Phys · neurological
Myasthenia Gravis
Also known as myasthenia gravis · MG · fatigable weakness · acetylcholine receptor antibody myasthenia · AChR-MG · MuSK antibody myasthenia · MuSK-MG · seronegative myasthenia · ocular myasthenia · myasthenic crisis · cholinergic crisis · penicillamine-induced myasthenia · immune checkpoint inhibitor myasthenia
Consultant-physician-depth guide to myasthenia gravis: autoimmune neuromuscular junction pathology (anti-AChR, anti-MuSK), fatigable weakness across ocular, bulbar, limb and respiratory muscles, bedside and serological diagnosis, pyridostigmine and immunosuppression, thymectomy (MGTX), and myasthenic crisis management for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Myasthenia Gravis

The answer first
Myasthenia gravis (MG) is an autoimmune disorder of the postsynaptic neuromuscular junction in which pathogenic antibodies reduce the number or function of acetylcholine receptors (AChRs), producing fatigable weakness — weakness that worsens with sustained activity and improves with rest. It is the commonest disorder of neuromuscular transmission. The two questions that drive every clinical decision in MG are: is this truly fatigable weakness (and not a fixed myopathy, a cranial nerve palsy, or Lambert-Eaton)? and which antibody subtype is it (AChR, MuSK, or seronegative)? — because the antibody determines the phenotype, the response to treatment, and the role of thymectomy. [1]
The clinical decision rules a registrar must own at viva: [1]
- Fatigability is the diagnosis. A single normal examination does not exclude MG — a patient with variable ptosis or diplopia must be tested under fatiguing conditions: sustained upgaze for one minute, repeated eye opening, or the ice pack test. The signature is variability with use.
- Antibody subtype changes management. Anti-AChR antibodies are present in about 85 per cent of generalized MG; anti-MuSK in about 5 to 8 per cent; the remainder are seronegative. MuSK-MG is a different disease — prominent bulbar and respiratory weakness, female predominance, poor response to pyridostigmine (which can worsen it), and no benefit from thymectomy [9]. Rituximab is particularly effective in MuSK-MG [10].
- Pyridostigmine is symptomatic, not disease-modifying. It improves most AChR-MG but does not alter the immune attack. Generalized MG needs immunosuppression (prednisolone plus a steroid-sparing agent) to achieve and maintain remission [2].
- Myasthenic crisis is a respiratory emergency. Assess vital capacity and negative inspiratory force early. A worsening patient needs IVIG or plasma exchange for rapid immunotherapy and ventilatory support — not more pyridostigmine, unless there are clear cholinergic signs (sweating, salivation, miosis, fasciculations) suggesting cholinergic crisis.
The organising principle is antibody-stratified, escalation-based management: confirm fatigability, identify the antibody, start symptomatic and immunosuppressive therapy, screen for thymoma, and reserve biological therapy (rituximab, eculizumab, FcRn inhibitors) for refractory disease. [1]
Pathophysiology — the neuromuscular junction under attack
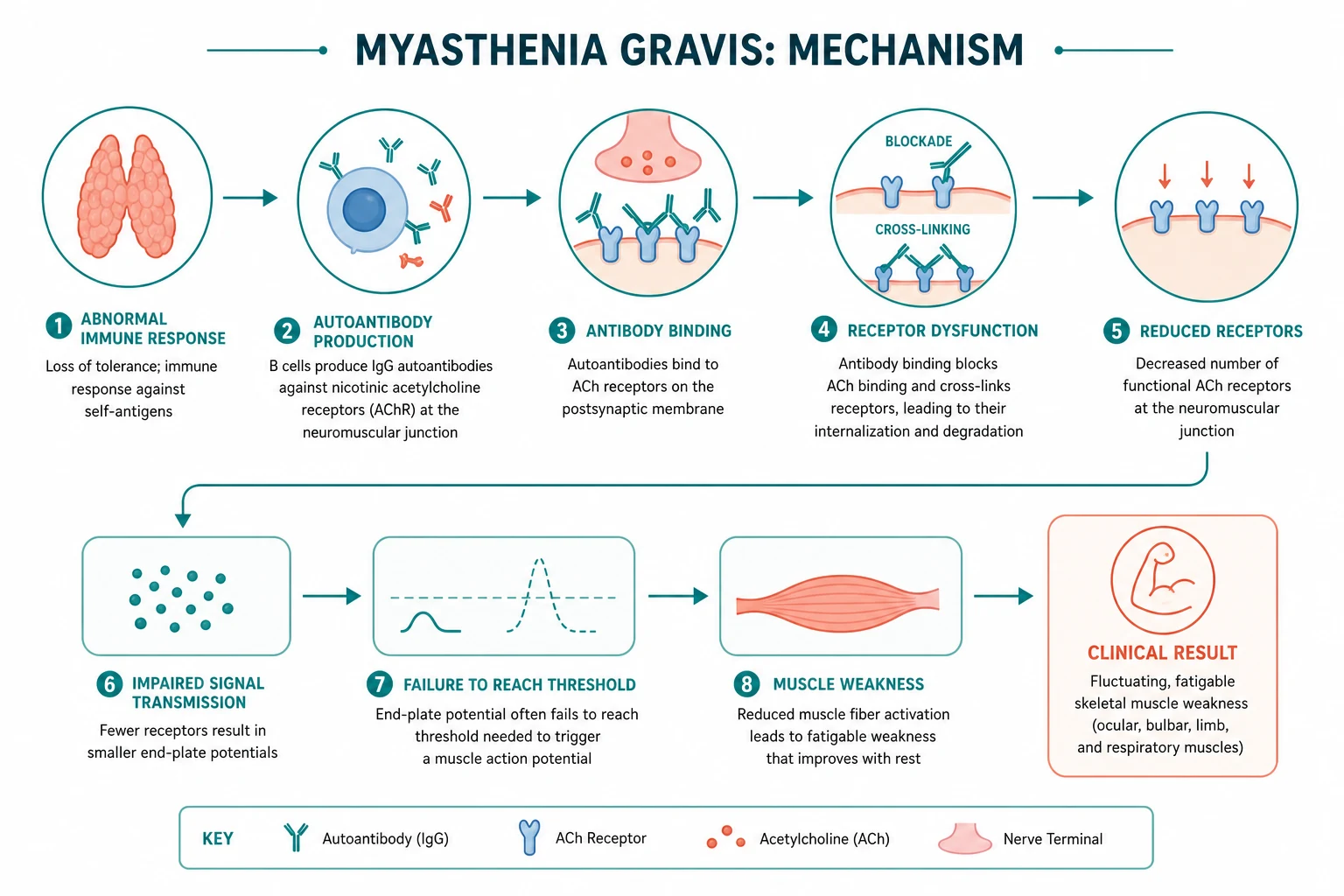

Normally, a motor nerve action potential releases acetylcholine (ACh) from presynaptic vesicles into the synaptic cleft. ACh binds nicotinic AChRs clustered in the deep junctional folds of the motor end plate, depolarising the muscle fibre. Crucially, the system has a large safety factor: far more ACh is released, and far more AChRs are available, than the minimum needed to trigger a muscle action potential. MG reduces that safety factor — so transmission succeeds at rest but fails after repeated firing, when ACh release falls and the depleted receptor reserve can no longer generate an end-plate potential. [1]
Anti-AChR antibody MG — the prototypic disease
About 85 per cent of generalized MG is driven by IgG1 and IgG3 antibodies against the postsynaptic AChR [5]. Three mechanisms operate together:
- Complement-mediated destruction of the junctional folds. IgG1 and IgG3 fix complement at the postsynaptic membrane, activating the membrane attack complex (C5b-9). This destroys the elaborate junctional folding that normally concentrates AChRs, flattening and simplifying the end plate.
- Receptor blockade. Some antibodies bind the ACh binding site directly and competitively block ACh.
- Receptor modulation (internalisation). Antibody cross-linking of adjacent AChRs accelerates their endocytosis and degradation, reducing receptor density further. [1]
The net effect is a reduced density of functional AChRs on a simplified postsynaptic membrane. The safety factor falls. At rest, transmission succeeds; after sustained activity, the falling ACh release is insufficient to depolarise the end plate, and transmission fails — weakness appears. This is the molecular basis of fatigability. [1]
Anti-MuSK antibody MG — a different disease
Anti-MuSK antibodies are found in about 5 to 8 per cent of MG, and in 30 to 40 per cent of AChR-negative generalized MG [8]. MuSK (muscle-specific tyrosine kinase) is part of the agrin-LRP4-MuSK signalling pathway that clusters and maintains AChRs at the junction during development and adult life. MuSK antibodies are predominantly IgG4 — they do not fix complement. Instead, they block the LRP4-MuSK interaction and disrupt agrin-dependent AChR clustering, dispersing AChRs without destroying the junctional folds. The result is a distinct phenotype: prominent bulbar, facial, neck and respiratory weakness with relative ocular sparing, a female predominance (about 85 per cent), earlier onset, and a higher rate of crisis [8]. Critically, MuSK-MG responds poorly — and sometimes worsens — with pyridostigmine, and does not benefit from thymectomy [9].
Seronegative MG
About 5 to 10 per cent of patients have no detectable AChR or MuSK antibodies on standard assays. Some have antibodies against LRP4 or agrin; in others, the antibodies are below the detection threshold. The phenotype usually resembles mild-to-moderate AChR-MG. Treatment follows the AChR-MG pathway. [1]
The thymus in MG
The thymus is central to AChR-MG. Thymic hyperplasia with germinal centres is found in about 65 per cent of AChR-MG patients — these germinal centres contain B cells producing anti-AChR antibodies. Thymoma is found in 10 to 15 per cent and is the source of autoreactive T cells. This explains why thymectomy is a rational and effective treatment in AChR-MG (the MGTX trial proved benefit) but is not indicated in MuSK-MG, where the thymus is histologically normal and not part of the pathology [4][9].
DWE high-yield: The antibody subclass matters. AChR-MG is complement-driven (IgG1/IgG3) — this is why eculizumab, a C5 complement inhibitor, works in refractory AChR-MG. MuSK-MG is IgG4-driven — complement blockade does not help, but B-cell depletion (rituximab) is highly effective. This mechanistic distinction is a classic viva and MCQ discriminator. [1]
Clinical spectrum — the signature is fatigability
The cardinal feature of MG is weakness that worsens with sustained activity and improves with rest. Every muscle group can be involved, but the distribution follows a recognisable hierarchy: ocular muscles are affected first and most often, then bulbar, then limb, then respiratory. [1]
Ocular MG
Ptosis and diplopia are the presenting symptoms in about half of patients and develop at some point in over 90 per cent [5]. The ocular signs are variable and asymmetric: ptosis may alternate sides or shift through the day, typically worsening toward evening or after reading. Diplopia is often vertical or oblique and may be elicited only on sustained gaze in a particular direction. Purely ocular MG (no generalized weakness for two or more years) carries a lower antibody titre and a different prognosis — about half of ocular MG will eventually generalise.
Bulbar MG
Bulbar involvement produces fatigable dysarthria (speech slurs and fades with prolonged conversation), fatigable chewing (the patient cannot finish a meal), dysphagia, and nasal regurgitation from palatal weakness. A nasal or "myasthenic" voice, pooling of secretions, and a weak cough are red flags for impending respiratory compromise. Bulbar weakness is prominent in MuSK-MG and predicts crisis. [1]
Limb MG
Limb weakness is proximal greater than distal and fatigable. Patients report difficulty lifting objects overhead, climbing stairs, or rising from a chair after the activity is sustained — but they can do it once. Head drop from neck extensor weakness is a characteristic and disabling sign. Examination must test for fatigability directly: ask the patient to hold the arms outstretched for a minute, or to repeatedly sit-to-stand, and watch power decline. [1]
Respiratory MG and crisis
Diaphragmatic and intercostal weakness produces breathlessness that worsens on exertion or lying flat, a weak voice, and an ineffective cough. When weakness progresses to ventilatory failure, the patient is in myasthenic crisis — the feared complication and the leading cause of MG mortality. Crisis is covered in detail below. [1]
DCE viva trap: Examiners want you to demonstrate that you understand fatigability as a dynamic phenomenon, not a fixed deficit. If you examine an MG short-case patient only at rest and report "power 5 out of 5", you have missed the diagnosis. You must provoke the weakness — sustained upgaze, repeated movement, prolonged speech — and show that it recovers with rest. [1]
MGFA clinical classification — severity stratification

The MGFA clinical classification, established in 2000, stratifies MG severity and is the common language of trials and clinic letters [1]. Learn it — examiners expect you to use it.
| Class | Definition |
|---|---|
| Class I | Any ocular muscle weakness; all other muscle strength normal |
| Class II | Mild weakness affecting other than ocular muscles; may have ocular weakness of any severity. IIa = predominantly limb/axial; IIb = predominantly bulbar/respiratory |
| Class III | Moderate weakness affecting other than ocular muscles. IIIa = limb/axial; IIIb = bulbar/respiratory |
| Class IV | Severe weakness affecting other than ocular muscles. IVa = limb/axial; IVb = bulbar/respiratory |
| Class V | Intubation required, with or without mechanical ventilation (except routine postoperative) |
DWE high-yield: Class V is defined by intubation — it is the operational definition of myasthenic crisis. A pre-intubation patient with FVC under 15 mL/kg is heading there; the role of the physician is to escalate before the patient crosses into Class V. [1]
Differentials — what else fatigues or fluctuates
Not all fatigable or fluctuating weakness is MG. The discriminating diagnosis is usually made at the bedside. [1]
| Condition | Key discriminator from MG |
|---|---|
| Lambert-Eaton myasthenic syndrome (LEMS) | Weakness improves with brief exercise (facilitation); reflexes depressed or absent but reappear after exercise; autonomic features (dry mouth, impotence, constipation); paraneoplastic (small cell lung cancer); anti-P/Q-type voltage-gated calcium channel antibody; presynaptic pathology |
| Chronic inflammatory demyelinating polyneuropathy (CIDP) | Symmetric distal and proximal weakness, sensory loss, areflexia; not fatigable in the MG sense; demyelinating on nerve conduction |
| Motor neuron disease (ALS) | Progressive, not fatigable; upper and lower motor neuron signs; tongue fasciculations; no fluctuation |
| Thyroid myopathy | Proximal weakness without fatigability; check TSH — autoimmune thyroid disease coexists in up to 15 per cent of MG |
| Oculopharyngeal muscular dystrophy | Adult onset, family history, symmetric ptosis and dysphagia, slowly progressive, no fluctuation |
| Chronic progressive external ophthalmoplegia (CPEO) | Mitochondrial; symmetric, slowly progressive ophthalmoplegia and ptosis, no fluctuation or fatigability, no response to pyridostigmine |
| Cranial nerve palsy | Fixed, follows a single nerve distribution (e.g. third nerve: ptosis, "down and out" eye, pupil involvement), not fatigable |
| Congenital myasthenic syndromes | Onset in infancy or childhood, family history, no antibodies; sodium-channel or AChR subunit mutations |
| Botulism | Descending symmetric paralysis with dilated pupils, autonomic failure; presynaptic ACh release blockade |
The single most important discriminator for the exam is MG versus LEMS. MG fatigues with repetition; LEMS facilitates (the patient is stronger after a few seconds of sustained contraction). LEMS has depressed reflexes that return with use and autonomic features; MG has normal reflexes and no autonomic features. LEMS is paraneoplastic (small cell lung cancer in about half) and requires an anti-P/Q-type voltage-gated calcium channel antibody and a CT chest. [1]
DCE short-case trap: When presented with "ptosis and ophthalmoplegia" in a short case, always ask: is it fatigable (MG), fixed and symmetric (CPEO, oculopharyngeal MD), or fixed and asymmetric with a nerve distribution (third nerve palsy)? The sustained-upgaze and ice-pack tests separate these at the bedside. [1]
Diagnosis — bedside, serology, electrophysiology, imaging

The diagnosis of MG rests on four pillars: clinical pattern of fatigability, serology, electrophysiology, and thymus imaging. [1]
Bedside tests — demonstrate fatigability at the bedside
- Sustained upgaze for one minute. Ask the patient to look up at the ceiling and hold the gaze. In MG, ptosis develops or worsens over 30 to 60 seconds as the levator palpebrae fatigues. A positive test reproduces or worsens the patient's ptosis.
- Ice pack test. Place a pack of ice over the ptotic eyelid for two minutes. Cold inhibits acetylcholinesterase and improves neuromuscular transmission locally — the ptosis improves visibly. Sensitivity for ocular MG is high (over 90 per cent) and it is simple, safe, and bedside.
- Cogan lid twitch. After sustained downgaze, rapid upgaze produces an excessive upward twitch of the upper lid before it settles — a sign of fatigable then briefly recovered levator function.
- Fatigability of speech. Ask the patient to count slowly to 100; a myasthenic voice fades and slurs with sustained phonation. [1]
The edrophonium (Tensilon) test — a short-acting acetylcholinesterase inhibitor given intravenously — is now rarely used because of the risk of bradycardia, syncope and bronchospasm, and because serology and bedside tests are usually sufficient. If performed, atropine and resuscitation equipment must be at the bedside. [1]
Serology — the antibody panel
| Antibody | Sensitivity | When to test |
|---|---|---|
| Anti-AChR (binding) | About 85 per cent in generalized MG; about 50 per cent in ocular MG | First-line in all suspected MG |
| Anti-MuSK | 5 to 8 per cent of all MG; 30 to 40 per cent of AChR-negative generalized MG | If anti-AChR is negative, especially with bulbar/respiratory phenotype |
| Anti-LRP4 | 2 to 5 per cent; some seronegative cases | Selected seronegative cases |
| Anti-striated muscle (anti-titin, anti-ryanodine receptor) | Marker of thymoma (especially under 50 years) | If thymoma suspected; also correlates with disease severity |
A negative AChR antibody does not exclude MG — always test anti-MuSK in an AChR-negative patient with a compatible phenotype, because the treatment algorithm diverges. [1]
Electrophysiology — confirm impaired transmission
- Repetitive nerve stimulation (RNS). Stimulate a motor nerve (typically a distal and a proximal muscle — facial or accessory nerve for ocular/bulbar disease) at 3 Hz. In MG, the compound muscle action potential amplitude falls by more than 10 per cent between the first and fourth or fifth stimulus — a decremental response. Sensitivity is moderate (around 75 per cent in generalized MG, lower in ocular), higher with proximal and facial muscles.
- Single-fibre EMG (SFEMG). The most sensitive test for MG (over 95 per cent for ocular MG when the appropriate muscles are studied). It measures jitter — the variability in time between two muscle fibres of the same motor unit — and blocking (failure of one fibre to fire). In MG, jitter is increased and blocking appears. The limitation is that it is technically demanding, operator-dependent, and non-specific (abnormal in any neuromuscular junction disorder, including LEMS and ALS). [1]
DWE high-yield: The exam-worthy sequence is: decrement on RNS confirms impaired neuromuscular transmission; increased jitter on SFEMG is the most sensitive but non-specific. Antibodies confirm the autoimmune, postsynaptic nature. LEMS shows a low compound muscle action potential amplitude that increments (facilitates) after brief exercise or high-frequency stimulation — the mirror image of MG. [1]
CT thorax — screen for thymoma
CT of the thorax is mandatory at diagnosis in every MG patient. Thymoma is present in 10 to 15 per cent; thymic hyperplasia in about 65 per cent of AChR-MG. A mediastinal mass in an MG patient is a thymoma until proven otherwise, and thymoma mandates thymectomy (it may be malignant). MRI may add detail for equivocal lesions. MuSK-MG is not associated with thymoma. [1]
Management — symptomatic, immunosuppression, thymectomy, crisis


Management is layered: symptomatic therapy first, immunosuppression to control the disease, thymectomy where indicated, and biological therapy or rapid immunotherapy (IVIG/plasma exchange) for refractory disease or crisis [2][3].
Symptomatic therapy — pyridostigmine
Pyridostigmine is an acetylcholinesterase inhibitor that prolongs ACh action at the junction. It is the first drug in essentially all MG and provides meaningful symptomatic benefit in most AChR-MG. [1]
- Dose: 30 to 60 mg orally, four times daily (typically 60 mg every 4 to 6 hours); can be titrated up to 120 mg per dose and a total of 600 to 1200 mg daily. A sustained-release formulation at bedtime helps morning weakness.
- Onset: 30 minutes; peak 1 to 2 hours; duration 3 to 6 hours.
- Adverse effects: cholinergic — diarrhoea, abdominal cramps, nausea, salivation, lacrimation, bronchorrhoea, miosis, fasciculations. Excess produces cholinergic excess (see crisis below).
- MuSK caveat: pyridostigmine is often ineffective or frankly worsens MuSK-MG (up to half of MuSK patients are non-responsive or worse) [8]. Do not escalate the dose in a deteriorating MuSK patient — reduce it.
Pyridostigmine does not modify the underlying immune disease. Patients with generalized MG need immunosuppression. [1]
Immunosuppression — prednisolone and steroid-sparing agents
Prednisolone is the mainstay of immunosuppression. It produces improvement in the majority, but two principles govern its use: [1]
- Start low and titrate up. Begin at 10 to 20 mg daily and increase gradually over 2 to 4 weeks toward a target of 0.75 to 1 mg/kg/day (typically 50 to 80 mg). The reason for slow initiation is that high-dose steroids can precipitate acute worsening of MG within the first 1 to 2 weeks — a well-recognised steroid-induced exacerbation. Once a response is achieved (usually 2 to 4 weeks at the target dose), taper slowly to the lowest effective maintenance dose.
- Add a steroid-sparing agent early. Because long-term prednisolone causes osteoporosis, diabetes, hypertension, cataracts, weight gain, infection and adrenal suppression, introduce a steroid-sparing immunosuppressant early so prednisolone can be tapered. The standard agents: [1]
| Agent | Key points and monitoring |
|---|---|
| Azathioprine | First-line steroid-sparer. Check TPMT activity before starting (deficiency causes fatal myelosuppression). Dose 1 to 3 mg/kg/day. Takes 6 to 12 months to work. Monitor FBC and LFTs monthly initially. Adverse effects: hepatitis, myelosuppression, pancreatitis, increased infection risk |
| Methotrexate | 7.5 to 15 mg weekly with folic acid. An alternative steroid-sparer; monitor LFTs |
| Tacrolimus / ciclosporin | Calcineurin inhibitors; nephrotoxicity and hypertension limit use. Tacrolimus preferred where rapid effect needed |
| Rituximab | Anti-CD20 B-cell depletion. Particularly effective in MuSK-MG [10] and refractory AChR-MG. Given as two infusions of 1 g two weeks apart; monitor CD19 and IgG |
The aim of immunosuppression is the MGFA post-intervention status of minimal manifestations (MM) or better — the patient has no functional limitation from MG — sustained on the lowest possible drug burden. [1]
Thymectomy — when and for whom
Thymectomy is a defining management decision in MG and a high-yield exam topic. The rules: [1]
- Thymoma: always. Any MG patient with a thymoma (10 to 15 per cent) undergoes thymectomy — the tumour may be malignant and the procedure is both diagnostic and therapeutic.
- Non-thymomatous AChR-positive generalized MG: consider. The MGTX trial (Wolfe et al., NEJM 2016) randomised 126 patients with non-thymomatous AChR-MG to extended transsternal thymectomy plus prednisone versus prednisone alone. Over 3 years, the thymectomy group had a lower time-weighted average QMG score, required a lower prednisone dose, and needed fewer azathioprine courses and hospitalisations [4]. Thymectomy is therefore recommended — particularly in young (under 50) AChR-positive generalized MG — as a strategy to improve outcome and reduce drug burden. It is not a cure, and benefit accrues over years, not weeks.
- MuSK-MG: not indicated. Thymectomy does not improve MuSK-MG [9] — the thymus is not part of the pathology. Do not offer it.
- Ocular-only MG and the elderly: thymectomy is generally not offered; the evidence does not support it.
Biological therapy for refractory disease
For patients refractory to prednisolone plus a steroid-sparing agent, targeted biologicals are now available, anchored by the pathophysiology: [1]
- Rituximab (anti-CD20) — depletes B cells. Particularly effective in MuSK-MG, with shorter time to remission than in AChR-MG [10], and useful in refractory AChR-MG.
- Eculizumab — a monoclonal antibody against complement C5, blocking formation of the membrane attack complex. Approved for refractory AChR-MG on the basis of the REGAIN trial (Howard et al., Lancet Neurology 2017) and its open-label extension, in which 57 per cent of patients achieved minimal-manifestation status by 130 weeks [6][7]. It does not work in MuSK-MG (IgG4 does not fix complement). Patients must be vaccinated against meningococcus at least two weeks before the first dose, because terminal complement blockade carries a 1 to 2 per cent risk of meningococcal infection.
- FcRn inhibitors (efgartigimod, rozanolixizumab) — block the neonatal Fc receptor that recycles IgG, accelerating degradation of pathogenic IgG including anti-AChR antibodies. Approved for generalized AChR-MG; rapid onset over weeks.
- Ravulizumab — a longer-acting C5 inhibitor (8-weekly dosing) alternative to eculizumab.
DWE high-yield: The mechanistic logic is examinable. Eculizumab works in AChR-MG because the pathogenic antibody is complement-fixing (IgG1/IgG3); it does not work in MuSK-MG because MuSK antibodies are IgG4 and do not fix complement. Rituximab works well in both, but especially MuSK-MG. [1]
Drugs to avoid in myasthenia gravis
A large number of drugs impair neuromuscular transmission and can precipitate worsening or crisis. Every MG patient should carry an alert card. The key classes: [1]
| Drug class | Examples | Mechanism / risk |
|---|---|---|
| Aminoglycosides | Gentamicin, neomycin, tobramycin | Reduce presynaptic ACh release |
| Fluoroquinolones | Ciprofloxacin, levofloxacin | Direct NMJ toxicity |
| Macrolides / ketolides | Azithromycin, clarithromycin, telithromycin | Reduce ACh release |
| Beta-blockers | Propranolol, timolol (eye drops count) | Reduce ACh release; worsen ptosis |
| Calcium channel blockers | Verapamil, diltiazem | Inhibit ACh release |
| Magnesium | IV magnesium sulphate | Inhibits ACh release — can precipitate crisis |
| Neuromuscular blockers | Curare, rocuronium, botulinum toxin | Prolonged paralysis |
| Iodinated contrast | High-dose IV contrast | Can transiently worsen MG |
| Immune checkpoint inhibitors | Pembrolizumab, nivolumab, ipilimumab | Can trigger de novo or worsen existing MG; may coexist with myocarditis |
DWE high-yield: The single most dangerous drug interaction in an MG patient is intravenous magnesium — given for pre-eclampsia, torsades, or hypomagnesaemia, it can precipitate catastrophic respiratory failure by inhibiting ACh release. If magnesium must be given, the patient needs intensive monitoring and may need ventilation. [1]
Myasthenic crisis
Myasthenic crisis is respiratory or bulbar failure from MG weakness severe enough to threaten ventilation — MGFA Class V (intubated) or impending. It is the feared complication and historically carried high mortality; with modern intensive care, mortality is now 4 to 8 per cent, but it remains a neurological emergency. [1]
Precipitants
A crisis is usually triggered by an identifiable precipitant. Find it. Common causes: infection (respiratory, most often), aspiration, medication non-adherence, steroid taper, surgery, pregnancy and the postpartum period, drug exacerbation (above), and immune checkpoint inhibitors. [1]
Assessment — the 20/30/30 rule
Assess respiratory and bulbar function objectively and frequently. [1]
- Forced vital capacity (FVC): thresholds for intubation are around 15 to 20 mL/kg (a 60 kg patient at 20 mL/kg is 1.2 L).
- Negative inspiratory force (NIF): below 30 cmH2O predicts ventilatory failure.
- Bulbar function: weak cough, pooled secretions, nasal voice, inability to protect the airway. [1]
A useful practical rule is the 20/30/30 rule: FVC below 20 mL/kg, NIF below 30 cmH2O, or a rapidly deteriorating bulbar picture triggers escalation to the intensive care unit for ventilatory support. Do not wait for the patient to tire — by the time the arterial blood gas shows CO2 retention, the patient is in trouble. [1]
Management
- Airway and ventilation. Non-invasive ventilation (BiPAP) can buy time and may avoid intubation in a cooperative patient without excessive secretions. If bulbar weakness prevents airway protection or FVC is critically low, intubate early.
- Rapid immunotherapy — IVIG or plasma exchange. Both are effective; choose by availability and comorbidity:
- IVIG: 0.4 g/kg/day for 5 days (total 2 g/kg). Easier to administer, fewer line complications. [1] - Plasma exchange (plasmapheresis): 5 exchanges over 1 to 2 weeks. Faster onset (days), preferred in crisis and in MuSK-MG (where IVIG is less effective). Requires central venous access.
- Treat the precipitant. Antibiotics for infection, aspirated secretions, withdraw offending drugs.
- Review immunosuppression. Continue or optimise long-term immunosuppression; consider rituximab or eculizumab after the acute episode to reduce recurrence. [1]
Myasthenic versus cholinergic crisis
A deteriorating MG patient raises the question: is the weakness from too little acetylcholine (myasthenic crisis) or too much (cholinergic crisis from pyridostigmine excess)? The distinction matters because the treatments are opposite. [1]
| Feature | Myasthenic crisis | Cholinergic crisis |
|---|---|---|
| Pupils | Normal or dilated | Constricted (miosis) |
| Skin | Dry, pale | Sweaty, flushed |
| Secretions | Dry mouth, weak cough | Salivation, lacrimation, bronchorrhoea |
| Gut | Normal or constipated | Abdominal cramps, diarrhoea |
| Fasciculations | Absent | Present |
| Action | IVIG or plasma exchange; ventilatory support | Stop or reduce pyridostigmine; atropine |
DWE high-yield: In a deteriorating MG patient without cholinergic signs (dry, dilated pupils, no fasciculations), the diagnosis is myasthenic crisis — give rapid immunotherapy and ventilatory support. Adding more pyridostigmine is wrong and can precipitate a cholinergic crisis on top. Cholinergic crisis is rare and almost always follows pyridostigmine escalation in a deteriorating patient. [1]
DCE long-case approach
Opening statement (SASPOP)
"Ms Papadopoulos is a 38-year-old accountant with a 2-year history of acetylcholine-receptor-antibody-positive generalized myasthenia gravis. She presents for review after a recent myasthenic crisis triggered by a chest infection, with a newly diagnosed thymoma on CT thorax. She is currently on pyridostigmine 90 mg five times daily and prednisolone 40 mg daily, and has developed steroid-induced osteoporosis. Her main concerns are preventing further crises, the timing of thymectomy, and managing the side effects of her medication. [1]
Her main problems are:
- AChR-positive generalized MG, recently in crisis — disease control inadequate
- Thymoma on CT thorax — requires thymectomy
- Steroid-induced osteoporosis — on prednisolone 40 mg daily
- Risk of recurrent myasthenic crisis — precipitant (infection) and drug interactions to address
- Psychological and occupational impact — time off work, anxiety about driving and relapse" [1]
Integrated management plan
- Optimise disease control. Add a steroid-sparing agent — azathioprine, after confirming TPMT activity — so prednisolone can be tapered. Continue pyridostigmine for symptom control. Consider biological therapy if refractory.
- Thymectomy. Thymoma mandates thymectomy (extended transsternal). The MGTX trial supports a meaningful benefit in non-thymomatous AChR-MG; with a thymoma the indication is absolute. Refer to thoracic surgery.
- Crisis prevention. Vaccinate (influenza, pneumococcus, COVID-19); provide a medical alert card listing drugs to avoid; educate on early warning signs and an action plan; review drug list for MG-worsening agents.
- Steroid complication management. Bone protection — bisphosphonate and vitamin D/calcium, bone densitometry; monitor glucose and blood pressure; gastric protection if indicated.
- Shared decision-making. Discuss the timing of thymectomy relative to disease stability, fertility considerations, and the realistic goal of minimal manifestations rather than cure. [1]
DCE short-case approach: neurological examination for fatigable weakness
Instruction: "Examine this patient's neurological system. They have a drooping eyelid and double vision." [1]
Systematic routine
- General inspection: Observe for ptosis (asymmetric, variable), facial weakness (myasthenic snarl), nasal voice, head posture (compensating for diplopia). Look for a sternotomy scar (previous thymectomy), Cushingoid features (chronic steroids), and a medical-alert bracelet.
- Cranial nerves — eyes first:
- Inspect for ptosis; assess lid position and levator function.
- Sustained upgaze for one minute — does ptosis develop or worsen? (The key provocative test.)
- Ice pack test — apply ice to the ptotic lid for two minutes; watch the ptosis improve.
- Test extraocular movements — variable ophthalmoplegia, often asymmetric, fatiguing on sustained gaze.
- Pupils — normal and reactive in MG (pupil sparing is a key point; pupillary involvement suggests another diagnosis).
- Cogan lid twitch.
- Bulbar: Ask the patient to count to 100 — does the voice fade and slur? Test palatal movement; look for nasal regurgitation history.
- Motor: Test power, then fatigability — arms outstretched for one minute, repeated sit-to-stand, neck flexion and extension against resistance (head drop). Power may be normal at rest and decline with use. Reflexes are normal or preserved (depressed reflexes suggest LEMS).
- Respiratory: Measure FVC at the bedside if available; assess cough strength. [1]
Presentation template
"I examined Mrs Papadopoulos' neurological system. She is a 38-year-old woman who is alert and cooperative. [1]
On inspection, there is asymmetric ptosis, worse on the right, and a mild facial weakness giving a myasthenic snarl. There is a well-healed midline sternotomy scar consistent with previous thymectomy. [1]
Cranial nerve examination of the eyes reveals ptosis that is variable. On sustained upgaze for one minute, the ptosis worsens bilaterally — fatigability of the levator palpebrae. The ice pack test, applying ice to the right eyelid for two minutes, improves the ptosis. Extraocular movements show a variable, asymmetric ophthalmoplegia that fatigues on sustained lateral gaze. Pupils are equal and reactive — there is no pupillary involvement. [1]
Bulbar assessment: her speech is initially clear but fades and becomes nasal after counting to 60. Palatal elevation is symmetrical but fatigues with repeated movement. [1]
Motor examination reveals normal tone. Power is 5 out of 5 in all four limbs at rest, but on holding the arms outstretched for one minute, the right arm drifts and the grip fatigues. Reflexes are symmetrical and present. Sensation is intact. Coordination is intact. [1]
In summary, this patient has fatigable weakness of the ocular, bulbar and limb muscles — a positive sustained-upgaze test and a positive ice-pack test. The pupils are spared and the reflexes are preserved, which together with the fatigability points to a disorder of the postsynaptic neuromuscular junction: myasthenia gravis. The sternotomy scar suggests she has had a thymectomy, consistent with AChR-antibody MG. I would confirm with anti-acetylcholine receptor antibodies and CT thorax." [1]
DCE short-case trap: Examiners are watching for two things: (1) that you provoke the weakness rather than examining only at rest, and (2) that you comment specifically on the pupils and the reflexes — normal pupils and preserved reflexes exclude LEMS and a third nerve palsy and anchor the diagnosis in the neuromuscular junction. Missing fatigability or failing to test the pupils is a viva failure. [1]
Key DWE MCQ patterns
- Fatigable ptosis improving with ice — the bedside diagnosis of MG. A positive ice pack test (ptosis improves after two minutes of cold) is the bedside discriminator; the diagnosis is confirmed by anti-AChR antibodies.
- MuSK versus AChR phenotype — bulbar/respiratory, female, poor pyridostigmine response. MuSK-MG is not helped by thymectomy and responds best to rituximab [9][10].
- MGTX trial — thymectomy benefit in non-thymomatous AChR-MG. Extended transsternal thymectomy plus prednisone was superior to prednisone alone (lower QMG, lower prednisone dose) [4].
- Myasthenic crisis versus cholinergic crisis. A weak, dry patient with dilated pupils is myasthenic (give IVIG/plasma exchange); a weak, sweaty patient with miosis and fasciculations is cholinergic (reduce pyridostigmine, give atropine).
- Respiratory thresholds — FVC 15 to 20 mL/kg or NIF below 30 cmH2O predicts the need for ventilation. Do not wait for CO2 retention.
- Eculizumab requires meningococcal vaccination. Terminal complement blockade carries a 1 to 2 per cent risk of meningococcal infection [6].
- Prednisolone can initially worsen MG — start low (10 to 20 mg) and titrate up. A patient started on high-dose prednisolone can deteriorate in the first 1 to 2 weeks.
- IV magnesium is dangerous in MG — it inhibits ACh release and can precipitate crisis.
- MG versus LEMS — fatigability versus facilitation. LEMS improves with exercise, has depressed reflexes that reappear with use, autonomic features, and is paraneoplastic (small cell lung cancer; anti-P/Q-type VGCC antibody).
- Azathioprine requires TPMT testing before starting — TPMT deficiency causes fatal myelosuppression. The drug takes 6 to 12 months to work.
References
[1] Jaretzki A III, Barohn RJ, Ernstoff RM, et al. (MGFA 2000) — Myasthenia gravis: recommendations for clinical research standards. Establishes the MGFA clinical classification (Class I ocular through Class V intubated/crisis) and the post-intervention status framework. The common language of MG trials and clinic letters. [2] Sanders DB, Wolfe GI, Benatar M, et al. (MGFA 2016) — International consensus guidance for management of myasthenia gravis: executive summary. Recommends symptomatic treatment first, early steroid-sparing immunosuppression, thymectomy in thymoma and selected AChR-MG, and IVIG or plasma exchange for crisis. [3] Narayanaswami P, Sanders DB, Wolfe G, et al. (MGFA 2020 Update) — Updated consensus guidance adding recommendations on rituximab, eculizumab and methotrexate, early immunosuppression in ocular MG, and checkpoint-inhibitor-associated MG. [4] Wolfe GI, Kaminski HJ, Aban IB, et al. (MGTX 2016) — Randomised trial of extended transsternal thymectomy plus prednisone versus prednisone alone in non-thymomatous AChR-MG. Thymectomy reduced time-weighted QMG score and prednisone requirement over 3 years. [5] Gilhus NE, Lefvert AK, Verschuuren J (Nat Rev Dis Primers 2019) — Comprehensive primer on MG: anti-AChR (IgG1/IgG3, complement) and anti-MuSK (IgG4) pathophysiology, clinical spectrum, and treatment. [6] Howard JF Jr, Utsugisawa K, Benatar M, et al. (REGAIN 2017) — Phase 3 RCT of eculizumab versus placebo in anti-AChR refractory generalized MG. Clinically meaningful improvement in secondary endpoints; approved for refractory AChR-MG. [7] Muppidi S, Utsugisawa K, Baggi F, et al. (2021) — Open-label extension of REGAIN: 57 per cent of patients achieved minimal-manifestation status by 130 weeks of eculizumab, with sustained benefit. [8] Pasnoor M, Wolfe GI, Nations S, et al. (2010) — Clinical findings in 53 US MuSK-antibody-positive MG patients. Predominantly female (85 per cent), prominent bulbar and respiratory weakness, and about half non-responsive to pyridostigmine. [9] Clifford KM, Hobson-Webb LD, Benatar M, et al. (2019) — Multicentre cohort showing thymectomy was not associated with clinical improvement in MuSK-MG; thymectomy is not indicated in MuSK disease. [10] Litchman T, Roy B, Kumar A, et al. (2020) — Single-centre retrospective study comparing rituximab response in AChR-positive and MuSK-positive MG. Both groups improved and achieved remission, but MuSK-MG reached remission faster (230 versus 441 days) with fewer exacerbations, supporting rituximab especially in MuSK-MG.
Myasthenia Gravis Foundation of America clinical classification and consensus guidance (MGFA, 2000 and 2020 updates); American Academy of Neurology practice resources; Muscle Study Group; Australian Association of Neurologists clinical guidance. [1]
References
- [1]Jaretzki A III, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America Neurology, 2000.PMID 10891897
- [2]Sanders DB, Wolfe GI, Benatar M, et al. International consensus guidance for management of myasthenia gravis: Executive summary Neurology, 2016.PMID 27358333
- [3]Narayanaswami P, Sanders DB, Wolfe G, et al. International Consensus Guidance for Management of Myasthenia Gravis: 2020 Update Neurology, 2021.PMID 33144515
- [4]Wolfe GI, Kaminski HJ, Aban IB, et al. Randomized Trial of Thymectomy in Myasthenia Gravis N Engl J Med, 2016.PMID 27509100
- [5]Gilhus NE, Lefvert AK, Verschuuren J Myasthenia gravis Nat Rev Dis Primers, 2019.PMID 31048702
- [6]Howard JF Jr, Utsugisawa K, Benatar M, et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study Lancet Neurol, 2017.PMID 29066163
- [7]Mantegazza R, Wolfe GI, Muppidi S, et al. Post-intervention Status in Patients With Refractory Myasthenia Gravis Treated With Eculizumab During REGAIN and Its Open-Label Extension Neurology, 2021.PMID 33229455
- [8]Pasnoor M, Wolfe GI, Nations S, et al. Clinical findings in MuSK-antibody positive myasthenia gravis: a U.S. experience Muscle Nerve, 2010.PMID 19882635
- [9]Clifford KM, Hobson-Webb LD, Benatar M, et al. Thymectomy may not be associated with clinical improvement in MuSK myasthenia gravis Muscle Nerve, 2019.PMID 30575980
- [10]Litchman T, Roy B, Kumar A, et al. Differential response to rituximab in anti-AChR and anti-MuSK positive myasthenia gravis patients: a single-center retrospective study J Neurol Sci, 2020.PMID 32028072