Phys · neurological
Multiple Sclerosis
Also known as multiple sclerosis · MS · relapsing-remitting multiple sclerosis · RRMS · secondary progressive multiple sclerosis · SPMS · primary progressive multiple sclerosis · PPMS · disseminated sclerosis · optic neuritis · transverse myelitis · clinically isolated syndrome
Consultant-physician-depth guide to multiple sclerosis — the demyelinating autoimmune disease of the CNS. Covers the clinical course (RRMS 85 per cent at onset, SPMS, PPMS 15 per cent), pathophysiology (T-cell mediated attack on CNS myelin; HLA-DRB1*15:01; EBV, vitamin D, latitude, smoking), the clinical syndromes (optic neuritis, transverse myelitis, brainstem, cerebellar; Lhermitte sign, Uhthoff phenomenon), the McDonald 2017 diagnostic criteria (dissemination in space and time on MRI, CSF oligoclonal bands, VEP delayed P100), acute relapse management (methylprednisolone, plasma exchange), the full DMT landscape (interferon, glatiramer, fingolimod, dimethyl fumarate, teriflunomide, cladribine, siponimod, natalizumab with PML risk, ocrelizumab for PPMS and RRMS, alemtuzumab, HSCT), symptom management (spasticity, fatigue, bladder, depression, pain), and pregnancy planning (washout, relapse rate in pregnancy, postpartum rebound, breastfeeding). Structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Multiple Sclerosis

The answer first
Multiple sclerosis is a chronic autoimmune inflammatory demyelinating disease of the central nervous system in which autoreactive T cells and B cells attack CNS myelin, producing relapsing episodes of neurological disability (the relapsing-remitting phase) and, over time, a progressive neurodegenerative decline driven by axonal loss (the progressive phase). Approximately 85 per cent of patients present with relapsing-remitting MS (RRMS) — discrete attacks of neurological dysfunction separated by periods of remission — while 15 per cent present with primary progressive MS (PPMS), an insidious decline without relapses from onset [2]. Over decades, most RRMS patients transition to secondary progressive MS (SPMS), in which steady disability accumulation replaces discrete relapses.
The clinical presentation is determined by the location of demyelinating lesions: optic neuritis (painful unilateral visual loss with a relative afferent pupillary defect), transverse myelitis (sensory level, weakness, sphincter disturbance), brainstem syndromes (internuclear ophthalmoplegia, vertigo, diplopia), and cerebellar syndromes (ataxia, nystagmus, scanning dysarthria). Two eponymous signs are high-yield: the Lhermitte sign (an electric shock radiating down the spine on neck flexion, from cervical cord demyelination) and the Uhthoff phenomenon (transient worsening of pre-existing symptoms with heat or exercise, from conduction block in demyelinated axons at higher body temperature). [1]
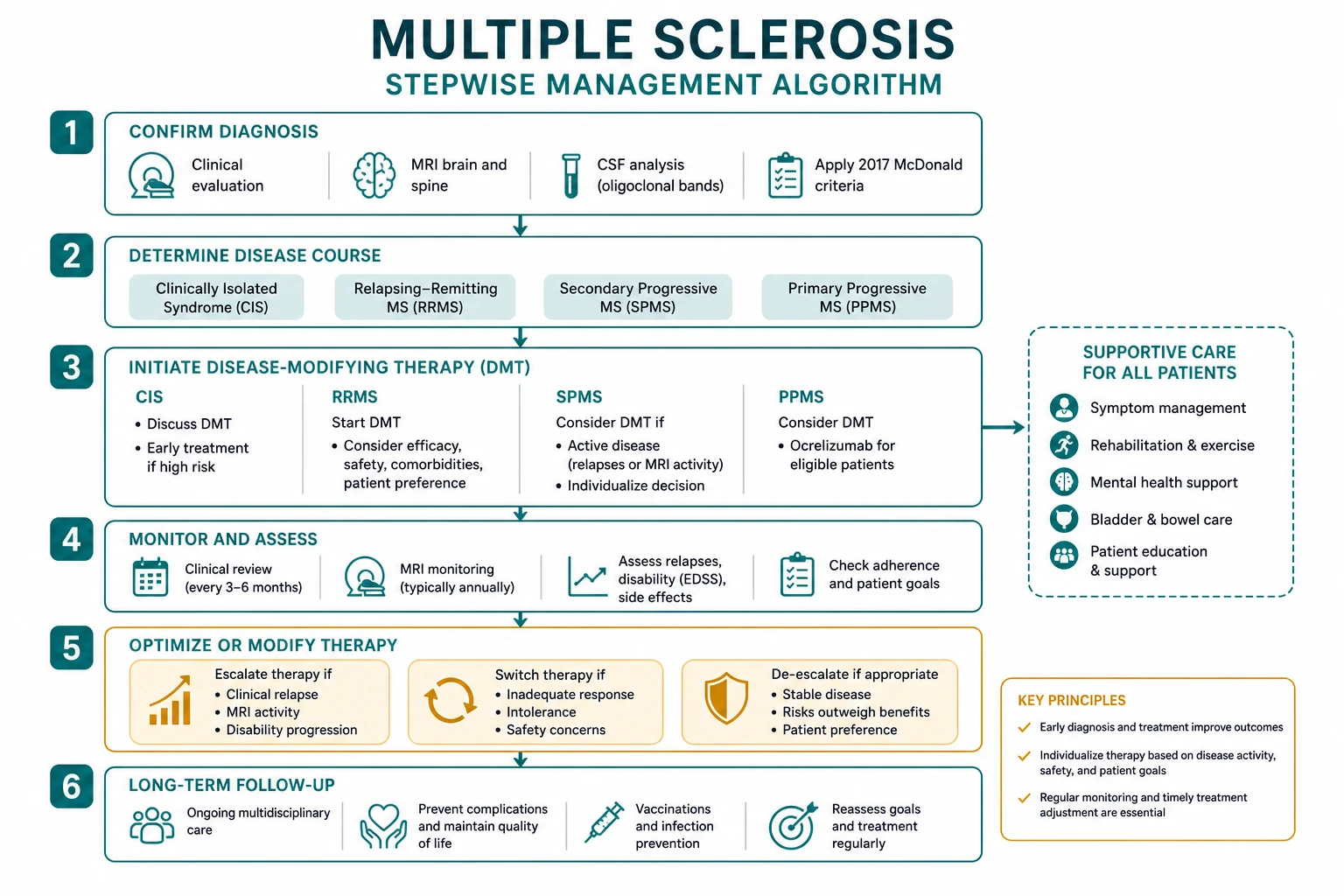
Diagnosis rests on the McDonald 2017 criteria, which require dissemination in space and time demonstrated either clinically or by MRI, with CSF oligoclonal bands able to substitute for dissemination in time [1]. Treatment has two arms: acute relapse management (methylprednisolone 1 g IV for 3 to 5 days; plasma exchange for steroid-refractory severe relapses) and disease-modifying therapy (DMT) — a rapidly expanding armamentarium ranging from injectable first-line agents (interferon-beta, glatiramer) through oral agents (dimethyl fumarate, fingolimod, teriflunomide, cladribine, siponimod) to high-efficacy monoclonal antibody infusions (natalizumab, ocrelizumab, alemtuzumab) and, for aggressive disease, haematopoietic stem cell transplantation [4] [5] [6].
DCE trap: The biggest pivot in the MS long case is recognising that management is no longer one-size-fits-all. The modern approach demands a risk-stratified DMT strategy — escalating from platform therapy to high-efficacy agents in active disease, while actively screening for the serious complications of each drug (PML with natalizumab, autoimmunity with alemtuzumab). The candidate who presents a coherent DMT strategy, names the monitoring requirements for each drug, and integrates symptom management and reproductive planning will pass the MS long case. [1]
Clinical course — the 2013 Lublin phenotype revisions
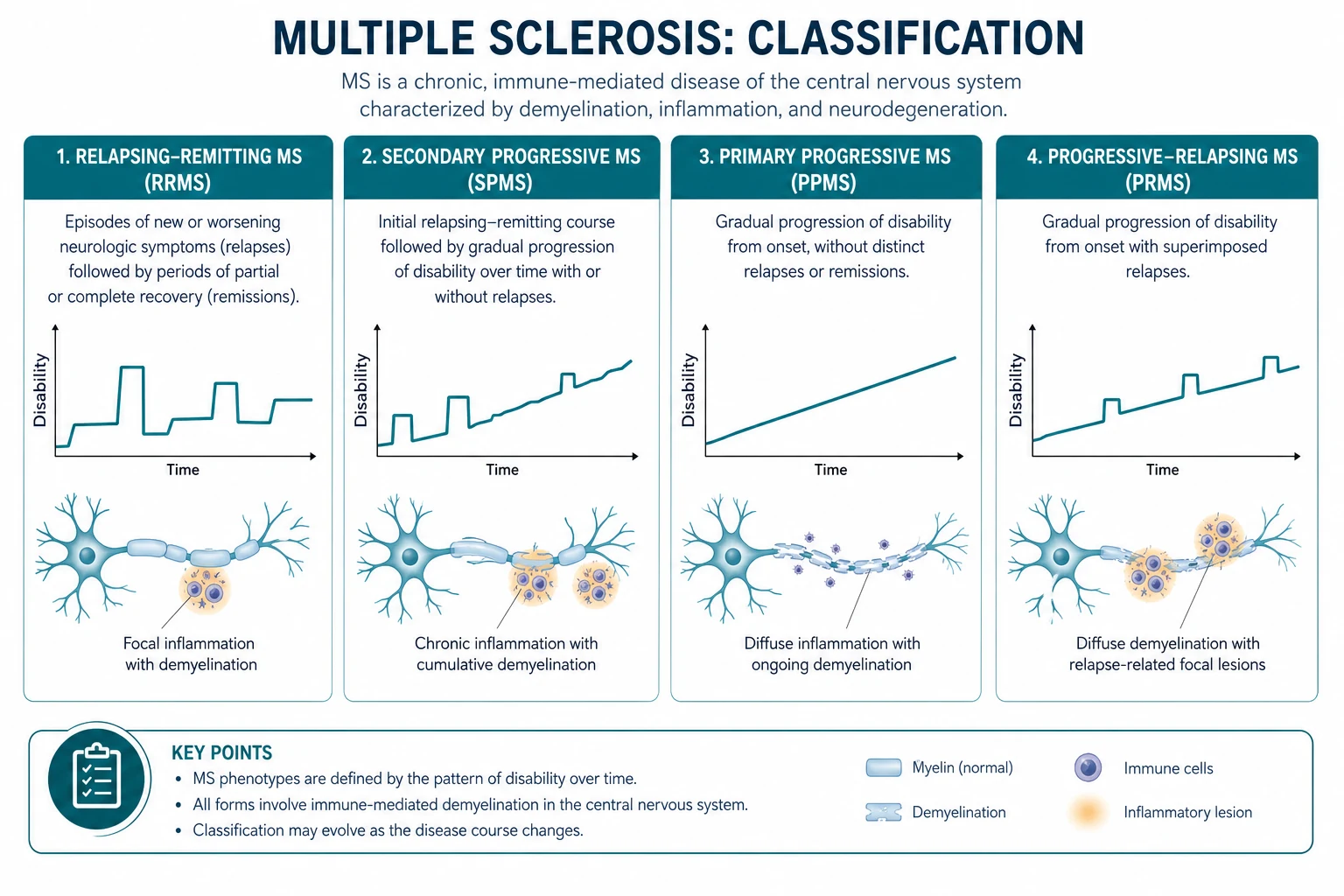
The 2013 International Advisory Committee revisions, led by Lublin, refined the original 1996 phenotype classification by adding two modifiers to each of the four traditional courses: disease activity (active or inactive, based on clinical relapses and MRI activity) and disease progression (progressing or not progressing, based on disability accumulation measured by EDSS) [2]. This matters because it allows clinicians to make treatment decisions based on whether a patient has ongoing inflammatory activity even if they are in a progressive phase.
| Phenotype | Definition | Proportion at onset | Key features |
|---|---|---|---|
| Relapsing-remitting MS (RRMS) | Clearly defined relapses with full or partial recovery, no progression between attacks | Approximately 85 per cent | Female predominance (3:1), onset typically 20 to 40 years, relapse rate 0.5 to 1 per year untreated |
| Secondary progressive MS (SPMS) | Progressive neurological decline following an initial relapsing-remitting course | Develops in most RRMS patients over 15 to 20 years | Transition is insidious; may be active (with relapses or MRI activity) or inactive, with or without progression |
| Primary progressive MS (PPMS) | Progressive neurological decline from onset, without relapses or remissions | Approximately 15 per cent | Equal sex ratio, older onset (around 40 years), motor and sphincter involvement, worse prognosis, fewer MRI lesions |
| Progressive-relapsing MS | Progressive disease from onset with superimposed relapses | Now subsumed under PPMS-active | Removed as a separate category in 2013 revisions; now classified as PPMS with disease activity |
DWE high-yield question: "What proportion of MS patients present with relapsing-remitting disease at onset, and how many eventually develop secondary progressive disease?" Answer: approximately 85 per cent present with RRMS. Over 15 to 20 years, the majority transition to SPMS — the progressive phase is driven by axonal loss and neurodegeneration that eventually outpaces the capacity for inflammatory repair [2].
Pathophysiology — the autoimmune, genetic, and environmental interplay
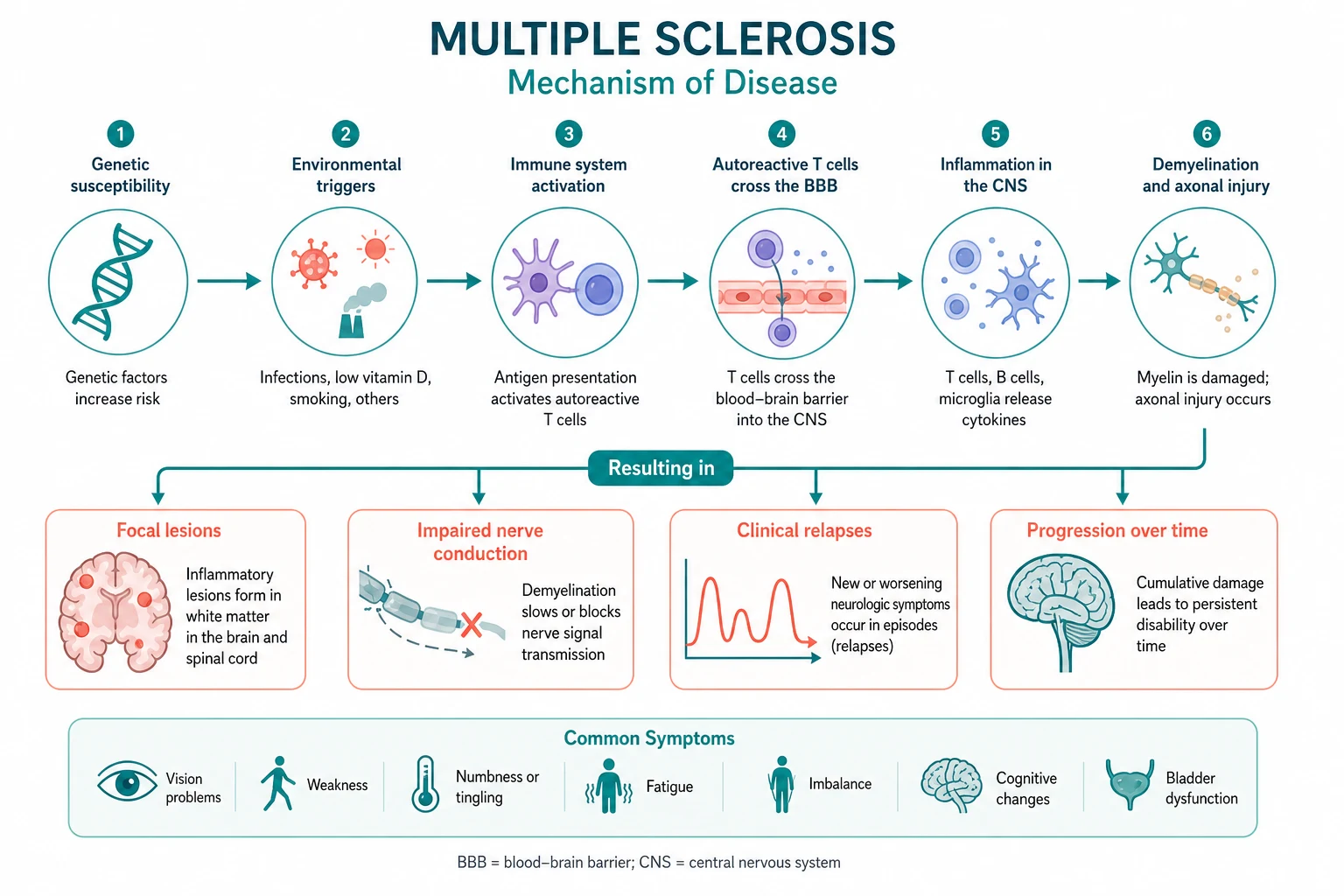
MS is an autoimmune disease driven by autoreactive T cells and B cells that attack CNS myelin. The pathogenesis involves both an inflammatory component (responsible for relapses and acute lesion formation) and a neurodegenerative component (responsible for progressive disability and axonal loss). These two processes are temporally linked — the transition from RRMS to SPMS reflects the point at which accumulated axonal loss and neurodegeneration outpace the capacity for inflammatory remyelination. [1]
The inflammatory cascade
Autoreactive CD4-positive T-helper-1 and T-helper-17 cells that are specific for myelin antigens (myelin basic protein, proteolipid protein, myelin oligodendrocyte glycoprotein) cross the blood-brain barrier, recognise myelin antigens presented by antigen-presenting cells, and initiate an inflammatory cascade. This recruits macrophages and B cells, activates complement, and produces the characteristic perivenular inflammatory infiltrate with active demyelination. B cells play a critical role not only as antibody-producing cells (oligoclonal bands are produced intrathecally by B-cell clones) but also as antigen-presenting cells that perpetuate T-cell activation — this is the mechanism that explains the dramatic efficacy of anti-CD20 therapies (ocrelizumab, rituximab) [5].
The neurodegenerative component
Even in acute lesions, there is axonal transection — the inflammatory process damages axons, not just myelin. Over time, the cumulative axonal loss produces irreversible disability. Chronic lesions show gliotic scarring (the hardened sclerotic plaque that gives the disease its name), and shadow plaques represent areas of attempted but incomplete remyelination. The progressive phase is driven by compartmentalised inflammation trapped behind a closed blood-brain barrier, mitochondrial dysfunction producing virtual hypoxia, and oxidative injury to axons. [1]
Genetic susceptibility
The strongest genetic association is with HLA-DRB1*15:01, which confers approximately a 3-fold increased risk. This class II MHC allele affects antigen presentation to CD4-positive T cells, consistent with the autoimmune nature of the disease. The concordance rate in monozygotic twins is approximately 25 to 30 per cent (compared with 3 to 5 per cent in dizygotic twins and siblings), confirming that genetic factors are necessary but not sufficient — the environment provides the trigger. [1]
The EBV trigger — the strongest environmental risk factor
The 2022 study by Bjornevik and colleagues, published in Science, provided the most compelling evidence to date that Epstein-Barr virus infection is a necessary trigger for MS [3]. In a cohort of more than 10 million US military personnel, EBV seroconversion was associated with a 32-fold increased risk of MS. Of the 801 individuals who developed MS, all but one were EBV-seropositive before the MS diagnosis. Critically, serum neurofilament light chain (a marker of neuroaxonal damage) rose only after EBV seroconversion, demonstrating that the neurological damage occurred after, not before, the viral infection. No other virus, including cytomegalovirus, increased MS risk. This finding has profound implications: an EBV vaccine could theoretically prevent MS, and anti-EBV therapies may become a future treatment strategy.
Other environmental factors
- Latitude gradient — MS prevalence increases with distance from the equator (from fewer than 5 per 100,000 in tropical regions to more than 200 per 100,000 in northern Europe and Canada), likely mediated by ultraviolet light exposure and vitamin D.
- Low vitamin D — prospective studies show that higher vitamin D levels are associated with lower MS risk and lower relapse rates. Vitamin D supplementation is now standard in MS management.
- Smoking — smoking increases MS risk by approximately 1.5-fold, accelerates progression to SPMS, and reduces the efficacy of some DMTs. Smoking cessation is a modifiable risk factor that every patient should be counselled on.
- Adolescent obesity — obesity in adolescence (particularly in females) increases MS risk by approximately 2-fold, likely mediated by low-grade chronic inflammation. [1]
Clinical presentation — the syndromes
The clinical presentation is determined entirely by the anatomical location of demyelinating lesions. MS can affect any part of the CNS white matter, and the classic syndromes reflect the most common lesion locations. [1]
Optic neuritis
The classic presentation is acute unilateral painful visual loss developing over hours to days, with pain on eye movement (because the inflamed optic nerve is tethered to the extraocular muscles). Examination reveals reduced visual acuity, a relative afferent pupillary defect (RAPD) on the swinging flashlight test, impaired colour vision (red desaturation is an early sign), and a visual field defect (usually central scotoma). Fundoscopy shows disc swelling in papillitis (anterior optic neuritis) or a normal disc in retrobulbar neuritis (posterior optic neuritis, more common in MS). Most patients recover substantially within weeks, but residual deficit (RAPD, impaired colour vision, optic atrophy on follow-up) is the rule rather than the exception. Bilateral simultaneous optic neuritis is atypical for MS and should prompt investigation for NMOSD or MOG-antibody disease. [1]
Transverse myelitis
MS-related transverse myelitis is typically partial and asymmetric — it affects a portion of the cord, producing a sensory level (often incomplete), unilateral or asymmetric leg weakness, and sphincter disturbance (urinary urgency or retention). The lesion is usually short (less than 2 vertebral segments) on MRI. This is a critical distinction from neuromyelitis optica spectrum disorder (NMOSD), in which transverse myelitis is longitudinally extensive (more than 3 segments), bilateral, and severe, with poor recovery. The Lhermitte sign (electric shock down the spine on neck flexion) indicates cervical cord demyelination and, while not specific to MS, is highly characteristic. [1]
Brainstem syndromes
Internuclear ophthalmoplegia (INO) is the classic brainstem sign of MS — impaired adduction of the eye on the side of the medial longitudinal fasciculus (MLF) lesion, with contralateral abducting nystagmus. Bilateral INO in a young patient is highly suggestive of MS. Other brainstem presentations include vertigo, diplopia (from sixth nerve palsy or skew deviation), facial sensory loss or trigeminal neuralgia (in a young patient, trigeminal neuralgia should prompt investigation for MS), dysarthria, and dysphagia. [1]
Cerebellar syndromes
Cerebellar involvement produces gait and limb ataxia, intention tremor (which can be severe and disabling), dysmetria on finger-nose testing, dysdiadochokinesia, and nystagmus (including gaze-evoked, downbeat, and pendular forms). Scanning dysarthria (exploded, syllable-by-syllable speech) is characteristic of cerebellar involvement. [1]
Paroxysmal and other symptoms
- Paroxysmal symptoms — brief (seconds to minutes), stereotyped, repetitive episodes of neurological dysfunction (tonic spasms, paroxysmal dysarthria, paroxysmal sensory disturbances), often triggered by movement or hyperventilation. They respond dramatically to carbamazepine.
- Fatigue — the most common and most disabling symptom, affecting 80 per cent of patients. It is distinct from depression and sleepiness, and partially responds to amantadine and exercise.
- Heat sensitivity (Uhthoff phenomenon) — transient worsening of pre-existing symptoms with heat, exercise, or fever. This is conduction block in previously demyelinated axons at higher body temperature — it is not a relapse.
- Cognitive impairment — affects 40 to 65 per cent, with slowed processing speed, impaired attention, and executive dysfunction, often disproportionate to physical disability.
- Depression — common (lifetime prevalence approximately 50 per cent), partly reactive and partly disease-related (CNS inflammation), and requires active screening and treatment.
- Bladder dysfunction — neurogenic bladder (urgency, frequency, incontinence from detrusor overactivity; or retention from detrusor-sphincter dyssynergia), affecting 75 per cent of patients with established disease.
- Pain — neuropathic pain (trigeminal neuralgia, Lhermitte sign, painful tonic spasms) and musculoskeletal pain (from spasticity and postural abnormalities). [1]
Diagnosis — the McDonald 2017 criteria
The McDonald 2017 criteria, published by Thompson and the International Panel on Diagnosis of MS, are the current diagnostic standard [1]. The core principle remains: MS is a clinical diagnosis requiring dissemination in space (DIS) and dissemination in time (DIT), demonstrated either clinically or by paraclinical evidence (MRI, CSF). The 2017 revision made three key changes from the 2010 criteria: CSF oligoclonal bands can substitute for DIT (if DIS is met clinically or radiologically), cortical lesions now count for DIS (previously only juxtacortical lesions counted), and symptomatic lesions now count for both DIS and DIT (previously only asymptomatic lesions were counted).
Dissemination in space (DIS)
DIS requires at least one T2-hyperintense lesion in at least two of the four characteristic CNS locations: [1]
- Periventricular — lesions adjacent to the lateral ventricles, often ovoid and perpendicular to the ventricular wall (Dawson fingers, representing perivenular inflammation along medullary veins).
- Juxtacortical or cortical — lesions adjacent to or within the cerebral cortex. The 2017 revision added cortical lesions (detected on specialised MRI sequences such as double inversion recovery).
- Infratentorial — lesions in the brainstem or cerebellum.
- Spinal cord — lesions in the cervical or thoracic cord, typically peripheral and occupying less than half the axial cord area. [1]
Dissemination in time (DIT)
DIT can be demonstrated in one of three ways:
- A new T2-hyperintense or gadolinium-enhancing lesion on follow-up MRI compared to baseline, regardless of timing.
- The simultaneous presence of gadolinium-enhancing and non-enhancing lesions on a single MRI scan (the enhancing lesion is acute, the non-enhancing lesion is older — demonstrating that lesions have occurred at different times).
- CSF oligoclonal bands (isoelectric focusing showing bands in CSF that are not present in matched serum) — the 2017 revision allows this to substitute for DIT if DIS is met. [1]
Gadolinium enhancement
Gadolinium enhancement indicates active inflammation — the contrast agent leaks across a disrupted blood-brain barrier into active lesions. An enhancing lesion is acute or subacute (weeks old), while a non-enhancing T2 lesion is older (months to years). The simultaneous presence of both on a single scan satisfies DIT. [1]
CSF examination
CSF analysis is not required in all cases — if the clinical picture and MRI are typical, the diagnosis can be made without it. However, CSF should be examined when the presentation is atypical, the MRI is non-diagnostic, or mimics need exclusion. The hallmark finding is oligoclonal bands detected by isoelectric focusing, present in the CSF but not in matched serum (indicating intrathecal antibody synthesis). They are found in approximately 95 per cent of MS patients. A CSF pleocytosis of more than 50 white cells, a neutrophilic predominance, or the absence of oligoclonal bands should prompt reconsideration of the diagnosis. [1]
Visual evoked potentials (VEP)
VEP demonstrates delayed P100 latency with preserved waveform amplitude, reflecting slowed conduction through a demyelinated (but not destroyed) optic nerve. VEP can reveal clinically silent optic nerve involvement and, under the McDonald criteria, can contribute to DIS by providing evidence of a lesion in the optic nerve pathway. [1]
Excluding the mimics
Before diagnosing MS and initiating lifelong DMT, the critical mimics must be excluded: [1]
- Neuromyelitis optica spectrum disorder (NMOSD) — AQP4-IgG positive, longitudinally extensive transverse myelitis (more than 3 segments), severe often bilateral optic neuritis with poor recovery, area postrema syndrome (intractable hiccups and vomiting), brain lesions in the hypothalamus and brainstem. MS DMTs (interferon, glatiramer, natalizumab) may be harmful in NMOSD.
- MOG-antibody disease (MOGAD) — MOG-IgG positive, often bilateral optic neuritis with prominent disc swelling, ADEM-like presentation in children, better recovery, typically steroid-responsive.
- Acute disseminated encephalomyelitis (ADEM) — monophasic, typically post-infectious or post-vaccination, with encephalopathy (uncommon in adult MS).
- Neurosarcoidosis — ACE elevation, meningeal enhancement, uveitis, hilar lymphadenopathy.
- Lyme disease, HIV, HTLV-1, syphilis — infectious mimics with characteristic serology.
- Vitamin B12 deficiency, copper deficiency — myelopathy with a peripheral neuropathy and characteristic blood tests.
- Cervical spondylotic myelopathy — structural cord compression, not demyelination.
- Susac syndrome — microvascular occlusions producing the triad of encephalopathy, branch retinal artery occlusions, and sensorineural hearing loss, with characteristic snowball white matter lesions in the corpus callosum. [1]
DWE trap: The most common diagnostic error is failing to check AQP4-IgG and MOG-IgG before labelling a demyelinating syndrome as MS. NMOSD and MOGAD have different prognoses, different treatments, and some MS DMTs may worsen NMOSD. Every patient with a first demyelinating event should have these antibodies checked. [1]
Acute relapse management
Corticosteroids
The first-line treatment for an acute MS relapse is high-dose methylprednisolone — 1 g IV daily for 3 to 5 days, or 500 mg orally daily for 5 days (oral is non-inferior to IV in most studies and is more convenient). Steroids accelerate recovery from a relapse but do not improve the long-term outcome or residual deficit. The mechanism is suppression of the inflammatory cascade and reduction of oedema around active lesions. Gastroprotection (a proton pump inhibitor) and bone protection (calcium and vitamin D) should be considered, and blood glucose should be monitored, particularly in patients with diabetes. [1]
Plasma exchange
For severe relapses that do not respond to steroids (particularly optic neuritis with threatened vision, severe transverse myelitis, or brainstem relapses), plasma exchange is the evidence-based second-line treatment — typically 5 to 7 exchanges over approximately 2 weeks. The mechanism is removal of pathogenic antibodies and inflammatory mediators. Repeating courses of steroids for a steroid-refractory relapse is not effective — plasma exchange is the correct escalation. [1]
When NOT to treat
Not every relapse requires steroids. Mild sensory relapses (numbness, tingling) that do not impair function can be managed expectantly. Steroids should be reserved for relapses that cause functional impairment (motor, visual, cerebellar, or sphincter involvement). Frequent courses of steroids (more than 3 to 4 per year) should prompt a review of the DMT strategy — breakthrough disease means the current DMT is inadequate. [1]
Disease-modifying therapy — the treatment landscape
The DMT landscape has transformed MS management over the past two decades. The goal of DMT is to reduce relapse frequency, prevent disability accumulation, and achieve no evidence of disease activity (NEDA) — a composite of no relapses, no new MRI lesions, no disability progression, and (increasingly) no brain volume loss. The choice of DMT is individualised based on disease activity, patient preference, comorbidities, reproductive plans, JC virus status, and monitoring capacity. [1]
Injectable first-line agents
Interferon-beta (several preparations: interferon beta-1a IM 30 micrograms weekly — Avonex; interferon beta-1a SC 44 micrograms three times weekly — Rebif; interferon beta-1b SC 250 micrograms alternate days — Betaferon) reduces the annualised relapse rate by approximately one-third. The PRISMS trial established the efficacy of subcutaneous interferon beta-1a in RRMS, demonstrating significant reductions in relapse rate, disability progression, and MRI disease activity [12]. Common side effects include flu-like symptoms (fever, myalgia, fatigue after each injection — managed with paracetamol and pre-dosing), injection-site reactions, depression, and liver enzyme elevation. Neutralising antibodies develop in 5 to 30 per cent of patients (depending on the preparation) and reduce efficacy.
Glatiramer acetate (20 mg SC daily) is a synthetic polymer of four amino acids designed to mimic myelin basic protein. It has similar efficacy to interferon-beta (approximately one-third reduction in relapse rate) and is an alternative first-line agent, particularly for patients who cannot tolerate interferon or who develop neutralising antibodies. Its side-effect profile is favourable (injection-site reactions and a transient systemic reaction with chest tightness, flushing, and dyspnoea). It is the DMT of choice in pregnancy (category B — no evidence of teratogenicity). [1]
Oral agents
Dimethyl fumarate (240 mg twice daily) activates the Nrf2 antioxidant pathway. The DEFINE trial demonstrated a significant reduction in relapse rate (approximately 50 per cent) and disability progression compared to placebo [11]. Side effects include flushing and gastrointestinal upset (nausea, diarrhoea — usually transient), and lymphopenia (requiring CBC monitoring every 3 to 6 months). Rare cases of PML have occurred with severe, prolonged lymphopenia.
Fingolimod (0.5 mg daily) is an S1P receptor modulator that sequesters lymphocytes in lymph nodes, preventing CNS entry. It was the first oral DMT approved for RRMS and reduces relapse rate by approximately 50 per cent compared to placebo. It requires first-dose observation for 6 hours for bradycardia and atrioventricular block (baseline ECG and observation with monitoring). Other risks include macular oedema (baseline and 3 to 4 month ophthalmology review), elevated liver enzymes, and a small PML risk. [1]
Teriflunomide (14 mg daily) inhibits pyrimidine synthesis in proliferating lymphocytes. It reduces relapse rate by approximately 30 per cent. It is teratogenic (requires pregnancy prevention and, if pregnancy is desired, an accelerated elimination procedure with cholestyramine or activated charcoal). Monitoring includes liver function and CBC. [1]
Cladribine (3.5 mg per kg total dose over 2 years, given as two annual short courses) is a purine analogue that selectively depletes lymphocytes. The CLARITY trial demonstrated significant reductions in relapse rate and disability progression [10]. It is a high-efficacy oral agent with a convenient dosing schedule but carries risks of severe lymphopenia, infections (including herpes zoster — varicella vaccination is required before treatment), and malignancy (a signal in early studies that requires ongoing surveillance).
Siponimod (2 mg daily, with dose titration) is an S1P receptor modulator selective for S1P1 and S1P5, approved for SPMS with active disease. The EXPAND trial demonstrated a 21 per cent reduction in disability progression compared to placebo in SPMS — the first DMT to show efficacy in the progressive phase [6]. Like fingolimod, it requires first-dose cardiac monitoring.
High-efficacy infusion therapies
Natalizumab (300 mg IV every 4 weeks) is a humanised monoclonal antibody against alpha-4 integrin that blocks T-cell adhesion and entry into the CNS. It is one of the most effective DMTs for RRMS, reducing relapse rate by approximately 68 per cent. However, it carries a risk of progressive multifocal leukoencephalopathy (PML) — a devastating brain infection caused by JC virus reactivation. The risk is stratified by three factors identified by Bloomgren and colleagues [7]:
- JC virus antibody positivity — all confirmed PML cases have occurred in JCV-positive patients. JCV-negative patients have the lowest risk (fewer than 0.1 per 1000).
- Prior immunosuppression — approximately doubles the risk.
- Treatment duration over 24 months — risk increases with duration, peaking at 25 to 48 months. [1]
In the highest-risk group (JCV-positive, prior immunosuppression, treatment over 24 months), the PML risk is up to 11 per 1000. JCV antibody status should be checked before starting natalizumab and monitored periodically (seroconversion occurs in approximately 2 to 3 per cent per year). Any new neurological symptom in a patient on natalizumab requires urgent MRI with diffusion-weighted imaging and CSF JCV PCR to exclude PML. [1]
Ocrelizumab (600 mg IV every 6 months, after two initial doses 2 weeks apart) is a humanised anti-CD20 monoclonal antibody that depletes B cells. It is the only DMT approved for both PPMS and RRMS. The ORATORIO trial demonstrated that ocrelizumab reduced disability progression by 24 per cent in PPMS compared to placebo — a landmark result in a disease that had been notoriously resistant to treatment [4]. The OPERA I and II trials in RRMS showed superior efficacy to interferon beta-1a, with approximately 46 to 47 per cent reduction in annualised relapse rate and 40 per cent reduction in disability progression [5]. Pre-treatment screening includes hepatitis B surface antigen and core antibody (reactivation risk), hepatitis C, quantiferon, and immunoglobulin levels. Side effects include infusion reactions, infections, and a possible signal for malignancy (breast cancer in the trials) that requires ongoing surveillance.
Alemtuzumab (12 mg IV daily for 5 days, then 3 days 12 months later) is an anti-CD52 monoclonal antibody that causes profound and prolonged lymphocyte depletion. It is one of the most effective DMTs, with the CARE-MS I trial (treatment-naive RRMS) and CARE-MS II trial (RRMS despite prior DMT) demonstrating significant reductions in relapse rate and disability compared to interferon beta-1a [8] [9]. However, its use is limited by secondary autoimmunity — thyroid disease (approximately 30 per cent), immune thrombocytopenia (approximately 2 per cent), and Goodpasture syndrome (less than 1 per cent). This requires monthly blood monitoring (CBC, creatinine, urinalysis) for 48 months after the last dose. Because of its risk profile, alemtuzumab is reserved for highly active RRMS and requires specialist centre administration.
Rituximab (anti-CD20, off-label in many countries but widely used) depletes B cells similarly to ocrelizumab. It is commonly used in NMOSD and in MS where ocrelizumab is not available or not affordable. [1]
Haematopoietic stem cell transplantation (HSCT)
For aggressive, highly active RRMS that is refractory to other DMTs, autologous HSCT may be considered. It involves immunoablation (typically with cyclophosphamide and anti-thymocyte globulin) followed by reinfusion of the patient's own haematopoietic stem cells, effectively "resetting" the immune system. It can produce dramatic and durable remission in selected patients but carries a treatment-related mortality of 1 to 2 per cent, risks of infertility and secondary autoimmunity, and its long-term efficacy relative to high-efficacy DMTs (ocrelizumab, alemtuzumab) is debated. It is reserved for specialist centres and carefully selected patients. [1]
Choosing a DMT — escalation versus induction
Two strategies exist:
- Escalation — start with a safe, moderately effective first-line agent (interferon, glatiramer, dimethyl fumarate) and escalate to high-efficacy therapy only if breakthrough disease occurs. This prioritises safety but risks early disability accumulation.
- Induction — start with a high-efficacy agent (natalizumab, ocrelizumab, alemtuzumab) from the outset in patients with poor prognostic features (frequent relapses, high MRI lesion burden, spinal cord involvement, incomplete recovery). This prioritises early disease control but accepts higher drug risk. [1]
The modern trend is towards earlier use of high-efficacy therapy, supported by evidence that early inflammatory control prevents long-term disability. The choice is individualised and shared with the patient. [1]
Symptom management
Symptom management is as important as DMT in determining quality of life. A multidisciplinary team (neurology, rehabilitation medicine, physiotherapy, occupational therapy, urology, neuropsychology, and social work) is the standard of care. [1]
Spasticity
Management is stepped:
- Physiotherapy and stretching — first-line for all patients.
- Oral antispasticity agents — baclofen (5 to 80 mg daily in divided doses, starting at 5 mg three times daily) and tizanidine (2 to 36 mg daily). Both cause sedation and weakness. Diazepam and dantrolene are alternatives (dantrolene causes generalised muscle weakness and carries hepatotoxicity risk).
- Botulinum toxin injections — for focal spasticity (e.g., a spastic flexed arm or equinovarus foot), with physiotherapy to prevent contracture.
- Intrathecal baclofen — for severe generalised spasticity refractory to oral therapy, delivered via an implanted pump. It dramatically reduces spasticity at a fraction of the oral dose, with fewer systemic side effects, but carries surgical and infection risks. [1]
Fatigue
Fatigue is the most common and most disabling symptom. Management includes:
- Ruling out treatable causes — anaemia, thyroid dysfunction, sleep apnoea, depression, vitamin D deficiency.
- Energy conservation strategies and graded exercise.
- Amantadine (100 mg twice daily) — modestly effective, with a dopamine-modulating mechanism. Side effects include livedo reticularis, insomnia, and (rarely) hallucinations.
- Modafinil — off-label, for selected patients. [1]
Bladder dysfunction
Neurogenic bladder affects 75 per cent of patients with established disease. Two patterns predominate:
- Detrusor overactivity (urge incontinence) — anticholinergic agents (oxybutynin, tolterodine, solifenacin) or mirabegron (beta-3 agonist). Monitor for urinary retention.
- Detrusor-sphincter dyssynergia (retention) — intermittent self-catheterisation is the definitive management. Post-void residual volume should be checked.
- Combined patterns are common and may require urodynamic studies to guide management. Botulinum toxin injection of the detrusor is effective for refractory overactivity. [1]
Neuropathic pain
- Trigeminal neuralgia in MS — carbamazepine is first-line (as for classic TN); alternatives include oxcarbazepine, gabapentin, pregabalin, and baclofen. MS-related TN may be bilateral and may respond to DMT escalation.
- Lhermitte sign and painful tonic spasms — carbamazepine, gabapentin, or pregabalin.
- Chronic neuropathic limb pain — gabapentin, pregabalin, amitriptyline, or duloxetine. [1]
Mood and cognition
- Depression — SSRIs are first-line (sertraline, escitalopram). Tricyclics should be used with caution in patients with bladder dysfunction (anticholinergic effects worsen retention). Active screening (PHQ-9) is essential.
- Cognitive impairment — neuropsychological assessment, cognitive rehabilitation, and ensuring DMT optimisation (cognitive decline may signal ongoing disease activity). [1]
Pregnancy and reproductive health
MS is twice as common in women as in men and typically presents in the childbearing years, so pregnancy planning is a core part of management. The landmark PRIMS study (Confavreux, 1998) prospectively followed 254 women through 269 pregnancies and established the natural history of MS in pregnancy [13]:
- Relapse rate drops during pregnancy — significantly in the first trimester and markedly in the third trimester (annualised relapse rate of 0.2 in the third trimester compared with 0.7 pre-pregnancy), likely due to the immunotolerant pregnancy state (shift from Th1 to Th2, regulatory T-cell expansion).
- Relapse rate rises postpartum — in the first 3 months postpartum, the annualised relapse rate is 1.2 (significantly higher than pre-pregnancy), reflecting immune system reactivation. However, 72 per cent of women do not experience a postpartum relapse.
- Breastfeeding — exclusive breastfeeding may be protective against postpartum relapse (the PRIMS study found no adverse effect, and subsequent studies suggest a benefit). If the patient chooses to breastfeed, DMTs are typically withheld (with the exception of glatiramer, which is safe in breastfeeding).
- Epidural analgesia — the PRIMS study found no adverse effect on relapse rate or disability progression.
- Long-term — pregnancy does not worsen long-term disability. [1]
DMT planning around pregnancy
- Interferon-beta and glatiramer — can be continued until pregnancy is confirmed (category B), and glatiramer can be continued throughout pregnancy. Interferon is typically stopped at confirmation.
- Dimethyl fumarate, fingolimod, teriflunomide, cladribine, siponimod — discontinue before conception. Washout periods: dimethyl fumarate 1 month; fingolimod 2 months; teriflunomide requires accelerated elimination (cholestyramine) or wait 2 years; cladribine 6 months; siponimod is contraindicated.
- Natalizumab — may be continued through pregnancy in patients with highly active disease, because stopping it risks severe rebound relapses. If stopped, restart promptly postpartum. The decision is individualised and made with MDT input.
- Ocrelizumab and rituximab — deplete B cells for 6 to 12 months, so discontinue 6 to 12 months before conception. If pregnancy occurs unexpectedly, the drug is already administered — B-cell depletion will continue, and the neonate should be monitored for B-cell depletion (vaccination with live vaccines should be deferred).
- Alemtuzumab — discontinue 4 months before conception; secondary autoimmunity monitoring must continue. [1]
Prognosis and the goals of care
MS is a lifelong disease with a highly variable course. Untreated, the median time from onset to needing a walking aid (EDSS 6) is approximately 15 years, and to wheelchair dependence (EDSS 8) approximately 25 years. However, the DMT era has fundamentally changed this trajectory — patients diagnosed and treated early with effective DMTs have substantially better outcomes. [1]
Favourable prognostic factors include female sex, young onset, relapsing-remitting course, optic neuritis or sensory presentation, high MRI lesion burden with good recovery from the first attack, and low disability at 5 years. Poor prognostic factors include male sex, older onset, motor or cerebellar presentation, primary progressive course, incomplete recovery from relapses, and early spinal cord or brainstem involvement. [1]
Life expectancy is reduced by approximately 7 years compared with the general population, with the principal causes of death being complications of advanced disease (aspiration pneumonia, urinary tract infections, pressure injuries) and cardiovascular disease. DMTs are progressively narrowing this gap. [1]
DCE integration point: In the MS long case, the candidate must present a coherent management plan that addresses four domains: (1) the DMT strategy (drug choice, efficacy, risk, monitoring, NEDA targets), (2) acute relapse management, (3) symptom management (spasticity, fatigue, bladder, mood, pain), and (4) reproductive and life planning (pregnancy, occupation, driving, advance care planning in advanced disease). A candidate who can name the specific DMT, its mechanism, its monitoring requirements, its PML or autoimmunity risk, and the washout period for pregnancy will pass the MS long case with distinction. [1]
High-yield DWE points and exam traps
- RRMS is 85 per cent at onset; PPMS is 15 per cent. SPMS develops from RRMS over 15 to 20 years in most patients. The 2013 Lublin revisions add activity and progression modifiers to each phenotype [2].
- McDonald 2017: DIS requires lesions in 2 of 4 areas (periventricular, juxtacortical/cortical, infratentorial, spinal cord). DIT requires a new lesion on follow-up, or simultaneous enhancing and non-enhancing lesions, or CSF oligoclonal bands. The 2017 revision allows CSF OCB to substitute for DIT and counts symptomatic and cortical lesions [1].
- EBV is a necessary trigger. Bjornevik 2022 — 32-fold increased risk of MS after EBV seroconversion; all but one of 801 MS cases were EBV-positive before diagnosis [3].
- HLA-DRB1*15:01 is the strongest genetic association (approximately 3-fold increased risk).
- Ocrelizumab is the only DMT approved for PPMS (ORATORIO trial, 24 per cent reduction in disability progression) and is also highly effective in RRMS (OPERA trials, superior to interferon) [4] [5].
- Natalizumab PML risk is stratified by JCV positivity, prior immunosuppression, and treatment duration over 24 months. JCV-negative patients have the lowest risk (fewer than 0.1 per 1000); the highest-risk group has up to 11 per 1000 [7].
- Alemtuzumab causes secondary autoimmunity in up to 30 per cent (thyroid disease, ITP, Goodpasture) — requires monthly blood monitoring for 48 months [8] [9].
- Siponimod is the first DMT for SPMS (EXPAND trial, 21 per cent reduction in disability progression) [6].
- Pregnancy: relapse rate drops (lowest in third trimester) and rebounds postpartum (first 3 months). Breastfeeding may be protective. DMT washout periods vary by drug [13].
- Plasma exchange (5 to 7 exchanges) is the treatment for steroid-refractory relapses — do not repeat steroid courses.
- The Uhthoff phenomenon is NOT a relapse — it is transient conduction block in previously demyelinated axons at higher body temperature.
- Always check AQP4-IgG and MOG-IgG before diagnosing MS — NMOSD and MOGAD have different treatments and some MS DMTs may be harmful.
- Internuclear ophthalmoplegia in a young patient is MS until proven otherwise — bilateral INO is highly suggestive.
- Lhermitte sign (electric shock down spine on neck flexion) indicates cervical cord demyelination — it is characteristic but not specific to MS.
- VEP shows delayed P100 latency with preserved waveform — demonstrating slowed conduction through a demyelinated (but not destroyed) optic nerve.
References
- [1]Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, Correale J, Fazekas F, Filippi M, Freedman MS, Fujihara K, Galetta SL, Hartung HP, Lublin FD, Marrie RA, Miller AE, Miller DH, Montalban X, Mowry EM, Sorensen PS, Tintore M, Traboulsee AL, Trojano M, Uitdehaag BMJ, Vukusic S, Waubant E, Weinshenker BG, Reingold SC, Cohen JA Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria Lancet Neurol, 2018.PMID 29275977
- [2]Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sorensen PS, Thompson AJ, Wolinsky JS, Balcer LJ, Banwell B, Barkhof F, Bebo B, Calabresi PA, Clanet M, Comi G, Fox RJ, Freedman MS, Goodman AD, Inglese M, Kappos L, Kieseier BC, Lincoln JA, Lubetzki C, Miller AE, Montalban X, O'Connor PW, Petkau J, Pozzilli C, Rudick RA, Sormani MP, Stuve O, Waubant E, Polman CH Defining the clinical course of multiple sclerosis: the 2013 revisions Neurology, 2014.PMID 24871874
- [3]Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, Elledge SJ, Niebuhr DW, Scher AI, Munger KL, Ascherio A Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis Science, 2022.PMID 35025605
- [4]Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G, de Seze J, Giovannoni G, Hartung HP, Hemmer B, Lublin F, Rammohan KW, Selmaj K, Traboulsee A, Sauter A, Masterman D, Fontoura P, Belachew S, Garren H, Mairon N, Chin P, Wolinsky JS Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis N Engl J Med, 2017.PMID 28002688
- [5]Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung HP, Hemmer B, Lublin F, Montalban X, Rammohan KW, Selmaj K, Traboulsee A, Wolinsky JS, Zangillo JC, Younes M, Arnold DL, Masterman D, Chin P, Mairon N, Garren H, Kappos L Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis N Engl J Med, 2017.PMID 28002679
- [6]Kappos L, Bar-Or A, Cree BAC, Hartung HP, Jeffery DR, Kappos L, Arnold DL, Bar-Or A, Cree B, Hartung HP, Jeffery D, Kapoor R, Kurukulasuriya N, Pellegrini F, Pirotta M, Pradhan A, Selmaj K, Shinohara T, Tomic D, Trojano M, Wolinsky JS Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study Lancet, 2018.PMID 29576505
- [7]Bloomgren G, Richman S, Hotermans C, Subramanyam M, Goelz S, Natarajan A, Lee S, Havrdova E, Selmaj K, Phillips JT, Sandrock E, Kappos L, Ticho B, Bozic C Risk of natalizumab-associated progressive multifocal leukoencephalopathy N Engl J Med, 2012.PMID 22591293
- [8]Cohen JA, Coles AJ, Arnold DL, Confavreux C, Fox EJ, Hartung HP, Havrdova E, Selmaj KW, Weiner HL, Fisher E, Brinar VV, Giovannoni G, Stojanovic M, Erian BO, Weber MS, Mairon N, Klingelschmitt G, Dangond F Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial Lancet, 2012.PMID 23122652
- [9]Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ, Hartung HP, Havrdova E, Selmaj KW, Weiner HL, Fisher E, Brinar VV, Giovannoni G, Stojanovic M, Erian BO, Weber MS, Mairon N, Klingelschmitt G, Dangond F Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial Lancet, 2012.PMID 23122650
- [10]Giovannoni G, Comi G, Cook S, Rammohan K, Rieckmann P, Soelberg Sorensen P, Vermersch P, Chang P, Hamlett A, Musch B, Greenberg SJ A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis N Engl J Med, 2010.PMID 20089960
- [11]Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, Tornatore C, Sweetser MT, Yang M, Sheikh SI, Dawson KT Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis N Engl J Med, 2012.PMID 22992073
- [12]PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group Lancet, 1998.PMID 9820297
- [13]Confavreux C, Hutchinson M, Hours MM, Cortinovis-Tourniaire P, Moreau T, the Pregnancy in Multiple Sclerosis Group Rate of pregnancy-related relapse in multiple sclerosis. Pregnancy in Multiple Sclerosis Group N Engl J Med, 1998.PMID 9682040