptoms of respiratory muscle weakness that must be asked about at every visit: orthopnoea (breathless lying flat), morning headache (nocturnal hypoventilation with carbon dioxide retention), poor sleep, daytime somnolence, weak cough, and difficulty clearing secretions. A weak, bovine cough is a red flag. [1]
Cognitive involvement — the ALS-FTD spectrum
MND is no longer considered a pure motor disease. About 15 per cent of ALS patients meet criteria for frontotemporal dementia (FTD), and up to 50 per cent have milder cognitive or behavioural impairment detectable on formal testing [9][11]. The features are frontal: executive dysfunction, personality change, apathy, perseveration, loss of insight, and impaired verbal fluency. C9orf72 expansion is strongly associated with the ALS-FTD phenotype. Cognitive impairment matters because it predicts poor adherence to NIV and shorter survival — a patient who cannot manage the mask, or whose family cannot help, will not benefit unless support is built in.
Clinical variants — the spectrum beyond classic ALS
Classic ALS (combined UMN and LMN) is one end of a spectrum. The variants differ in prognosis and in which neuron population dominates [9][10]:
| Variant | Neuron population | Key features | Prognosis |
|---|---|---|---|
| Classic ALS | UMN and LMN | Combined signs, progressive | Median 3 to 5 years |
| Bulbar-onset ALS | UMN and LMN, brainstem | Dysarthria, dysphagia, tongue wasting | Worse — median 2 to 3 years |
| Primary lateral sclerosis (PLS) | UMN only | Slowly progressive spasticity, hyperreflexia, pseudobulbar; no LMN signs for at least 3 to 4 years | Much slower — normal or near-normal lifespan; about 1 per cent per year convert to ALS |
| Progressive muscular atrophy (PMA) | LMN only | Wasting, weakness, fasciculations, areflexia; no UMN signs | Intermediate — median 5 years; many develop UMN signs and reclassify as ALS |
| Progressive bulbar palsy | Bulbar UMN and/or LMN | Predominantly bulbar; often progresses to classic ALS | Poor — median under 2 years from onset |
Viva trap: PLS and PMA are diagnoses made only after prolonged observation. The rule of thumb is that PLS requires UMN-only disease for at least 3 to 4 years before the diagnosis is secure, because most patients who develop LMN signs within that window simply have slowly evolving classic ALS. Do not label a patient "PLS" after 6 months of spasticity. [1]
Differentials — what else wastes and stiffens
The differentials fall into three groups: mimics with a UMN component, mimics with an LMN component, and treatable causes of motor neuropathy that must not be missed [8][9].
The treatable mimics — diagnose or exclude these first
| Mimic | Discriminating feature | Confirmatory test | Why it matters |
|---|---|---|---|
| Cervical myelopathy | LMN signs at the level of the lesion (arms), UMN signs below (legs); neck pain; no bulbar involvement | MRI cervical spine | The most common mimic; surgical decompression may halt progression. Combined arm wasting and leg spasticity can look exactly like ALS |
| Multifocal motor neuropathy (MMN) with conduction block | Asymmetric, predominantly distal LMN weakness; no UMN signs; often affects one arm; slowly progressive; male predominance | NCS shows motor conduction block; anti-GM1 antibodies; normal sensory studies | Treatable with intravenous immunoglobulin. Missing this denies a patient years of function |
| Kennedy disease (X-linked bulbospinal muscular atrophy) | Male, midlife onset; slowly progressive LMN bulbar and limb weakness; gynaecomastia; sensory loss; hand tremor; no UMN signs | Androgen receptor gene CAG repeat expansion (X-linked); NCS shows sensory involvement | Slowly progressive, near-normal lifespan; avoiding unnecessary riluzole and counselling on X-linked inheritance |
| Vitamin B12 deficiency | Subacute combined degeneration: posterior column (vibration/proprioception loss), corticospinal, and neuropathy; sensory signs present | Serum B12, homocysteine, methylmalonic acid | Fully reversible if treated early; always check B12 in a progressive weakness workup |
| Lead toxicity | Motor neuropathy (wrist drop), abdominal pain, gum lead line, microcytic anaemia | Serum lead level; blood film (basophilic stippling) | Treatable with chelation; occupational history is the clue |
| Inclusion body myositis | Older patient; finger flexor and knee extensor weakness; dysphagia; not UMN | Creatine kinase; muscle biopsy; MRI | Steroid-resistant but distinct; misdiagnosis leads to futile immunosuppression |
How to discriminate at the bedside
The single most useful discriminator is sensation. ALS is a pure motor disease — significant sensory loss, a sensory level, or sphincter disturbance points away from ALS and toward myelopathy, neuropathy, or a structural lesion. The second is the distribution: MMN is asymmetric and predominantly upper-limb distal without UMN signs; Kennedy disease is symmetric, slowly progressive, with gynaecomastia and sensory loss; cervical myelopathy spares the bulbar muscles and the legs' LMN signs. [1]
DWE high-yield: A patient with progressive asymmetric hand and arm weakness, no sensory loss, no UMN signs, and a normal MRI spine has multifocal motor neuropathy with conduction block until proven otherwise — send anti-GM1 antibodies and request motor nerve conduction studies looking for conduction block. MMN responds to intravenous immunoglobulin; ALS does not. Confusing the two is a catastrophic diagnostic error. [1]
Diagnosis — clinical, then confirm denervation and exclude mimics

The diagnosis of MND is clinical, made on the basis of progressive motor impairment with combined UMN and LMN signs in multiple body regions, supported by electrophysiology and after exclusion of mimics [1][2][4].
El Escorial criteria (1994) and the Airlie House revision (2000)
The El Escorial criteria classify diagnostic certainty by the number of body regions showing UMN and LMN signs [1][2]:
| Category | Definition |
|---|---|
| Clinically Definite ALS | UMN and LMN signs in three or more regions (bulbar, cervical, thoracic, lumbar) |
| Clinically Probable ALS | UMN and LMN signs in at least two regions, with some UMN signs rostral (above) to the LMN signs |
| Clinically Possible ALS (laboratory-supported) | UMN and LMN signs in one region only, or UMN signs in two or more regions, or LMN signs rostral to UMN — with EMG evidence of denervation and exclusion of mimics |
The requirement for UMN signs to be rostral to LMN signs reflects the direction of corticospinal tract travel — a UMN sign in the leg with an LMN sign in the arm is anatomically consistent with ALS, whereas the reverse is more consistent with a cervical lesion. [1]
Awaji-Shima revision (2008) — increases sensitivity
The Awaji criteria gave electrophysiological evidence of LMN degeneration equal weight to clinical signs and established that fasciculation potentials in an affected muscle count as evidence of active denervation (equivalent to fibrillation potentials and positive sharp waves), provided there are chronic neurogenic changes [3]. This increased diagnostic sensitivity, particularly for bulbar-onset disease and early presentations, and effectively abolished the awkward "laboratory-supported probable" category by allowing EMG to substitute for clinical LMN signs within a region.
Gold Coast criteria (2020) — simplify to ALS or not-ALS
The Gold Coast criteria abandon the categorical (definite / probable / possible) scheme and return a single binary answer: ALS or not ALS [4]. A diagnosis of ALS requires:
- Progressive motor impairment (by history or examination) in regions previously normally functioning.
- UMN and LMN dysfunction in at least one body region — or UMN signs in two or more regions if LMN signs are absent.
- Investigations that exclude other causes (the mimics above). [1]
The Gold Coast criteria were designed for earlier diagnosis and earlier trial entry; many clinics now use them in parallel with the El Escorial categories. [1]
Investigations — what to send and why
The investigation strategy is to confirm widespread denervation and actively exclude mimics [8][9]:
| Investigation | Purpose and interpretation |
|---|---|
| Nerve conduction studies (NCS) | Normal sensory studies (rules out a sensory neuropathy). Motor studies rule out conduction block (MMN) and demyelination (CIDP). Reduced compound muscle action potential amplitudes reflect motor axon loss |
| Electromyography (EMG) | The key confirmatory test. Active denervation: fibrillations and positive sharp waves. Chronic reinnervation: large, long-duration, polyphasic motor unit potentials with reduced recruitment. Must be found in at least two of the four body regions to support the diagnosis (per Awaji). Fasciculation potentials in affected muscles are supportive |
| MRI brain and cervical spine | Excludes cervical myelopathy (the commonest mimic), structural brainstem lesions, and rarely MS. May show increased T2 signal along the corticospinal tracts in advanced ALS |
| Bloods | Full blood count, electrolytes, creatine kinase (mildly elevated in ALS, markedly elevated suggests myopathy), B12, thyroid function, glucose. Exclude reversible causes |
| Anti-GM1 antibodies | If MMN is suspected (asymmetric, no UMN signs, upper-limb distal) — though absence does not exclude MMN |
| Genetic testing | C9orf72, SOD1, TARDBP, FUS — increasingly offered, especially with a family history or young onset, and increasingly relevant since the arrival of gene-specific therapy (tofersen for SOD1) |
| Respiratory function | Forced vital capacity (erect and supine), sniff nasal inspiratory pressure — from diagnosis and at every visit. The single most important prognostic and management test |
DWE high-yield: The EMG in ALS shows active denervation (fibrillations, positive sharp waves) together with chronic reinnervation (large motor units, reduced recruitment) in a widespread distribution, with normal sensory studies and no conduction block. The combination of denervation and reinnervation in the same muscle — the disease is active and chronic at once — is the electrophysiological signature. Conduction block is the feature of MMN and its absence is essential to the ALS diagnosis. [1]
Management — multidisciplinary, disease-modifying, and symptom-led


The evidence-based management of MND rests on three pillars: multidisciplinary care (the framework that delivers every other intervention), disease-modifying drugs (modest but real), and symptom management (where most of the quality-of-life benefit is won) [8][14].
Pillar 1 — Multidisciplinary clinic care
Attendance at a specialist multidisciplinary ALS clinic independently prolongs survival (by a median of about 7 to 8 months in population studies) and improves quality of life, almost certainly by ensuring timely and coordinated intervention [14]. The team includes a neurologist, respiratory physician, speech and language therapist, dietitian, occupational therapist, physiotherapist, specialist MND nurse, palliative care, clinical psychology, and social work. The patient should be referred to a specialist clinic at diagnosis.
Pillar 2 — Disease-modifying drugs
| Drug | Mechanism | Benefit | Dose and monitoring | Key teaching point |
|---|---|---|---|---|
| Riluzole | Glutamate antagonist — inhibits presynaptic glutamate release | Median survival benefit of about 3 months | 50 mg orally twice daily. Monitor liver function (transaminitis) and full blood count (neutropenia) at baseline, monthly for 3 months, then 3-monthly. Reduce dose in hepatic impairment | The first and best-established disease-modifying drug. Start early; the benefit is greater the earlier it is begun. Modest, but real and reproducible [5] |
| Edaravone | Free-radical scavenger — reduces oxidative stress | Modest slowing of functional decline on the ALSFRS-R in a selected early-stage population (2.49-point difference over 24 weeks in the Study 19 trial) | Intravenous infusion 60 mg over 60 minutes, daily for 14 days then a 14-day rest cycle; oral formulation available in some regions. Monitor for hypersensitivity | Benefit confined to a well-defined early-stage group (within 2 years of diagnosis, mild-moderate severity, FVC above 80 per cent). Not a respiratory drug; does not extend survival in advanced disease [6] |
Emerging and gene-specific therapies: tofersen, an antisense oligonucleotide targeting SOD1 mRNA, slows biomarker and functional decline in SOD1-mutant ALS and is a model for gene-targeted treatment. It is not yet routine practice for all MND but is transforming the approach to SOD1-confirmed disease. Stem cell, immunomodulatory and anti-inflammatory trials have not shown benefit and are not recommended outside trials [10].
Pillar 3 — Symptom management
Symptom control is the bulk of day-to-day MND care and delivers most of the measurable quality-of-life benefit [8]:
| Symptom | First-line management | Notes |
|---|---|---|
| Sialorrhoea | Hyoscine hydrobromide patch (1 mg every 72 hours), glycopyrrolate, or sublingual atropine. Refractory: botulinum toxin injection into the salivary glands, or saliva suction devices | Avoid anticholinergics if thick secretions or urinary retention predominate. Radiotherapy to salivary glands is a last resort |
| Pseudobulbar affect | Dextromethorphan-quinidine 20 mg/10 mg twice daily (or dextromethorphan alone where combined formulation unavailable); SSRIs (e.g. citalopram) as an alternative [13] | Distressing and under-recognised. Quinidine carries QT-prolongation risk — check ECG |
| Spasticity | Baclofen (oral; start 5 mg three times daily, titrate to 60 to 80 mg daily); tizanidine; physiotherapy and stretching. Severe: intrathecal baclofen pump | Avoid over-treatment that converts useful tone into disabling flaccidity |
| Cramps | Quinine sulphate (limited by cardiac and haematological toxicity), magnesium, baclofen, physiotherapy | Often the earliest symptom; may precede weakness by months |
| Pain | Simple analgesia first; neuropathic agents (gabapentin, pregabalin, amitriptyline) for neuropathic pain; opioids for pain and for dyspnoea in advanced disease | Pain is common (up to two-thirds) and often musculoskeletal, from immobility and loss of joint range |
| Constipation | Adequate hydration (where safe), dietary fibre, osmotic laxatives (macrogol), stimulants if required | Multifactorial: immobility, weak abdominal muscles, anticholinergics, low fluid intake |
| Dysarthria / communication | Speech and language therapy; augmentative and alternative communication (AAC) — from alphabet boards to eye-gaze voice-output devices | Plan early; set up AAC before speech is lost. Voice banking is increasingly offered |
| Anxiety and depression | SSRIs, psychological support, MND association peer support | Common and treatable; do not assume distress is solely a reaction to diagnosis |
| Cognitive impairment / FTD | Cognitive screening (ECAS — Edinburgh Cognitive and Behavioural ALS Screen); carer education and support; involve family in decision-making | Impairs adherence to NIV and PEG; a major negative prognostic factor |
Respiratory failure and non-invasive ventilation — the single biggest survival gain
Respiratory muscle weakness is the central clinical problem in MND because it is the usual cause of death. The intervention with the largest single survival benefit is non-invasive ventilation (NIV), which extends median survival by 7 to 13 months and improves sleep quality, daytime symptoms and quality of life [7].
When to start NIV — the thresholds
NIV should be offered (not merely discussed) when any of the following is present [7][8]:
- Symptoms of respiratory muscle weakness (orthopnoea, morning headache, poor sleep, daytime somnolence, weak cough) and evidence of respiratory muscle weakness on testing.
- Sniff nasal inspiratory pressure (SNIP) below 40 cmH2O — the most sensitive measure of diaphragmatic strength and the preferred surveillance test, falling before FVC.
- Forced vital capacity (FVC) below 50 per cent predicted (or below 1.5 L), or a supine FVC significantly lower than erect FVC (a fall of more than 25 per cent suggests diaphragmatic weakness).
- Symptomatic nocturnal hypoventilation or daytime hypercapnia (pCO2 above 45 mmHg) — a late sign indicating established failure. [1]
Viva trap: Do not wait for daytime hypercapnia — it is a late and ominous sign. The point of surveillance (SNIP every 3 months, FVC erect and supine every 3 months) is to detect and treat respiratory failure before the patient decompensates. A patient with daytime hypercapnia is heading for an emergency intubation or a comfort-care pathway, depending on goals of care. [1]
How NIV is delivered
Bilevel positive airway pressure (BiPAP), titrated to control symptoms and correct nocturnal hypoventilation, delivered via a nasal or full-face mask, increasing from a few hours overnight to daytime use as the disease progresses. Patient and family education, mask fitting, and anticipatory troubleshooting are essential — NIV fails most often because of poor tolerance or carer overload, not because of respiratory deterioration. Patients with severe bulbar weakness benefit less from NIV in survival terms but may still gain symptomatic relief. Tracheostomy ventilation is an option in selected, well-counselled patients but shifts the burden of care profoundly and is chosen by a minority. [1]
Nutrition and percutaneous endoscopic gastrostomy (PEG)
Weight loss and dysphagia predict shorter survival independently of respiratory function. Nutritional support improves quality of life and may extend survival [8].
PEG (or radiologically inserted gastrostomy, RIG) is indicated when there is symptomatic dysphagia, accelerating weight loss (more than 10 per cent of body weight), or dehydration. The critical principle is timing: PEG is safest and most effective when performed before respiratory function declines below an FVC of 50 per cent predicted (or a SNIP below 40 cmH2O), because the procedure and sedation carry higher risk in established respiratory failure. This means respiratory function must be checked at every clinic visit so that PEG, if it is wanted, is placed while it is still safe. Where respiratory function has already declined, NIV during the procedure (periprocedural NIV) reduces risk. RIG is preferred over PEG in patients with significant respiratory compromise. [1]
Advance care planning and end-of-life care
MND is a life-limiting diagnosis, and advance care planning must begin early, not at the terminal phase, while the patient retains the capacity to decide [8].
The conversation covers: the disease trajectory and likely future decisions; the patient's priorities and fears; advance directives and the appointment of a substitute decision-maker; the role and limits of NIV, PEG and tracheostomy; and the transition to palliative care. Palliative care is involved from diagnosis in many services and intensifies as the disease advances. At the end of life, the priorities are relief of dyspnoea (opioids — they relieve breathlessness without shortening survival when titrated to symptom), secretions (hyoscine), pain, and anxiety, and support for the family. The principle is that withholding life-prolonging treatment that the patient does not want is not euthanasia — it is good palliative care. [1]
Prognosis — be honest and specific
Median survival from symptom onset is 3 to 5 years; about 10 per cent of patients survive 10 years or more [9][15]. Adverse prognostic factors:
- Bulbar onset — median 2 to 3 years; worse than limb onset.
- Older age at onset and short diagnostic delay (a short delay reflects aggressive disease, not late presentation).
- Respiratory involvement early, or a low FVC/SNIP at diagnosis.
- Frontotemporal dementia or significant cognitive impairment — halves survival, partly through poor NIV adherence.
- Poor nutritional status and weight loss.
- C9orf72 expansion — slightly better or similar to sporadic ALS for motor outcomes but with the added FTD burden. [1]
Favourable factors include limb (especially lower-limb) onset, younger age, PLS phenotype (UMN only, slowly progressive), and engagement with a multidisciplinary clinic [14].
DCE long-case approach
Opening statement (SASPOP)
"This is Mrs R, a 64-year-old retired teacher who presents with a 14-month history of progressive difficulty walking and weakness of the right hand, and a 3-month history of slurred speech and weight loss. Her main problems are a clinically definite amyotrophic lateral sclerosis with bulbar involvement, established but under-monitored respiratory muscle weakness, nutritional decline, and the psychological impact of a new diagnosis." [1]
Problem list
- Clinically definite ALS (combined UMN and LMN signs in bulbar, cervical and lumbar regions) — confirmed by EMG, MRI spine and exclusion of mimics.
- Respiratory muscle weakness — orthopnoea, SNIP 38 cmH2O, FVC 48 per cent predicted. Urgent NIV initiation required.
- Bulbar involvement — dysarthria, dysphagia for liquids, sialorrhoea; weight loss 12 per cent in 6 months. PEG indicated and must precede further respiratory decline.
- Pseudobulbar affect and reactive depression.
- Reduced functional independence — mobility, transfers, self-care; home modifications and equipment needed.
- Advance care planning — not yet commenced; capacity currently intact. [1]
Integrated management plan
- Confirm diagnosis (done), communicate clearly and compassionately, refer to specialist MND clinic and MND association.
- Start riluzole 50 mg twice daily with baseline and monitoring liver function and full blood count [5].
- Initiate NIV — SNIP below 40 cmH2O and symptomatic; this is the single most important intervention for survival [7].
- Insert PEG before respiratory function declines further — FVC already below 50 per cent predicted, so coordinate with respiratory team for periprocedural NIV and consider RIG [8].
- Symptom control: hyoscine patch for sialorrhoea, baclofen for spasticity, dextromethorphan-quinidine for pseudobulbar affect [13].
- Allied health: speech therapy (AAC and voice banking), dietitian, occupational therapist (home assessment, equipment), physiotherapy (maintain mobility, prevent contractures), specialist nurse.
- Begin advance care planning now: goals, advance directive, substitute decision-maker, discussion of future mechanical ventilation decisions [8].
- Palliative care involvement from diagnosis; psychological support for patient and family.
- Screen for cognitive impairment (ECAS) and counsel on the ALS-FTD spectrum.
Probing questions and model answers
"Why did you choose riluzole and what will you tell the patient about its benefit?" — Riluzole is a glutamate antagonist shown in the Bensimon 1994 trial and subsequent Cochrane meta-analyses to extend median survival by about 3 months [5]. I will tell the patient honestly that it is not a cure, that it modestly prolongs life and is generally well tolerated, and that it is most effective started early. I monitor liver function and full blood count.
"When will you start NIV and how did you decide?" — She has symptoms (orthopnoea) and objective respiratory muscle weakness (SNIP 38 cmH2O, FVC 48 per cent predicted), both of which cross guideline thresholds for offering NIV [7][8]. I will not wait for daytime hypercapnia. I will titrate bilevel pressure to symptoms and nocturnal oxygenation, and educate the patient and family on managing the mask, because tolerance and carer capacity — not physiology — usually determine success.
"Why must the PEG go in now and not later?" — Because her FVC is already below 50 per cent predicted, the procedural risk of sedation is rising; if I wait, PEG may become unsafe. I will coordinate periprocedural NIV and discuss RIG with the interventional team. The principle is that PEG is timed by respiratory function, not by appetite. [1]
DCE short-case approach: neurological examination for UMN and LMN signs
Instruction: "Examine this patient's upper and lower limbs neurologically. They have been having difficulty with their hands and walking." [1]
Systematic examination routine
- Inspection — look for wasting (thenar eminence, first dorsal interosseous, intrinsic foot muscles, quadriceps, tongue), fasciculations (observe the tongue at rest in the mouth, never protruded), muscle bulk, and posture.
- Tone — assess upper and lower limb tone; look for spasticity (the catch) in the legs and arms.
- Power — test each muscle group (MRC 0 to 5), proximal and distal, comparing sides. Note the pattern of weakness.
- Reflexes — biceps, triceps, supinator, finger jerks, knee and ankle jerbs; plantar responses. Document brisk versus absent, and look for the paradox of a wasted muscle with a preserved or brisk reflex.
- Coordination — rapid alternating movements, finger-nose, heel-shin. Note that spasticity slows these, not cerebellar ataxia.
- Sensation — normal in MND. Test all modalities to confirm.
- Bulbar and cranial nerves — examine the tongue (wasting, fasciculations), palate movement, jaw jerk (brisk in pseudobulbar), and gag. Assess speech.
- Walk — spastic gait, foot drop. [1]
Key signs the patient demonstrates
A wasted, fasciculating tongue with a brisk jaw jerk; wasted first dorsal interosseous and thenar muscles with fasciculations in the forearms; brisk reflexes in wasted, weak arms; spasticity and hyperreflexia in the legs with extensor plantar responses; preserved sensation throughout; a weak, slow, spastic gait. [1]
Presentation template
"I have examined Mr P's neurological system. The key findings are of combined upper and lower motor neuron signs in the same body regions: there is wasting and fasciculation of the tongue and intrinsic hand muscles with a brisk jaw jerk, and spasticity, hyperreflexia and extensor plantar responses in the legs. Sensation is intact and there are no cerebellar or extrapyramidal features. The most likely diagnosis is motor neuron disease (amyotrophic lateral sclerosis). I would confirm this with electromyography to demonstrate widespread active denervation and chronic reinnervation, MRI of the cervical spine to exclude cervical myelopathy, and a screen for treatable mimics including anti-GM1 antibodies and vitamin B12." [1]
Discussion questions
"What is the differential from these signs?" — The combined UMN and LMN pattern in the same region, with bulbar involvement and spared sensation, is highly specific for ALS. The principal mimic is cervical myelopathy (which would not explain the bulbar signs), multifocal motor neuropathy (no UMN signs), and Kennedy disease (slower, sensory loss, gynaecomastia). The preserved sensation argues against B12 deficiency and a sensory neuropathy. [1]
"What would the EMG show?" — Active denervation (fibrillations and positive sharp waves) together with chronic reinnervation (large, polyphasic motor unit potentials with reduced recruitment) in at least two of the four body regions, with normal sensory studies and no conduction block [3].
Key DWE MCQ patterns
- Combined UMN and LMN signs in the same region — the bedside diagnosis of ALS. A wasted, fasciculating tongue with a brisk jaw jerk, or a spastic hyperreflexic arm that is also wasted, is MND until proven otherwise [2].
- Multifocal motor neuropathy — the treatable mimic. Asymmetric, predominantly distal, upper-limb weakness, no UMN signs, normal sensation, and a response to intravenous immunoglobulin. Look for conduction block on NCS and anti-GM1 antibodies [9].
- Cervical myelopathy — the structural mimic. LMN signs in the arms at the level of the lesion and UMN signs in the legs, with neck symptoms and no bulbar involvement. The discriminating feature from ALS is the absence of bulbar and rostral signs.
- NIV is the single biggest survival intervention. A symptomatic patient with a SNIP below 40 cmH2O or an FVC below 50 per cent predicted should be offered NIV; median survival benefit 7 to 13 months [7].
- Riluzole extends survival by about 3 months — start early, monitor liver function and full blood count. Edaravone gives modest functional benefit in a selected early-stage group, not a survival benefit [5][6].
- C9orf72 is the commonest genetic cause of ALS and the ALS-FTD overlap; SOD1 was first discovered and is the target of tofersen; the TDP-43 inclusion is the pathological hallmark in 97 per cent [11][12].
- Pseudobulbar affect is treatable with dextromethorphan-quinidine — distinct from depression; check QT interval before starting [13].
- Kennedy disease — the slowly progressive X-linked mimic. Male, midlife, gynaecomastia, sensory loss, hand tremor, no UMN signs; androgen receptor CAG repeat expansion [9].
References
[1] Brooks BR (1994) — El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. The original criteria classifying ALS as definite, probable or possible based on UMN and LMN signs across four body regions (bulbar, cervical, thoracic, lumbar). The framework still underpinning MND diagnosis.
[2] Brooks BR, Miller RG, Swash M, Munsat TL (2000) — El Escorial revisited (Airlie House). The revised criteria refining the regions, requiring UMN signs rostral to LMN signs for probable ALS, and introducing the laboratory-supported category. Examiners still test the categorical scheme.
[3] de Carvalho M, Dengler R, Eisen A, et al. (2008) — Electrodiagnostic criteria for diagnosis of ALS (Awaji consensus). Gave EMG equal weight to clinical LMN signs and established that fasciculation potentials count as evidence of active denervation. Increased sensitivity, especially for bulbar and early disease.
[4] Shefner JM, Al-Chalabi A, Baker MR, et al. (2020) — Gold Coast criteria for ALS. A proposal that abandons the definite/probable/possible categories for a binary ALS-or-not-ALS outcome, requiring progressive motor impairment with UMN and LMN dysfunction in at least one region and exclusion of mimics. The current direction of travel.
[5] Bensimon G, Lacomblez L, Meininger V (1994) — A controlled trial of riluzole in ALS. The landmark trial showing that riluzole 100 mg daily extended survival. The basis for riluzole as the first disease-modifying drug in MND.
[6] Writing Group, Edaravone ALS 19 Study Group (2017) — Safety and efficacy of edaravone in well defined ALS patients. A 24-week randomised trial showing a 2.49-point ALSFRS-R benefit in a selected early-stage population. Defines the narrow group in whom edaravone helps.
[7] Bourke SC, Tomlinson M, Williams TL, et al. (2006) — Effects of non-invasive ventilation on survival and quality of life in ALS. The randomised trial establishing NIV as the single most powerful survival intervention (median benefit 205 days in good-bulbar-function patients) and the foundation of respiratory surveillance thresholds.
[8] Andersen PM, Abrahams S, Borasio GD, et al. (2012) — EFNS guidelines on the clinical management of ALS (MALS), revised. The European guideline covering diagnosis, multidisciplinary care, riluzole, respiratory support (NIV), nutritional support (PEG), symptom management and palliative care. Now supplemented by the 2024 EAN/ERN EURO-NMD guideline.
[9] Kiernan MC, Vucic S, Cheah BC, et al. (2011) — Amyotrophic lateral sclerosis. The Lancet seminar: clinical features (combined UMN/LMN, bulbar, respiratory, cognitive), diagnosis (criteria, EMG), pathophysiology and management. The single best overview for fellowship preparation.
[10] Brown RH, Al-Chalabi A (2017) — Amyotrophic Lateral Sclerosis (NEJM). Authoritative review of pathophysiology (glutamate excitotoxicity, oxidative stress, protein aggregation, neuroinflammation), genetics, clinical spectrum and therapy, including gene-targeted treatment.
[11] DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. (2011) — Expanded GGGGCC hexanucleotide repeat in C9ORF72 causes chromosome 9p-linked FTD and ALS. The discovery that the C9orf72 expansion is the commonest genetic cause of ALS and of the ALS-FTD overlap, the genetic bridge between the two diseases.
[12] Rosen DR, Siddique T, Patterson D, et al. (1993) — Mutations in Cu/Zn superoxide dismutase gene are associated with familial ALS. The first ALS gene identified, establishing SOD1 and oxidative stress in ALS pathogenesis and founding the gene-targeted therapy era.
[13] Brooks BR, Thisted RA, Appel SH, et al. (2004) — Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine: a randomised trial. The evidence that dextromethorphan-quinidine (AVP-923) reduces pseudobulbar affect episodes in ALS, establishing a specific treatment for a distressing and under-recognised symptom.
[14] Traynor BJ, Alexander M, Corr B, Frost E, Hardiman O (2003) — Effect of a multidisciplinary ALS clinic on ALS survival. The population-based study showing that multidisciplinary clinic attendance independently prolongs survival (median about 7.5 months), the evidence base for the multidisciplinary model as standard care.
[15] Hardiman O, Al-Chalabi A, Brayne C, et al. (2017) — The changing picture of amyotrophic lateral sclerosis: lessons from European registers. Epidemiology and prognosis from population registers, including median survival 3 to 5 years and the 10 per cent 10-year survival figure, plus the recognised phenotypic and cognitive spectrum.
References
- [1]Brooks BR El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial Clinical limits of amyotrophic lateral sclerosis workshop contributors J Neurol Sci, 1994.PMID 7807156
- [2]Brooks BR, Miller RG, Swash M, Munsat TL El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis Amyotroph Lateral Scler Other Motor Neuron Disord, 2000.PMID 11464847
- [3]de Carvalho M, Dengler R, Eisen A, et al. Electrodiagnostic criteria for diagnosis of ALS Clin Neurophysiol, 2008.PMID 18164242
- [4]Shefner JM, Al-Chalabi A, Baker MR, et al. A proposal for new diagnostic criteria for ALS Clin Neurophysiol, 2020.PMID 32387049
- [5]Bensimon G, Lacomblez L, Meininger V A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group N Engl J Med, 1994.PMID 8302340
- [6]Writing Group on behalf of the Edaravone ALS 19 Study Group (Okita M, Abe K, Itoyama Y, et al.) Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial Lancet Neurol, 2017.PMID 28522181
- [7]Bourke SC, Tomlinson M, Williams TL, Bullock RE, Shaw PJ, Gibson GJ Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trial Lancet Neurol, 2006.PMID 16426990
- [8]Andersen PM, Abrahams S, Borasio GD, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)--revised report of an EFNS task force Eur J Neurol, 2012.PMID 21914052
- [9]Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis Lancet, 2011.PMID 21296405
- [10]Brown RH, Al-Chalabi A Amyotrophic Lateral Sclerosis N Engl J Med, 2017.PMID 28700839
- [11]DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS Neuron, 2011.PMID 21944778
- [12]Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis Nature, 1993.PMID 8446170
- [13]Brooks BR, Thisted RA, Appel SH, et al. Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine: a randomized trial Neurology, 2004.PMID 15505150
- [14]Traynor BJ, Alexander M, Corr B, Frost E, Hardiman O Effect of a multidisciplinary amyotrophic lateral sclerosis (ALS) clinic on ALS survival: a population based study, 1996-2000 J Neurol Neurosurg Psychiatry, 2003.PMID 12933930
- [15]Hardiman O, Al-Chalabi A, Brayne C, et al. The changing picture of amyotrophic lateral sclerosis: lessons from European registers J Neurol Neurosurg Psychiatry, 2017.PMID 28285264