Phys · neurological
Movement Disorders
Also known as Parkinson disease · Parkinson's disease · parkinsonism · atypical parkinsonism · multiple system atrophy · progressive supranuclear palsy · corticobasal degeneration · dementia with Lewy bodies · essential tremor · dystonia · Huntington disease · Wilson disease · restless legs syndrome · Tourette syndrome · tic disorders · chorea
Consultant-physician-depth guide to parkinsonism and the hyperkinetic movement disorders for FRACP DWE and DCE — Parkinson's disease pathology and the levodopa escalation ladder, atypical parkinsonism (MSA, PSP, CBD, DLB), drug-induced parkinsonism, essential tremor, dystonia, Huntington disease, Wilson disease, restless legs syndrome and tic disorders.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Movement Disorders
The answer first
Movement disorders divide into two families by the dominant motor sign: hypokinetic-rigid (too little movement — parkinsonism) and hyperkinetic (too much movement — tremor, chorea, dystonia, tics, myoclonus). The single most discriminating bedside observation is whether the problem is under-activity with slowness and stiffness, or excess, involuntary movement. [1]
For parkinsonism, the three questions that determine everything are: [1]
- Is it idiopathic Parkinson's disease (PD), or one of the atypical, secondary, or mimicking disorders? The most common diagnostic error at the bedside is calling everything parkinsonian "Parkinson's disease." Roughly 1 in 4 patients labelled as PD in the community do not have it.
- What is the dominant functional problem — motor (bradykinesia, gait, tremor, dyskinesia) or non-motor (cognition, mood, sleep, autonomic, hallucinations)? Management follows the dominant problem.
- Are there red flags for an atypical or secondary cause? Early falls, rapid progression, symmetrical signs, absent rest tremor, early autonomic failure, early dementia, poor levodopa response — each of these is a flag to stop, reconsider, and not simply escalate levodopa. [1]
For PD, levodopa (combined with a peripheral dopa-decarboxylase inhibitor — carbidopa or benserazide) remains the single most effective symptomatic therapy and the gold standard. There is no role for "levodopa-sparing" fear — delaying levodopa does not prevent motor complications and denies the patient the best treatment available. [1]
DWE high-yield trap: When asked "best initial treatment for a 70-year-old with disabling motor features of Parkinson's disease," the answer is levodopa, not a dopamine agonist or MAO-B inhibitor. The PD MED trial (PMID 24928805) settled this — levodopa gave the best mobility and quality-of-life outcomes. Reserve agonists for younger patients with mild symptoms or to delay levodopa where dyskinesia risk is the dominant concern. [1]
Classification
Hypokinetic-rigid (parkinsonian)
| Syndrome | One-line discriminator |
|---|---|
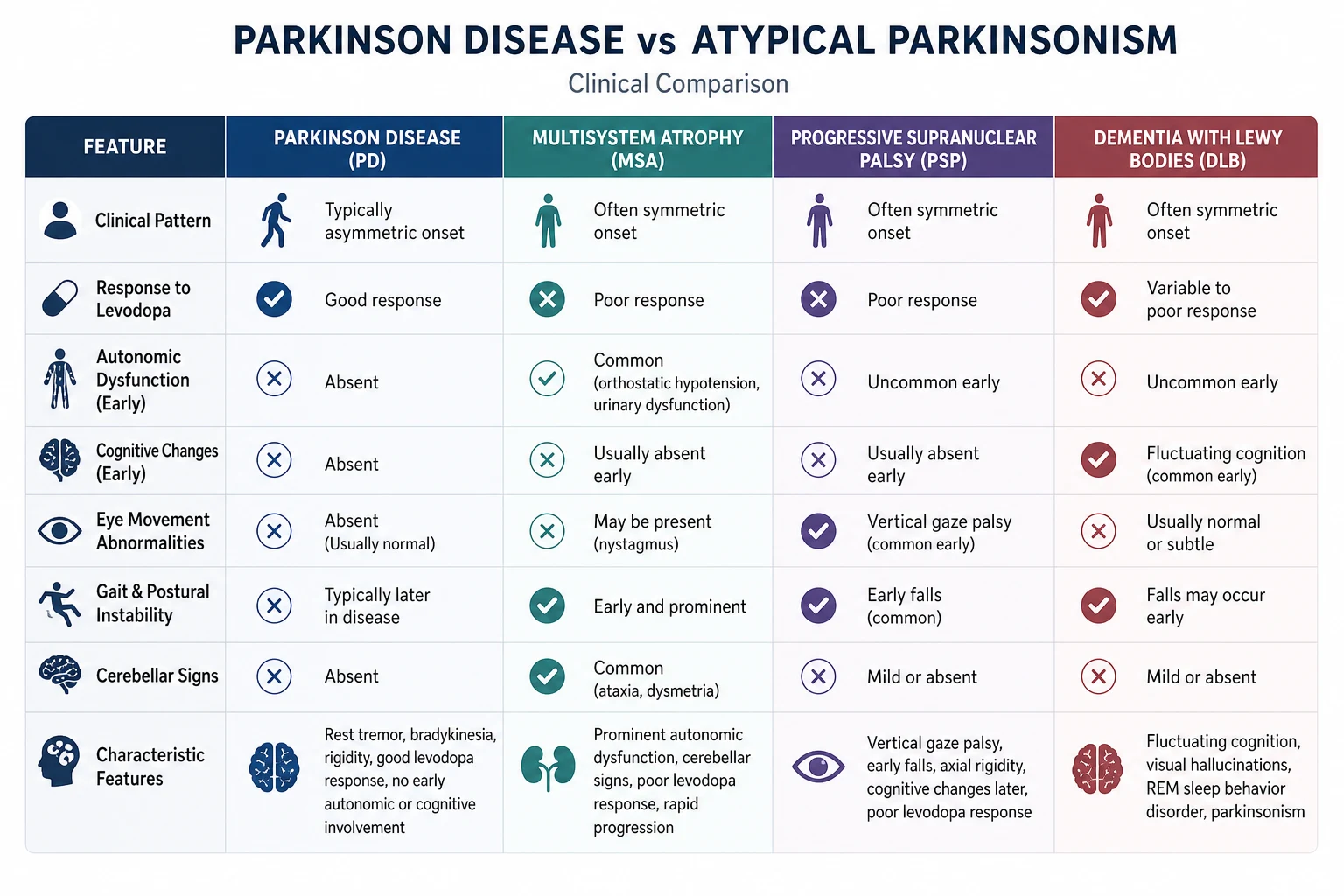
| Idiopathic Parkinson's disease | Asymmetric rest tremor, good sustained levodopa response, slow progression |
| Multiple system atrophy (MSA) | Parkinsonism plus cerebellar and/or prominent autonomic failure (orthostatic hypotension, urinary urgency/incontinence); cerebellar subtype MSA-C has prominent ataxia; parkinsonian subtype MSA-P resembles PD but responds poorly to levodopa |
| Progressive supranuclear palsy (PSP) | Vertical supranuclear gaze palsy, early axial rigidity, early recurrent falls (within first year), poor levodopa response |
| Corticobasal degeneration (CBD) | Marked asymmetric cortical signs — alien limb, apraxia, cortical sensory loss, myoclonus; asymmetric parkinsonism |
| Dementia with Lewy bodies (DLB) | Fluctuating cognition, detailed visual hallucinations, spontaneous parkinsonism, REM sleep behaviour disorder |
| Vascular parkinsonism | Lower body parkinsonism, gait apraxia, pyramidal signs, stepwise course, vascular risk factors |
| Drug-induced parkinsonism | Exposure to dopamine blocker (antipsychotic, metoclopramide, prochlorperazine); usually symmetric; resolves over weeks to months after withdrawal |
Hyperkinetic
| Movement | Description | Classical disorder |
|---|---|---|
| Tremor | Rhythmic oscillation of a body part | PD (rest), essential tremor (postural/action), enhanced physiological tremor |
| Chorea | Brief, irregular, non-rhythmic, flowing movements that move from one body part to another | Huntington disease, Sydenham chorea, drug-induced (levodopa), metabolic |
| Dystonia | Sustained co-contraction of agonist and antagonist muscles causing abnormal postures | Cervical dystonia, task-specific (writer's cramp), generalised dystonia |
| Tics | Brief, sudden, stereotyped movements or vocalisations; suppressible but with internal urge | Tourette syndrome, chronic tic disorders |
| Myoclonus | Sudden, shock-like jerks | Cortical, subcortical, spinal, metabolic (hepatic/renal failure) |
| Athetosis | Slow, writhing, distal movements | Cerebral palsy, Wilson disease |
Parkinson's disease — pathology and pathophysiology
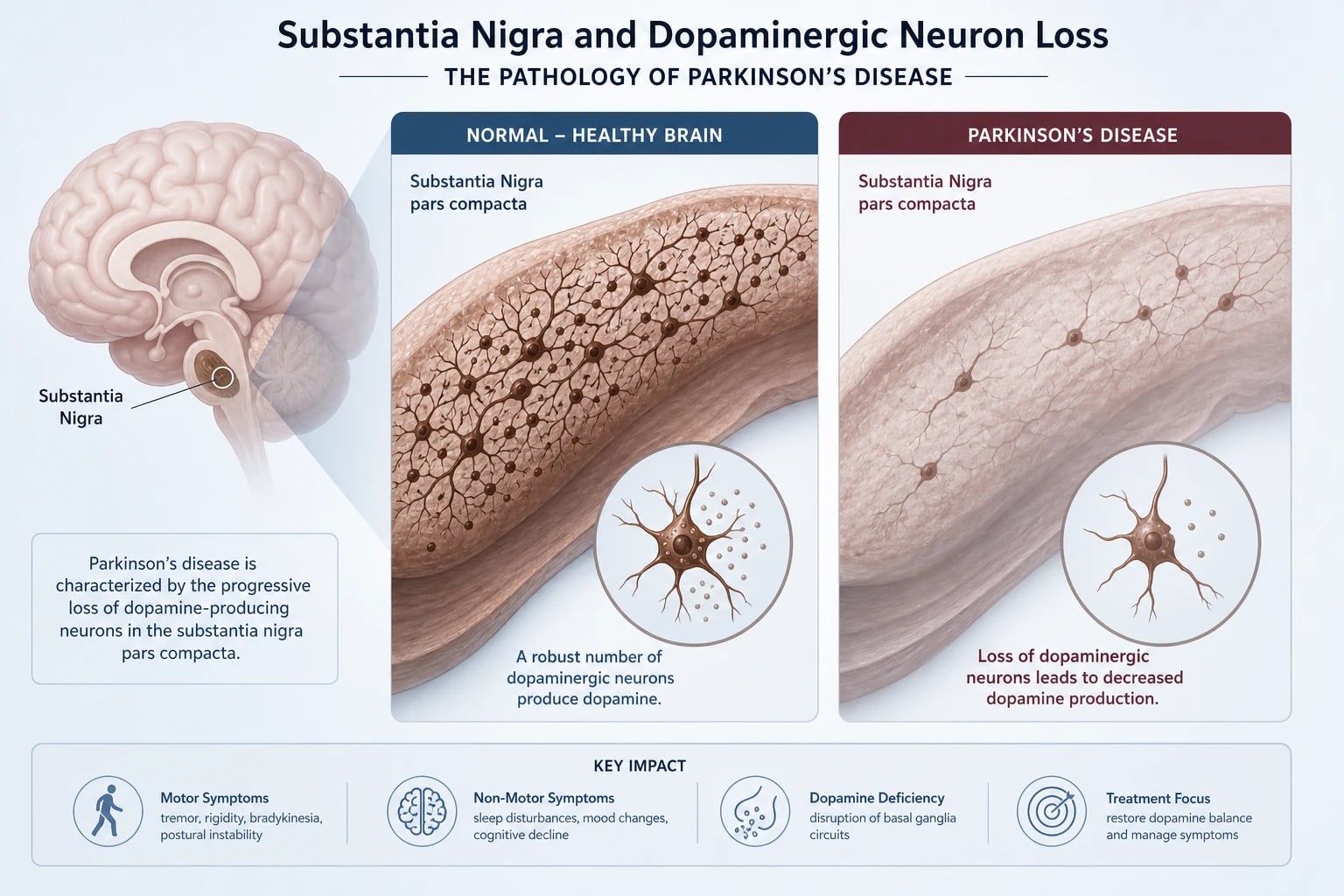
The substantia nigra and dopaminergic neuron loss
PD is defined pathologically by progressive loss of dopaminergic neurons in the pars compacta of the substantia nigra (in the midbrain). These neurons project via the nigrostriatal pathway to the striatum (putamen and caudate) and normally maintain a steady tonic release of dopamine that facilitates voluntary movement through the direct pathway (D1 receptor, facilitatory) and inhibits the indirect pathway (D2 receptor, inhibitory). [1]
When these neurons die, the direct pathway is underactive (less facilitation of movement) and the indirect pathway is overactive (excessive inhibition of the thalamocortical drive). The clinical consequence is bradykinesia — slowness and decrement of voluntary movement, the single mandatory feature of parkinsonism. [1]
Symptoms typically emerge only after approximately 60 to 80 percent of nigral dopaminergic neurons have been lost — which is why by the time of diagnosis, the disease is well established and the underlying pathology is widespread. [1]
Lewy bodies and alpha-synuclein
The pathological hallmark is the Lewy body — an eosinophilic, intracytoplasmic inclusion found in surviving neurons. Its main constituent is misfolded alpha-synuclein, a presynaptic protein that aggregates into insoluble fibrils. The discovery that alpha-synuclein (SNCA gene) mutations cause familial PD established alpha-synuclein aggregation as central to PD biology. [1]
Lewy body pathology does not begin in the substantia nigra. Braak staging describes a caudal-to-rostral spread beginning in the dorsal motor nucleus of the vagus and the olfactory bulb (stage 1), ascending through the pons and midbrain (stages 2 to 3, where nigral involvement produces motor symptoms), then spreading to the limbic system and cortex (stages 4 to 6, where dementia and psychiatric features emerge). This topographic progression explains the prodromal non-motor symptoms that precede motor PD by years — olfactory loss, constipation, REM sleep behaviour disorder — and the late emergence of dementia. [1]
Why pathology matters for management
The pathology is neurodegenerative and progressive. No currently available therapy slows neuron loss. All symptomatic treatments — levodopa, agonists, MAO-B inhibitors, deep brain stimulation — replace or modulate the lost dopamine signal but do not halt the underlying process. This is the framing for every management discussion: we are treating symptoms over a decades-long disease, not curing it. [1]
Cardinal motor features of parkinsonism
Parkinsonism (the syndrome) requires bradykinesia plus at least one of rigidity, rest tremor, or postural instability (MDS criteria, PMID 26474316). PD is the most common cause. [1]
Bradykinesia (mandatory)
Slowness of movement with decrement and fatiguing of amplitude or speed during repetitive action. Tested at the bedside with finger tapping, hand grips, pronation-supination, and foot tapping. Look for the progressive reduction in amplitude as the patient repeats the movement — this is the key. Bradykinesia also manifests as reduced facial expression (hypomimia), reduced blink rate, soft voice (hypophonia), and micrographia (handwriting that becomes progressively smaller). [1]
Rigidity
Velocity-independent resistance to passive movement throughout the range, felt in flexors and extensors equally. When combined with a co-existing tremor, the superimposed ratchet-like resistance is called cogwheel rigidity. Test by passively rotating the wrist, flexing-extending the elbow, and flexing-extending the neck — the latter is often the most sensitive early sign. Distinguish from spasticity (velocity-dependent, "clasp-knife", pyramidal) and from paratonia (gegenhalten, variable resistance proportional to the examiner's effort, frontal lobe). [1]
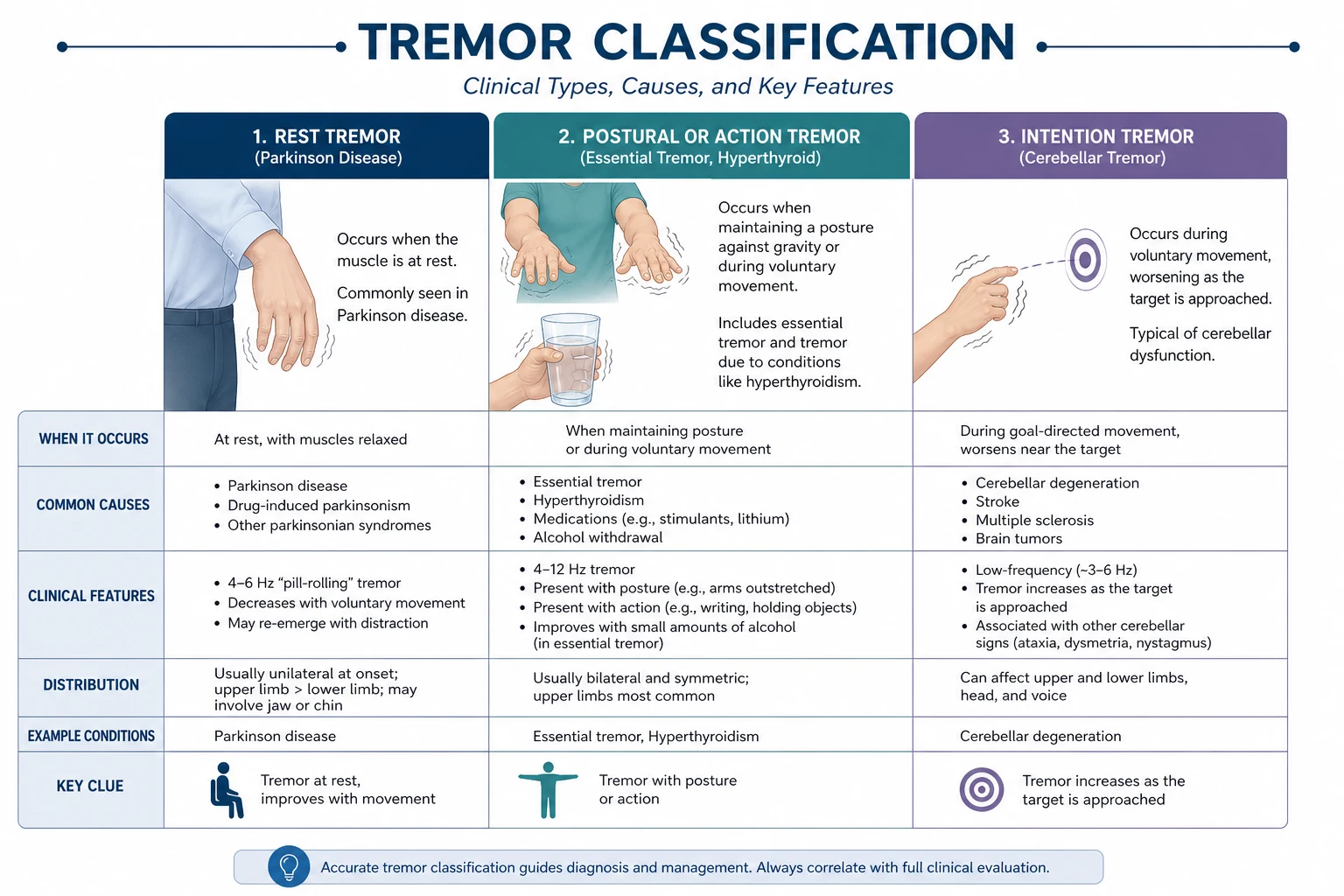
Rest tremor
A 4 to 6 Hz tremor present at rest, abolished or attenuated by action, and typically asymmetric at onset. Classic "pill-rolling" appearance in the hands. Best elicited by having the patient rest the hands in the lap with the mind distracted (counting backwards). About 70 percent of PD patients have a rest tremor at some point; tremor-dominant PD is a recognised subtype with a more benign long-term motor trajectory and fewer gait and cognitive complications than the postural instability and gait difficulty (PIGD) subtype. [1]
Postural instability
A late feature in PD — its presence early is a red flag for PSP or other atypical parkinsonism. Tested by the pull test (retropulsion): stand behind the patient, warn them, pull firmly backwards on the shoulders, and be ready to catch. A normal patient recovers in one step. A parkinsonian patient takes multiple steps (retropulsion) or falls (without correction). Postural instability reflects loss of postural righting reflexes and is the major contributor to falls and loss of independence. [1]
Non-motor features — the part that breaks the long case
Non-motor symptoms are the dominant determinant of quality of life and institutionalisation in PD, and they are the features examiners probe heavily in the DCE long case. They cluster into: [1]
- Prodromal (often precede motor symptoms by years): hyposmia (olfactory loss — present in 90 percent, useful discriminator from atypical parkinsonism and essential tremor), REM sleep behaviour disorder (RBD — acting out dreams, often reported by the bed partner), constipation.
- Neuropsychiatric: depression (in up to 50 percent, often predating motor symptoms), anxiety, apathy, psychosis (visual hallucinations — formed, detailed, often people or animals; delusions), and dementia (PD dementia, PDD — develops late, typically more than one year after motor onset, which distinguishes it from DLB).
- Autonomic: orthostatic hypotension (also worsened by dopaminergic therapy), urinary urgency and frequency, erectile dysfunction, constipation, seborrhoea, sweating dysfunction.
- Sleep: RBD, fragmented sleep, periodic limb movements, daytime somnolence.
- Sensory: pain, paraesthesia. [1]
DCE long-case trap: A candidate who presents only the motor aspects of a PD patient has failed the long case. The non-motor burden — cognition, mood, falls, autonomic failure, hallucinations, sleep — is where the integrated management plan earns its marks. [1]
Diagnosis of Parkinson's disease
PD is a clinical diagnosis. There is no confirmatory blood test or imaging in routine use. [1]
MDS clinical diagnostic criteria (2015, PMID 26474316)
The diagnosis requires: [1]
- Parkinsonism — bradykinesia PLUS rest tremor OR rigidity.
- Absence of absolute exclusion criteria — for example: cerebellar abnormalities, downward vertical gaze palsy (PSP), frontotemporal dementia, lower body only parkinsonism (vascular), drug-induced (dopamine blocker exposure), cortical sensory loss (CBD), normal dopaminergic imaging.
- At least two supportive criteria — clear beneficial response to dopaminergic therapy, levodopa-induced dyskinesia, rest tremor of a limb, olfactory loss, cardiac sympathetic denervation on MIBG scintigraphy.
- No red flags (features suggesting alternative diagnosis) — rapid gait impairment, no progression, early bulbar dysfunction, early severe autonomic failure, falls within first three years, anterocollis/contractures, absence of non-motor features, pyramidal signs, bilateral ptomyoclonus. [1]
Role of investigations
Routine neuroimaging (MRI brain) is performed to exclude structural lesions (vascular, NPH, mass) and is normal in early PD. DAT-SPECT (dopamine transporter imaging with ioflupane) visualises the presynaptic dopaminergic terminal and is abnormal in all parkinsonian syndromes (PD, MSA, PSP, CBD, DLB) but normal in essential tremor, drug-induced parkinsonism, vascular parkinsonism, and psychogenic. Its role is therefore narrow: to distinguish PD from essential tremor or drug-induced parkinsonism when clinically uncertain — not to distinguish PD from the atypical parkinsonisms (it cannot). [1]
Exam trap: A common MCQ asks "best investigation to distinguish PD from essential tremor" — the answer is DAT-SPECT. If the question asks "best investigation to distinguish PD from PSP/MSA/DLB" — the answer is clinical assessment and MRI, not DAT-SPECT (DAT is abnormal in all of them). [1]
Management of Parkinson's disease
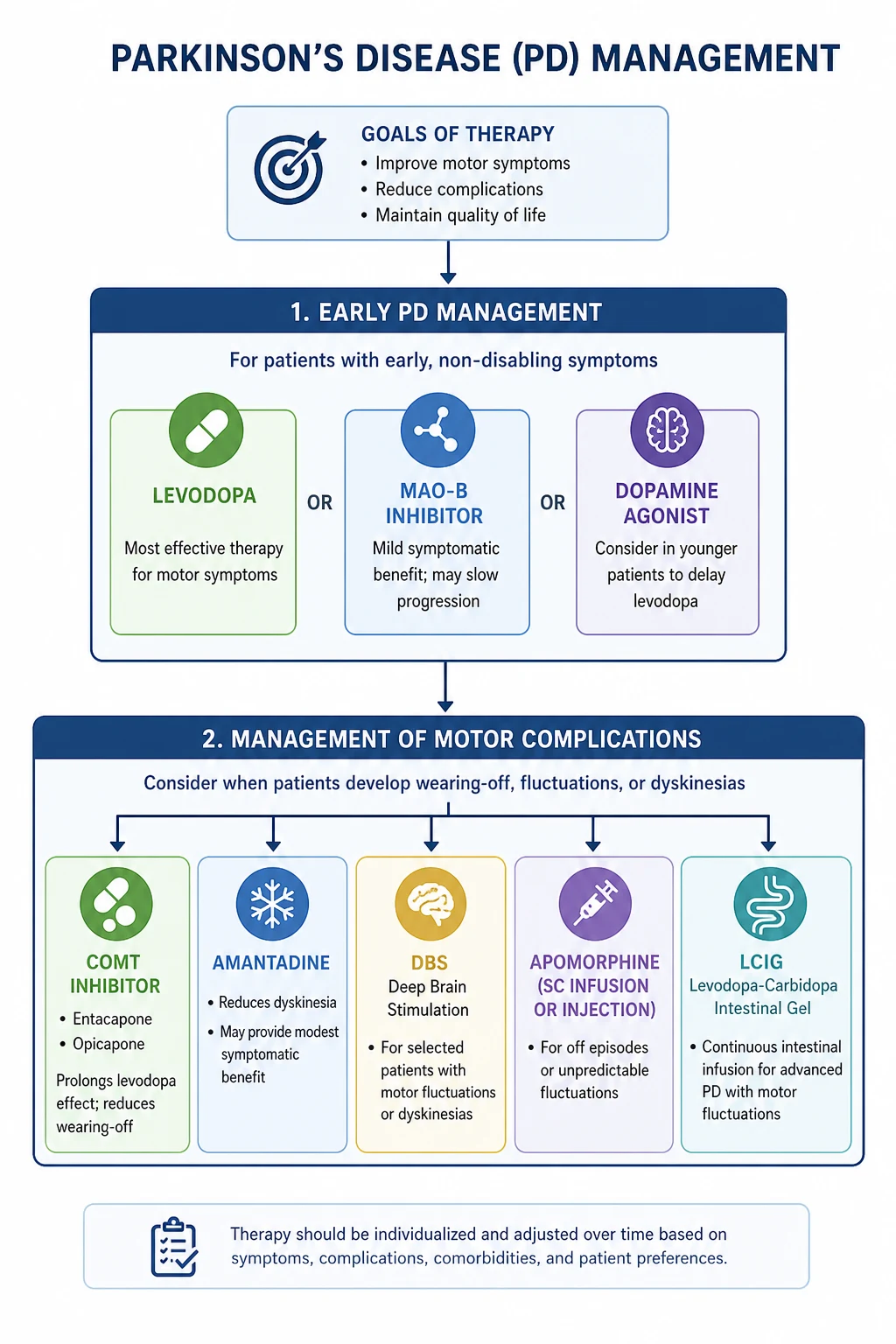
Principles
- Symptomatic only — no disease-modifying therapy exists. Do not promise slowing of progression.
- Individualise — age, subtype (tremor-dominant vs PIGD), cognitive/psychiatric comorbidity, occupation, and patient goals determine therapy.
- Time therapy to functional need — do not treat the diagnosis, treat the disability. Mild signs without functional impairment do not require symptomatic therapy.
- Levodopa is the most effective agent — use it when motor disability warrants treatment, especially in older patients.
- Non-motor symptoms need active treatment — depression, psychosis, constipation, orthostatic hypotension, RBD, dementia. [1]
Levodopa — the gold standard
Levodopa is always combined with a peripheral dopa-decarboxylase inhibitor (carbidopa in the US; benserazide in Australia/UK) to prevent peripheral conversion to dopamine and the resultant nausea and hypotension. Standard formulations: levodopa/carbidopa 100/25 or levodopa/benserazide 100/25 (Sinemet, Madopar, Kinson, Sinemet CR). Starting dose is typically one half or one tablet two to three times daily, titrated to motor response. Usual maintenance is 300 to 800 mg levodopa per day in divided doses, 3 to 5 times daily, timed to maximise "on" time. [1]
Acute adverse effects: nausea, postural hypotension, somnolence. Long-term: motor fluctuations and dyskinesia (see below). Do not use controlled-release formulations to initiate therapy — they have erratic absorption. [1]
The fear that "levodopa stops working" or "should be delayed to spare the patient dyskinesia" is a disproven myth. The PD MED trial (PMID 24928805) showed that early levodopa produced better mobility and quality of life than initial levodopa-sparing therapy, with motor complications that are manageable. [1]
MAO-B inhibitors (selegiline, rasagiline, safinamide)
Block central monoamine oxidase B, prolonging the action of endogenous and exogenous dopamine. Used as monotherapy in early mild PD (delaying the need for levodopa) or as an adjunct in fluctuating PD to reduce off time. [1]
- Selegiline 5 to 10 mg orally in the morning (give second dose at midday, not evening — it is an amphetamine metabolite and causes insomnia).
- Rasagiline 1 mg orally once daily (no amphetamine metabolite, preferred; TEMPO trial PMID 12470183).
- Safinamide 50 to 100 mg orally once daily (also modulates glutamate release). [1]
Adverse effects: insomnia, orthostatic hypotension, hallucinations. Avoid with pethidine, tramadol, dextromethorphan, and SSRIs/SNRIs because of the risk of serotonin syndrome (the classic exam interaction). [1]
Dopamine agonists (pramipexole, ropinirole, rotigotine)
Directly stimulate post-synaptic dopamine receptors (D2-like), independent of the levodopa pathway. Two subtypes: non-ergot (pramipexole, ropinirole, rotigotine — preferred, no fibrotic or valvular risk) and ergot (bromocriptine, cabergoline — now rarely used because of cardiac valve fibrosis and pulmonary/retroperitoneal fibrosis; require echocardiographic monitoring). [1]
- Pramipexole — start 0.088 mg three times daily (immediate release) or 0.09 mg once daily (extended release), titrate weekly.
- Ropinirole — start 0.25 mg three times daily, titrate weekly; prolonged-release once-daily form available.
- Rotigotine — transdermal patch 2 to 16 mg over 24 hours, useful for continuous delivery, poor gut absorption, or swallowing difficulty. [1]
Dopamine agonists are preferred as initial therapy in younger patients (under 60 to 65) with mild symptoms, where the goal is to delay levodopa and reduce the early dyskinesia risk that is higher in young-onset PD. They are not preferred in older patients or those with cognitive impairment — they cause significantly more neuropsychiatric side effects (hallucinations, confusion, somnolence, impulse control disorders) than levodopa. [1]
The impulse control disorder warning — high-yield exam point
Dopamine agonists carry a significant risk of impulse control disorders (ICDs) — pathological gambling, hypersexuality, compulsive shopping, binge eating, and punding (repetitive purposeless behaviour). The Weintraub DOMINION study (PMID 20457959) found an ICD prevalence of 13.6 percent in PD patients on dopamine agonists, with an odds ratio of 2.7 versus non-users. Risk factors: younger age, male, premorbid impulsivity, family history of gambling, and higher agonist dose. [1]
Every patient commenced on a dopamine agonist must be counselled about ICDs, and the family should be asked specifically about new or escalating behaviours at every review. Management is to reduce and wean the agonist (often switching the dopaminergic load to levodopa). ICDs can persist or worsen briefly after withdrawal (the dopamine agonist withdrawal syndrome, which can mimic drug craving and depression — wean slowly). [1]
COMT inhibitors (entacapone, opicapone)
Catechol-O-methyltransferase inhibitors block peripheral (entacapone) and central (opicapone) metabolism of levodopa, prolonging its half-life and smooth plasma levels. Used as adjuncts in fluctuating PD to reduce off time — added to each levodopa dose (entacapone 200 mg with each dose; combined as Stalevo — levodopa/carbidopa/entacapone) or once daily (opicapone 50 mg at night). Adverse effects: worsening dyskinesia (reduce levodopa dose by 25 to 30 percent when adding), orange-brown discolouration of urine, diarrhoea. Tolcapone is hepatotoxic and reserved for severe cases with LFT monitoring. [1]
Amantadine for dyskinesia
Amantadine (100 mg once or twice daily), originally an antiviral with NMDA-receptor antagonist properties, is the only oral agent with good evidence for reducing levodopa-induced dyskinesia (PMID 10803797). Extended-release amantadine (Gocovri) at 274 mg once nightly is specifically approved for dyskinesia and off-time reduction. Also useful as mild monotherapy in early PD. Adverse effects: confusion, hallucinations, livido reticularis, ankle oedema, orthostatic hypotension — limit use in cognitively impaired patients. [1]
Anticholinergics (trihexyphenidyl, benzatropine)
Now used rarely and only for tremor-dominant PD in younger patients (cognitive side effects make them unsuitable for older patients). Trihexyphenidyl 1 to 2 mg twice daily, titrated slowly. Adverse effects: dry mouth, blurred vision, urinary retention, constipation, confusion, hallucinations. [1]
Apomorphine
A potent non-ergot dopamine agonist given subcutaneously. Used in two ways: rescue injections (intermittent apomorphine pen — 2 to 10 mg subcutaneous for sudden off periods) and continuous subcutaneous infusion (apomorphine pump — device-aided therapy for advanced PD with refractory fluctuations, alternative to DBS or levodopa-carbidopa intestinal gel). Requires pre-treatment with domperidone (10 mg three times daily for two days) to control nausea — domperidone (unlike metoclopramide) does not cross the blood-brain barrier. [1]
Levodopa-carbidopa intestinal gel (LCIG / carbidopa-levodopa enteral suspension)
Continuous jejunal infusion of levodopa via a percutaneous endoscopic gastrostomy with a jejunal extension (PEG-J), achieving steady plasma levodopa levels. Indicated for advanced PD with severe motor fluctuations refractory to oral therapy. Highly effective for both off time and dyskinesia, but invasive and carries PEG-related complications. [1]
Deep brain stimulation (DBS)
Bilateral high-frequency stimulation of the subthalamic nucleus (STN) or internal globus pallidus (GPi), with an implanted pulse generator. Indicated for advanced PD with motor fluctuations or dyskinesia despite optimised medical therapy, provided the patient: (a) has a robust, clear levodopa response; (b) has preserved cognition; (c) has no untreated depression; (d) is medically fit for stereotactic neurosurgery. [1]
STN-DBS and GPi-DBS are equally effective for motor outcomes (Follett, PMID 20519680). STN allows greater reduction in dopaminergic medication (important where drug-induced dyskinesia or hallucinations are dose-limiting); GPi is preferred where there is pre-existing depression or concern about cognitive decline, as it has a more favourable neuropsychiatric profile. [1]
The EARLYSTIM trial (PMID 23406026) extended DBS to patients with early motor complications (rather than only very advanced disease) and showed superior quality-of-life and motor outcomes versus best medical therapy at two years — DBS is now considered earlier in the disease trajectory for suitable candidates. [1]
DBS does not help (and may worsen): axial symptoms that are levodopa-resistant (speech, freezing of gait, postural instability), cognition, dysphagia — these reflect non-dopaminergic disease progression. Contraindications: significant dementia, active untreated psychiatric disease, and a poor levodopa response (suggests atypical parkinsonism). [1]
Motor complications
After 5 years of levodopa therapy, approximately 50 percent of PD patients develop motor complications; after 10 years, this exceeds 80 percent. These are the dominant management problem in established PD. [1]
Wearing off (end-of-dose deterioration)
The duration of benefit from each levodopa dose shortens from the original 4 to 6 hours to 2 to 3 hours or less, so the patient "wears off" before the next dose. Often manifests first as non-motor wearing off (anxiety, sweating, pain, urinary urgency before the next dose is due). Management: shorten dosing interval, add a COMT inhibitor (entacapone/opicapone), add or increase MAO-B inhibitor, switch to a sustained-release strategy, consider device-aided therapy (LCIG, apomorphine pump, DBS). [1]
Dyskinesia
Involuntary choreiform or dystonic movements, most often at peak dose (when levodopa levels are highest, typically 1 to 2 hours after a dose). Reflects pulsatile stimulation of supersensitive post-synaptic dopamine receptors. Management: reduce individual levodopa dose size but increase frequency (smoothing plasma levels), add amantadine, switch to continuous delivery (LCIG), or consider GPi-DBS. Diphasic dyskinesia (occurring at the beginning and end of each dose) is harder to treat — increase levodopa dose or change to continuous delivery. [1]
On-off fluctuations
Sudden, unpredictable switches between "on" (mobile) and "off" (frozen, immobile), often not clearly related to dose timing. The most difficult motor complication. Management: continuous dopaminergic delivery (LCIG, apomorphine pump), STN-DBS. [1]
Freezing of gait (FOG)
Sudden, brief episodes where the patient feels their feet are "glued to the floor," typically on initiation of walking, turning, passing through doorways, or when approaching a destination. Often a levodopa-resistant feature reflecting non-dopaminergic disease progression. Management: optimise dopaminergic therapy (reduces off-period freezing), cueing strategies (visual lines on the floor, laser cane, marching to a metronome, counting), physiotherapy, avoid polypharmacy that worsens gait. FOG is a major falls risk and contributes to loss of independence. [1]
Atypical parkinsonism — "the levodopa-resistant syndromes"
The atypical parkinsonisms are alpha-synucleinopathies (MSA, DLB) or tauopathies (PSP, CBD) that share parkinsonian features with PD but progress faster, respond poorly to levodopa, carry additional neurological features, and have a much worse prognosis (median survival 6 to 10 years from onset, versus near-normal life expectancy in PD). [1]
Multiple system atrophy (MSA)
A sporadic alpha-synucleinopathy with combined parkinsonism, cerebellar dysfunction, autonomic failure and pyramidal signs (the "M" — multiple — systems). Two motor subtypes: MSA-P (parkinsonian, 80 percent, resembles PD but poorly levodopa-responsive) and MSA-C (cerebellar, 20 percent, with prominent ataxia). [1]
Diagnostic features (MDS criteria 2022, PMID 35445419):
- Autonomic failure: orthostatic hypotension (drop of at least 20/10 mmHg within 3 minutes of standing), urinary incontinence or retention (often erectile dysfunction in men).
- Cerebellar signs (MSA-C): ataxic gait, dysmetria, nystagmus, dysarthria.
- Pyramidal signs: brisk reflexes, extensor plantar response.
- Distinctive features: anterocollis (severe neck-flexor dystonia), inspiratory stridor (a poor prognostic sign — sudden death risk), cold hands and feet with discoloration (raynaud-like), rapid eye movement sleep behaviour disorder, emotional incontinence (pseudobulbar affect).
- Levodopa response is poor or absent — a key discriminator from PD.
- Cognition typically preserved (early dementia suggests DLB or PSP, not MSA). [1]
Median survival 6 to 9 years from onset. [1]
Progressive supranuclear palsy (PSP)
A tauopathy. The classic Richardson's syndrome (PSP-RS) presents with:
- Vertical supranuclear gaze palsy — the hallmark. Downward gaze is affected before upward. The deficit is supranuclear (overcome by the doll's-eye manoeuvre — the eyes move when the head is turned, proving the oculomotor nuclei and nerves are intact; the problem is loss of cortical control). Square-wave jerks on fixation.
- Early postural instability with falls — typically within the first year, often backwards ("the astasia champion").
- Axial rigidity greater than limb rigidity (rigid, extended posture — "rocket man").
- Pseudobulbar palsy: dysarthria, dysphagia, emotional lability.
- Frontal/executive cognitive dysfunction, apathy.
- Levodopa response is poor. [1]
MDS criteria (PMID 28467028) define four functional domains — ocular motor, postural instability, akinesia, cognitive — with three certainty levels (probable, possible, suggestive of PSP). Variant phenotypes include PSP-parkinsonism (resembles PD initially), PSP-corticobasal syndrome, and PSP-pure akinesia with gait freezing. Median survival 6 to 10 years. [1]
DCE short-case discriminator: "The patient has a parkinsonian look but falls within a year and cannot look down at their feet." That is PSP until proven otherwise. Test vertical eye movements. [1]
Corticobasal degeneration (CBD)
A tauopathy with the most asymmetric cortical presentation in the parkinsonian spectrum. Features include:
- Asymmetric parkinsonism (rigid, dystonic, poorly levodopa-responsive limb).
- Alien limb phenomenon — the limb moves on its own or interferes with the actions of the other limb (intermanual conflict), a striking sign.
- Cortical sensory loss — astereognosis, agraphesthesia, extinction, with intact primary sensation.
- Limb apraxia (ideomotor and ideational).
- Myoclonus (stimulus-sensitive).
- Cortical dementia with progressive aphasia or behavioural change. [1]
CBD is the hardest atypical parkinsonism to diagnose in life because it overlaps clinically with PSP, frontotemporal dementia, and posterior cortical atrophy. Median survival 6 to 8 years. [1]
Dementia with Lewy bodies (DLB)
An alpha-synucleinopathy. The central clinical feature is dementia — and it precedes or appears within one year of the parkinsonian features (if parkinsonism precedes dementia by more than one year, the diagnosis is PD dementia, PDD, not DLB — the "one-year rule"). The DLB Consortium criteria (McKeith 2017, PMID 28592453): [1]
Central feature: dementia with prominent attention, executive, and visuospatial dysfunction (rather than the memory-predominant pattern of Alzheimer disease). [1]
Core clinical features (two or more for probable DLB, one for possible):
- Fluctuating cognition with pronounced variation in attention and alertness.
- Recurrent, well-formed, detailed visual hallucinations.
- Spontaneous features of parkinsonism.
- REM sleep behaviour disorder (often the earliest feature, preceding dementia by years). [1]
Supportive biomarkers: reduced dopamine transporter uptake in basal ganglia on SPECT/PET, abnormal (low) uptake on MIBG myocardial scintigraphy, polysomnographic confirmation of REM sleep without atonia. [1]
The critical pharmacological trap: Never give a typical antipsychotic (haloperidol, chlorpromazine) or high-potency dopamine blocker to a patient with DLB. Up to 50 percent develop severe neuroleptic sensitivity — dramatic worsening of parkinsonism, immobility, obtundation, and a markedly increased mortality. Even olanzapine and risperidone carry significant risk. If an antipsychotic is unavoidable, use pimavanserin (a selective 5-HT2A inverse agonist, FDA-approved for PD psychosis; Cummings PMID 24183563) or low-dose quetiapine (with extreme caution and family counselling). Acetylcholinesterase inhibitors (rivastigmine) are first-line for DLB cognitive and psychotic features and are often remarkably effective. [1]
Drug-induced parkinsonism
Drug-induced parkinsonism (DIP) is the second most common cause of parkinsonism after PD. It is caused by dopamine D2 receptor blockade, most commonly: [1]
- Antipsychotics — typical (haloperidol, chlorpromazine) more than atypical, but risperidone is a frequent offender.
- Anti-emetics/prokinetics — metoclopramide, prochlorperazine, promethazine.
- Others — some calcium channel blockers (cinnarizine, flunarizine), tetrabenazine, reserpine (historical). [1]
Clinical features: more symmetric than PD, less likely to have rest tremor (more postural), may have co-existing tardive dyskinesia (the other face of chronic dopamine blockade), typically subacute onset. It is levodopa-unresponsive — do not add levodopa; the problem is downstream of the receptor. [1]
Management: withdraw the offending agent (substitute the antipsychotic with clozapine or quetiapine; substitute metoclopramide with domperidone or ondansetron). Improvement occurs over weeks to months (the dopamine receptor needs to up-regulate). Some cases (10 to 25 percent) persist and reflect unmasking of underlying idiopathic PD. [1]
DWE MCQ trap: A patient who develops parkinsonism on metoclopramide. The best next step is withdraw the metoclopramide, not add levodopa or a dopamine agonist. Also recognise that metoclopramide is a frequent cause of drug-induced parkinsonism in elderly patients and should be prescribed with caution in this group — domperidone (peripheral D2 blocker, no CNS penetration) is the safer prokinetic if one is needed, but note its QT prolongation risk. [1]
Essential tremor
The most common movement disorder. Postural and action tremor (worsens with sustained posture or goal-directed movement, e.g. holding a cup, pouring water), typically 4 to 12 Hz, affecting the hands (90 percent), head (voice tremor common), and rarely the legs. Absent at rest — the single most important discriminator from PD. Often improves transiently with alcohol (a useful clue and a social hazard). Family history is positive in 50 to 70 percent (autosomal dominant with incomplete penetrance). [1]
Distinguishing from PD rest tremor: PD has tremor at rest that improves with action; essential tremor has tremor with action (and posture) that is absent at rest. Essential tremor does not cause bradykinesia, rigidity, or non-motor features. Some patients have both (essential tremor may be a risk factor for PD). [1]
Management
First-line: propranolol (a non-selective beta-blocker; start 40 mg twice daily, titrate to 120 to 320 mg/day; long-acting preparation available) or primidone (an anticonvulsant; start very low at 25 mg at night to avoid acute nausea and sedation, titrate slowly to 250 mg three times daily). About 50 percent respond to either agent. The two can be combined. [1]
If refractory: gabapentin, topiramate, or botulinum toxin (for head/voice tremor). Thalamic DBS (VIM nucleus of the thalamus) or focused ultrasound thalamotomy (MR-guided) are highly effective for severe, medication-refractory essential tremor. [1]
Dystonia
Sustained or repetitive co-contraction of agonist and antagonist muscles producing abnormal postures or repetitive movements. Classify by distribution (focal, segmental, generalised, hemidystonia), by trigger (task-specific dystonia like writer's cramp), or by cause (primary/idiopathic — usually genetic, e.g. DYT1/TOR1A; secondary — structural, drug-induced, neurodegenerative). [1]
Common focal dystonias: cervical dystonia (spasmodic torticollis — the most common focal dystonia in adults), blepharospasm (forceful eye closure, can cause functional blindness), oromandibular dystonia, spasmodic dysphonia (laryngeal dystonia), and task-specific hand dystonia (writer's cramp, musician's dystonia). [1]
Management of focal dystonia: botulinum toxin injections into the affected muscles — the first-line and most effective therapy, repeated every 3 to 4 months. Oral agents (anticholinergics — trihexyphenidyl; baclofen; clonazepam) are less effective and limited by side effects. DBS (GPi) is reserved for severe generalised or refractory dystonia. In a child or young adult with generalised dystonia, a trial of levodopa is mandatory to exclude dopa-responsive dystonia (Segawa disease) — a rare but eminently treatable cause, dramatic levodopa response, often misdiagnosed as cerebral palsy. [1]
Huntington disease
An autosomal dominant neurodegenerative disorder caused by an expanded CAG trinucleotide repeat in the HTT gene on chromosome 4. Normal is fewer than 27 repeats; 36 repeats or more is pathogenic, with an inverse correlation between repeat length and age of onset (longer repeats — earlier onset, the "anticipation" phenomenon across generations, especially through paternal transmission). Prevalence is higher in populations of European descent. [1]
The classic triad is chorea, cognitive decline, and psychiatric disturbance (often depression, apathy, irritability, or psychosis). Motor onset is typically in the 30s to 50s. The chorea is gradual, distal, and flows across body parts. As the disease progresses, chorea gives way to bradykinesia, rigidity, and dystonia, with severe gait impairment and dysphagia. Frontal-subcortical dementia follows. Death typically occurs 15 to 20 years from onset. [1]
Diagnosis: genetic testing (HTT CAG repeat), guided by family history and compatible clinical picture. Genetic counselling is mandatory before testing — both for symptomatic patients and (with even greater care) for at-risk asymptomatic relatives, given the implications of a 50 percent inheritance and no disease-modifying therapy. Predictive genetic testing in asymptomatic at-risk adults follows international guidelines with a structured pre- and post-test counselling protocol. [1]
Management: symptomatic only. Tetrabenazine (a vesicular monoamine transporter 2 inhibitor, depleting central monoamines) is approved for chorea (Huntington Study Group, PMID 16476934) — start 12.5 mg once daily, titrate slowly to a maximum of 100 mg/day in divided doses. Watch for depression (dose-related, sometimes severe), sedation, parkinsonism, and akathisia. Chorea often becomes less prominent later in the disease as the patient converts to a rigid-bradykinetic phenotype, at which point tetrabenazine should be reduced. Depression needs active treatment. Multidisciplinary input (neurology, psychiatry, neuropsychology, speech pathology, dietetics, social work, genetic counselling) is essential. [1]
Wilson disease
A treatable autosomal recessive disorder of copper metabolism caused by mutations in the ATP7B gene on chromosome 13, which encodes a copper-transporting ATPase. Copper accumulates in liver, brain (basal ganglia), and eye. Wilson disease must be excluded in every patient presenting with a movement or psychiatric disorder under the age of 40 — the diagnosis is so consequential because it is treatable, and untreated disease is fatal. [1]
Presentation is hepatic (acute hepatitis, chronic hepatitis, fulminant liver failure, cirrhosis — typically in childhood/adolescence) or neurological/psychiatric (typically in the late teens to 30s). Neurological features: parkinsonism, tremor (a characteristic coarse "wing-beating" tremor), dystonia, ataxia, dysarthria, drooling. Psychiatric: behavioural change, depression, psychosis — may precede neurological signs. [1]
Diagnostic workup (Leipzig scoring, Ferenci 2003, PMID 12955875):
- Serum ceruloplasmin — typically low (under 0.2 g/L) in 85 to 95 percent, but can be normal (ceruloplasmin is an acute-phase reactant, raised in inflammation, pregnancy, oestrogen). A normal ceruloplasmin does not exclude Wilson disease. [1]- 24-hour urinary copper — elevated (over 100 micrograms/24 hours is highly suggestive; over 40 is suggestive and warrants further testing). This is the most useful single biochemical test.
- Slit-lamp examination for Kayser-Fleischer rings — golden-brown copper deposits in Descemet's membrane of the cornea, present in 95 percent of neurological Wilson disease (less often in hepatic presentations). Pathognomonic but not sensitive early.
- Liver biopsy hepatic copper — gold standard (over 250 micrograms/g dry weight is diagnostic; normal is under 50).
- ATP7B genetic testing — confirmatory, with full gene sequencing now first-line in many centres. [1]
Management: lifelong chelation therapy. First-line is penicillamine (250 to 500 mg four times daily, copper chelator, but with a high rate of adverse effects — initial neurological deterioration in 20 to 50 percent, skin changes, nephrotic syndrome, bone marrow suppression; requires pyridoxine supplementation) or trientine (better tolerated, increasingly preferred first-line). Zinc acetate (50 mg three times daily) impairs intestinal copper absorption and is used for maintenance and for pre-symptomatic patients. Dietary copper restriction (avoid shellfish, liver, nuts, chocolate). Liver transplantation is curative for decompensated liver disease and can stabilise neurological disease. Family screening of first-degree relatives is mandatory. [1]
The golden rule: A 25-year-old with a new movement disorder has Wilson disease until proven otherwise. The finding that transforms a patient with parkinsonism from "young-onset idiopathic PD" to "treatable Wilson disease" is the Kayser-Fleischer ring or a low ceruloplasmin — look actively. [1]
Restless legs syndrome (RLS)
A common (5 to 10 percent of adults) sensorimotor disorder defined by an irresistible urge to move the legs, usually accompanied by uncomfortable sensations (crawling, creeping, aching), with four essential clinical features:
- Urge to move worsens during rest or inactivity.
- Partially or totally relieved by movement (walking, stretching).
- Onset or worsening in the evening or night.
- Occurs or worsens at rest, not solely at night — circadian peak. [1]
Strongly associated with iron deficiency (check ferritin, treat if under 75 micrograms/L with oral iron; intravenous iron for refractory cases with low ferritin). Common in pregnancy, end-stage kidney disease, and iron deficiency anaemia. A positive family history is common. [1]
Management:
- Alpha-2-delta ligands (gabapentin, pregabalin) are now first-line (superior long-term safety to dopamine agonists because of the augmentation risk — see below). Start gabapentin 300 mg at night, titrate. [1]- Dopamine agonists (pramipexole, ropinirole, rotigotine) are highly effective short-term but carry a major long-term risk of augmentation — symptoms worsen, spread earlier in the day, and to other body parts. Montplaisir et al (PMID 16874755) documented this clearly. Limit agonist use where possible; if augmentation occurs, switch to an alpha-2-delta ligand and wean the agonist (slowly, to avoid withdrawal).
- Opioids (low-dose oxycodone, methadone) for refractory cases.
- Treat iron deficiency. Avoid caffeine, antihistamines, antidepressants (especially SSRIs, mirtazapine) that worsen RLS. [1]
Tic disorders and Tourette syndrome
Tics are sudden, brief, stereotyped, suppressible movements (motor) or vocalisations (vocal), associated with a premonitory urge (an internal sensation relieved by performing the tic). They are common in childhood; most resolve by adulthood. Tourette syndrome is defined by multiple motor tics and at least one vocal tic, persisting for over one year, with onset before age 18. [1]
Tourette syndrome is commonly comorbid with attention-deficit/hyperactivity disorder (ADHD) and obsessive-compulsive disorder (OCD) — the comorbidity, not the tics, is usually the main management problem. [1]
Management: most patients require no drug treatment — reassurance and education suffice. For severe, disabling tics: behavioural therapy (CBIT — comprehensive behavioural intervention for tics) is first-line. Pharmacotherapy: alpha-2 agonists (clonidine, guanfacine — also help comorbid ADHD), topiramate, or antipsychotics (risperidone, haloperidol, aripiprazole — most effective but reserved for refractory cases due to side-effect burden). Botulinum toxin for focal distressing tics. DBS is experimental for severe refractory cases. [1]
The DCE short case — parkinsonian examination
A parkinsonian patient is one of the most common DCE short cases and PACES stations because the signs are reproducible, the system teaches a structured routine, and the differential from signs is rich. [1]
Structured routine (examiner instruction: "Examine this patient's neurological system")
- Observe from the end of the bed — posture (stooped, flexed), masked face, reduced blink, tremor (pill-rolling), drooling, pill-rolling, festination.
- Face — expression, glabellar tap (persistent blink — Myerson's sign, now historical), examine cranial nerves including eye movements (look for vertical gaze restriction — PSP), and jaw jerk (brisk in pseudobulbar).
- Tone in the arms — wrist, elbow, with and without activation (tap the contralateral hand or count backwards to elicit activated rigidity).
- Tremor — at rest in the lap; with the arms outstretched (postural); with finger-nose (intention). Compare.
- Bradykinesia — finger taps, hand grips, pronation-supination. Look for decrement.
- Sensation — cortical sensory testing if asymmetric (CBD): astereognosis, graphesthesia.
- Reflexes — including plantar response.
- Legs — tone, power, taps, heel-shin (for ataxia — MSA-C).
- Gait — initiation (start hesitation — freezing), arm swing, stride length, turning (takes multiple steps, en bloc), and the pull test (postural stability).
- Stand back and present. [1]
Presentation template
"I examined Mr X's neurological system. The key findings are of a bradykinetic-rigid syndrome with an asymmetric rest tremor affecting the right hand, cogwheel rigidity at the right wrist and elbow, bradykinesia with fatiguing on repetitive finger tapping, reduced facial expression and arm swing, and a flexed, festinating gait. There is no cerebellar ataxia, no pyramidal signs, vertical eye movements are full, and cognitive testing was normal. These findings are consistent with idiopathic Parkinson's disease, asymmetric, tremor-predominant. I would like to assess the non-motor features — olfaction, sleep, mood, autonomic function — and review medication." [1]
Discussion from signs
- Asymmetric rest tremor with full vertical gaze, preserved cognition, no cerebellar or pyramidal signs, good levodopa response = idiopathic PD.
- Vertical gaze palsy, early falls, axial rigidity, frontal signs = PSP.
- Ataxia, dysarthria, autonomic failure, pyramidal signs, anterocollis = MSA.
- Alien limb, apraxia, cortical sensory loss, marked asymmetry, myoclonus = CBD.
- Symmetric, drug history positive = drug-induced. [1]
Long-term outcomes, complications and follow-up
PD is a chronic, progressive disease with a near-normal life expectancy but accumulating disability. The trajectory is dominated over time by levodopa-resistant features — freezing of gait, falls, dysarthria, dysphagia, autonomic failure, and dementia — which reflect non-dopaminergic (Braak stage 4 to 6) disease progression and are not improved by escalating dopaminergic therapy. [1]
Follow-up: regular (6 to 12 monthly, more often during dose titration or instability) review by a neurologist or physician with movement-disorders expertise. Review should include a structured motor assessment (MDS-UPDRS where available), a medication review (especially for polypharmacy and dopaminergic side effects), non-motor symptom screening, falls risk, cognition (Montreal Cognitive Assessment annually), mood, and a social/functional review. Multidisciplinary input from physiotherapy, occupational therapy, speech pathology (for voice and swallowing), dietetics, and a Parkinson's disease nurse specialist is associated with better outcomes. [1]
Advance care planning should be initiated early in the disease trajectory — addressing goals of care, advance directives, and the role of palliative care input for end-stage disease. Hospice and palliative care referral is appropriate for end-stage PD with recurrent aspiration pneumonia, weight loss, and end-stage dementia. [1]
Communication and shared decision-making
PD management is a decades-long partnership. The clinician's role is to set expectations honestly (symptomatic therapy, no cure, slow progression), individualise therapy to the patient's goals (independence at work, driving, family life, fall avoidance), and actively screen for non-motor symptoms that patients often do not volunteer (depression, hallucinations, sleep, sexual and urinary dysfunction, ICDs). Decisions about DBS, device-aided therapies, and the management of advanced disease require a structured multidisciplinary discussion with patient and family, weighing invasiveness, cognitive reserve, and quality-of-life goals. [1]
A particular communication challenge is the new diagnosis conversation. The patient has often catastrophised the diagnosis. Frame it honestly: most patients retain functional independence for many years, effective symptomatic therapy exists, and the disease is actively research-active. Avoid false reassurance ("it will be fine") and false pessimism ("this will only get worse"). Offer the Parkinson's support organisation (Parkinson's Australia in ANZ; Parkinson's UK; Michael J Fox Foundation in the US). [1]
Common exam traps
- Calling drug-induced parkinsonism "PD" — always take a drug history (metoclopramide, prochlorperazine, antipsychotics, cinnarizine). The clue is symmetric signs and a subacute onset.
- Missing Wilson disease in a young patient with a movement disorder — examine for Kayser-Fleischer rings, check ceruloplasmin and 24-hour urinary copper, and check LFTs.
- Prescribing a dopamine agonist to an older patient or one with cognitive impairment — high risk of hallucinations, confusion, and impulse control disorders. Use levodopa first.
- Failing to ask about impulse control disorders at follow-up — they destroy families and finances and are entirely iatrogenic.
- Giving haloperidol to a confused parkinsonian or DLB patient — potentially catastrophic. Use quetiapine (low dose) or pimavanserin, or treat the underlying cause of the delirium.
- Confusing DLB with PD dementia — the one-year rule. Dementia before or within one year of parkinsonism = DLB; more than one year later = PDD. The distinction matters for prognosis and antipsychotic sensitivity.
- Attributing PSP falls to "advanced PD" — early falls with vertical gaze palsy is PSP. Calling it "advanced PD" denies the patient and family an accurate prognosis.
- Stopping levodopa abruptly — causes dopamine withdrawal syndrome, akinetic crisis, and occasionally neuroleptic-malignant-like syndrome. Always wean dopaminergic drugs slowly.
- DAT-SPECT for the wrong question — it distinguishes PD from essential tremor and drug-induced parkinsonism; it does not distinguish PD from MSA, PSP, CBD or DLB.
- Treating every motor fluctuation with more levodopa — sometimes the answer is continuous delivery (LCIG, apomorphine), a COMT/MAO-B inhibitor, amantadine for dyskinesia, or DBS. Match the therapy to the pattern of the complication. [1]
Guidelines and regional deltas
- NICE NG71 (2017, updated 2024) — Parkinson's disease in adults. UK primary guidance; levodopa-sparing considered only for younger patients; emphasises physiotherapy, occupational therapy, speech and language therapy; recommends avoiding default antipsychotics in PD psychosis.
- Movement Disorder Society evidence-based medicine reviews — the international benchmark for PD treatment efficacy, periodically updated.
- Australian and New Zealand Association of Neurologists (ANZAN) — local guidance, with regional variations in DBS funding pathways and access to device-aided therapies.
- American Academy of Neurology (AAN) — practice advisories on dopamine agonist withdrawal, DBS candidacy, and PD psychosis.
- European Academy of Neurology / MDS-ES — European movement disorders guidance. [1]
Regional drug differences: benserazide (rather than carbidopa) is the common dopa-decarboxylase inhibitor combined with levodopa in Australia (Madopar); both are used in the UK. Apomorphine and LCIG access varies by region. Pimavanserin is FDA-approved but not available in Australia or the UK; in those regions quetiapine (off-label) or clozapile (clozapine, with monitoring) are used for PD psychosis, alongside cholinesterase inhibitors. [1]
References and further reading
Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord 2015;30:1591-1601 (PMID 26474316). PD MED Collaborative Group. Long-term effectiveness of dopamine agonists and monoamine oxidase B inhibitors compared with levodopa as initial treatment for Parkinson's disease (PD MED). Lancet 2014;384:1196-1205 (PMID 24928805). Schuepbach WMM, et al. Neurostimulation for Parkinson's disease with early motor complications (EARLYSTIM). N Engl J Med 2013;368:610-622 (PMID 23406026). Follett KA, et al. Pallidal versus subthalamic deep-brain stimulation for Parkinson's disease. N Engl J Med 2010;362:2095-2104 (PMID 20519680). Weintraub D, et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol 2010;67:589-595 (PMID 20457959). Cummings J, et al. Pimavanserin for patients with Parkinson's disease psychosis. Lancet 2014;383:533-540 (PMID 24183563). Hoglinger GU, et al. Clinical diagnosis of progressive supranuclear palsy: The Movement Disorder Society criteria. Mov Disord 2017;32:853-864 (PMID 28467028). Wenning GK, et al. The Movement Disorder Society Criteria for the Diagnosis of Multiple System Atrophy. Mov Disord 2022;37:1131-1148 (PMID 35445419). McKeith IG, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology 2017;89:88-100 (PMID 28592453). Huntington Study Group. Tetrabenazine as antichorea therapy in Huntington disease. Neurology 2006;66:366-372 (PMID 16476934). Parkinson Study Group. A controlled trial of rasagiline in early Parkinson disease: the TEMPO Study. Arch Neurol 2002;59:1937-1943 (PMID 12470183). Ferenci P, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int 2003;23:139-142 (PMID 12955875). Snow BJ, et al. Amantadine for levodopa-induced dyskinesia. Neurology 2000;54(7 Suppl 4):13S (PMID 10803797). Montplaisir J, et al. Ropinirole for restless legs syndrome: long-term effectiveness and augmentation. Sleep Med 2006;7:S26 (PMID 16874755). NICE NG71 — Parkinson's disease in adults (2017, updated 2024); Movement Disorder Society Clinical Diagnostic Criteria for Parkinson's Disease (2015); AAN Practice Advisories on Parkinson's Disease; Australian and New Zealand Association of Neurologists (ANZAN). [1]
References
- [1]Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson's disease Mov Disord, 2015.PMID 26474316
- [2]PD MED Collaborative Group; Gray R, Ives N, Rick C, et al. Long-term effectiveness of dopamine agonists and monoamine oxidase B inhibitors compared with levodopa as initial treatment for Parkinson's disease (PD MED): a large, open-label, pragmatic randomised trial Lancet, 2014.PMID 24928805
- [3]Schuepbach WMM, Rau J, Knudsen K, et al. Neurostimulation for Parkinson's disease with early motor complications N Engl J Med, 2013.PMID 23406026
- [4]Weintraub D, Koester J, Potenza MN, et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients Arch Neurol, 2010.PMID 20457959
- [5]Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson's disease psychosis: a randomised, placebo-controlled phase 3 trial Lancet, 2014.PMID 24183563
- [6]Hoglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria Mov Disord, 2017.PMID 28467028
- [7]Wenning GK, Stankovic I, Vignatelli L, et al. The Movement Disorder Society Criteria for the Diagnosis of Multiple System Atrophy Mov Disord, 2022.PMID 35445419
- [8]McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium Neurology, 2017.PMID 28592453
- [9]Huntington Study Group Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial Neurology, 2006.PMID 16476934
- [10]Parkinson Study Group A controlled trial of rasagiline in early Parkinson disease: the TEMPO Study Arch Neurol, 2002.PMID 12470183
- [11]Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease Liver Int, 2003.PMID 12955875
- [12]Follett KA, Weaver FM, Stern M, et al. Pallidal versus subthalamic deep-brain stimulation for Parkinson's disease N Engl J Med, 2010.PMID 20519680
- [13]Snow BJ, Calne DB, Lang AE, et al. The effect of amantadine on levodopa-induced dyskinesias in Parkinson's disease: a double-blind, placebo-controlled study Clin Neuropharmacol, 2000.PMID 10803797
- [14]Montplaisir J, Fantini L, Desautels A, et al. Ropinirole is effective in the long-term management of restless legs syndrome: a randomized controlled trial Mov Disord, 2006.PMID 16874755