Phys · neurological
Peripheral Neuropathy
Also known as peripheral neuropathy · polyneuropathy · distal symmetric polyneuropathy · DSPN · diabetic neuropathy · mononeuritis multiplex · Guillain-Barre syndrome · GBS · chronic inflammatory demyelinating polyradiculoneuropathy · CIDP · Charcot-Marie-Tooth disease · CMT · vasculitic neuropathy · small fibre neuropathy · critical illness polyneuropathy
Consultant-physician-depth systematic approach to peripheral neuropathy. Covers the four classification axes — fibre type (motor, sensory, autonomic, mixed), distribution (distal symmetric, mononeuropathy, mononeuritis multiplex, polyradiculoneuropathy), time course (acute, subacute, chronic), and pathology (axonal versus demyelinating — the key NCS discriminator). The full differential by cause: diabetes (the most common, with DSPN and cranial and lumbosacral variants), alcohol and thiamine, B12 and folate, B1 beriberi, B6 toxicity, vitamin E, drug-induced (vincristine, cisplatin, isoniazid, amiodarone, metronidazole, phenytoin, statins), toxins (lead, arsenic, mercury, organophosphates), hereditary (CMT, HNPP), autoimmune (GBS, CIDP, vasculitis), paraproteinaemic (MGUS, myeloma, amyloid, POEMS), uraemic, and critical illness (CIP/CIM). Investigation pathway (NCS/EMG, bloods, CSF, genetics, biopsy) and management (treat the cause, neuropathic pain, foot care, falls and burns prevention). Structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Peripheral Neuropathy

The answer first
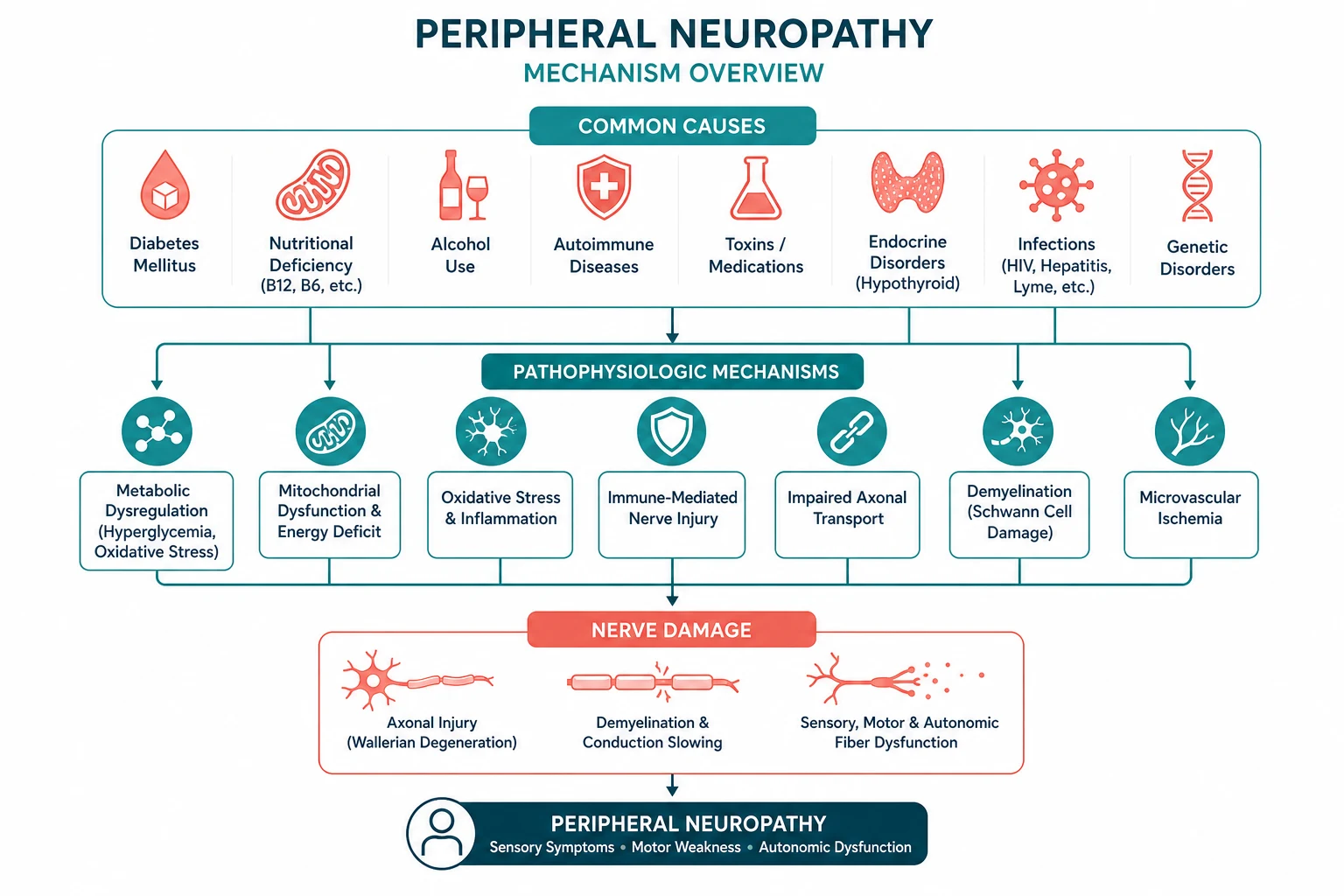
Peripheral neuropathy is a disorder of the peripheral nerves that produces a stereotyped constellation of sensory disturbance (numbness, tingling, burning, pain), motor loss (weakness, wasting, reduced reflexes), and autonomic dysfunction (orthostatic hypotension, gastroparesis, bladder and bowel and sexual dysfunction). It is among the most common problems in general neurology and general medicine — up to 8 per cent of the general population and over 50 per cent of patients with long-standing diabetes have some degree of peripheral neuropathy. [1]
The key to the systematic approach is classification along four axes — fibre type, distribution, time course, and pathology. Of these, two dominate the diagnostic reasoning. Distribution — distal symmetric, mononeuropathy, mononeuritis multiplex, or polyradiculoneuropathy — is the single most powerful clinical clue to the cause. And pathology — axonal versus demyelinating, determined by nerve conduction studies — is the single most powerful paraclinical discriminator, because it partitions the differential into two non-overlapping lists [3].
The most common pattern is distal symmetric polyneuropathy (DSPN), and the most common cause of DSPN is diabetes mellitus, producing a length-dependent, slowly progressive, mixed sensorimotor neuropathy with loss of ankle reflexes and vibration sense [1]. The most common hereditary neuropathy is Charcot-Marie-Tooth disease (CMT) [7]. The two neuropathies that demand emergency recognition are Guillain-Barre syndrome (GBS) — an acute demyelinating polyradiculoneuropathy that can cause respiratory failure within days [5] — and vasculitic neuropathy, presenting as mononeuritis multiplex, which causes irreversible nerve infarction unless treated urgently with immunosuppression [9].
Management rests on three pillars: treat the underlying cause (glycaemic control, immunotherapy, vitamin replacement, toxin removal, dialysis), control neuropathic pain (gabapentinoids, serotonin-noradrenaline reuptake inhibitors, tricyclic antidepressants) [10], and prevent complications (foot care and ulceration, falls, burns, and Charcot neuroarthropathy).
DCE trap: The candidate who, faced with a neuropathy long case, immediately generates a list of twenty causes has already failed. The correct approach is to classify first — state the distribution pattern and the axonal-versus-demyelinating status from the nerve conduction studies — and only then generate a focused, structured differential. This single discipline separates a pass from a distinction. [1]
The four classification axes
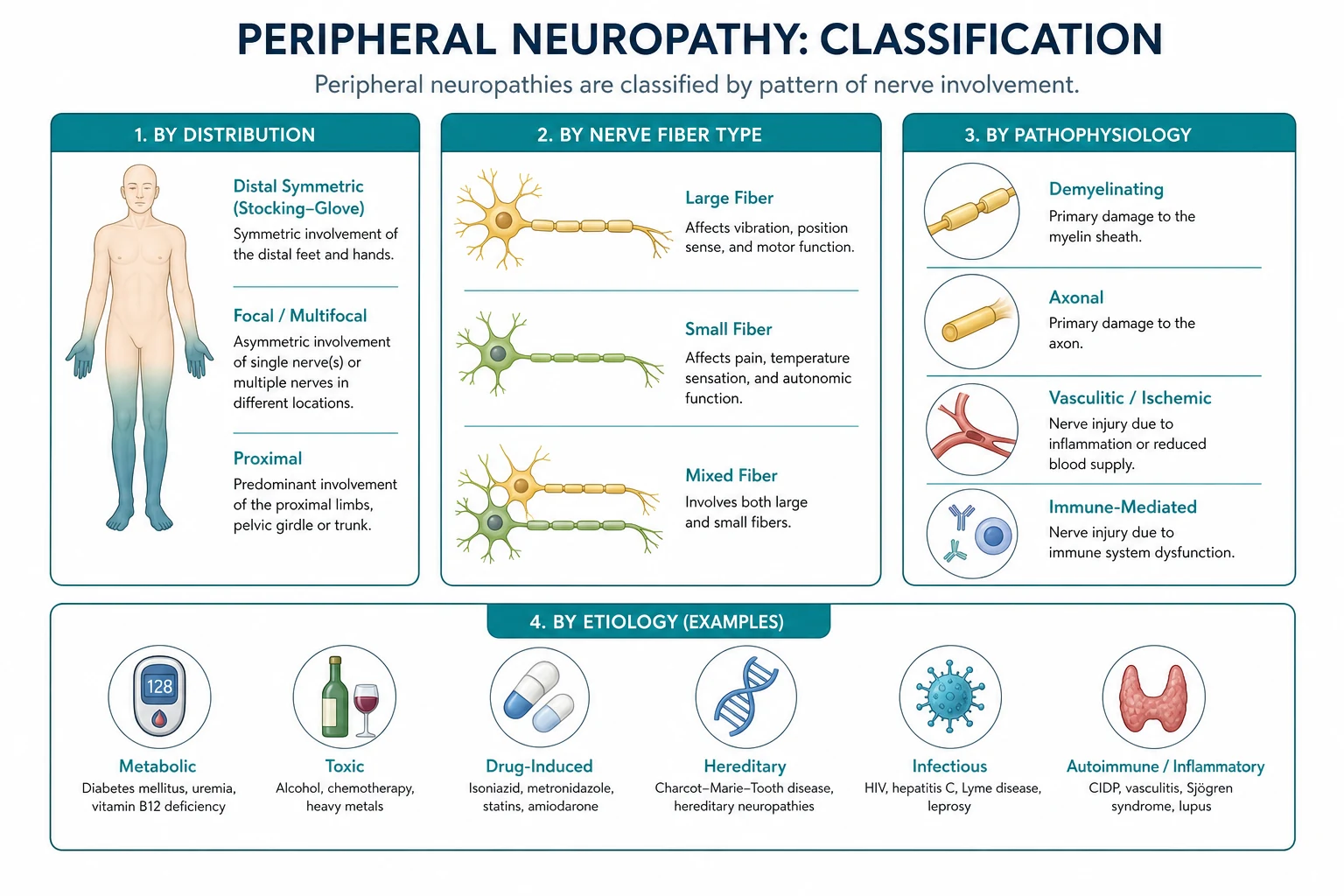
Axis 1 — Fibre type
Peripheral nerves contain motor, sensory, and autonomic fibres. The clinical syndrome reflects which fibre populations are damaged: [1]
- Motor fibre involvement — weakness (typically distal), muscle wasting, fasciculations, and reduced or absent reflexes. Distal leg weakness produces foot drop and a stepping gait; hand weakness produces impaired grip and fine motor tasks.
- Sensory fibre involvement — the most common manifestation. Large myelinated fibres carry vibration and proprioception; their loss produces numbness, loss of balance (sensory ataxia with a positive Romberg sign), and loss of ankle reflexes. Small fibres carry pain and temperature; their loss produces a loss of protective sensation (the basis of painless foot ulceration), while small fibre irritation produces the positive sensory symptoms — burning, pins and needles, electric shocks, and allodynia (pain from normally non-painful stimuli such as bedclothes).
- Autonomic fibre involvement — orthostatic hypotension (a systolic drop of more than 20 mmHg or diastolic drop of more than 10 mmHg on standing for 3 minutes), gastroparesis (early satiety, nausea, erratic glycaemic control), constipation or diabetic diarrhoea (typically nocturnal), urinary retention or incontinence, erectile dysfunction, gustatory sweating (sweating of the face and upper body on eating), anhidrosis or hyperhidrosis, and abnormal pupillary responses. [1]
Most neuropathies are mixed, but the fibre predilection is itself a clue. Diabetes and alcohol produce mixed sensorimotor and autonomic neuropathy; hereditary CMT is predominantly motor with some sensory loss; amyloidosis and diabetes are the principal causes of prominent autonomic neuropathy; and small fibre neuropathy (painful burning with preserved reflexes and strength) is characteristic of early diabetes, glucose intolerance, and some toxins. [1]
Axis 2 — Distribution (the most powerful clinical clue)
| Pattern | Description | Classic causes |
|---|---|---|
| Distal symmetric (length-dependent) | Symmetrical, ascending from the toes, stocking-glove distribution | Diabetes, alcohol, B12, uraemia, drugs, toxins, CMT |
| Mononeuropathy | Single nerve, in its anatomical territory | Entrapment (carpal tunnel, ulnar at elbow, peroneal at fibular head), compression, trauma, diabetes (cranial nerve III, VI) |
| Mononeuritis multiplex | Asymmetric, asynchronous, multiple individual nerves, painful, stepwise | Vasculitis (PAN, ANCA-associated, cryoglobulinaemia, rheumatoid), nonsystemic vasculitic neuropathy, diabetes, leprosy, amyloid, HNPP |
| Polyradiculoneuropathy | Proximal and distal, often with cranial nerve and respiratory involvement, diffuse areflexia | GBS (acute), CIDP (chronic) |
DWE high-yield point: Distal symmetric polyneuropathy is the most common pattern. Mononeuritis multiplex — painful, asymmetric, stepwise involvement of multiple named nerves — is the signature of vasculitis until proven otherwise, and is a neurological emergency. Polyradiculoneuropathy with both proximal and distal weakness suggests an autoimmune demyelinating process (GBS if acute, CIDP if chronic). [1]
Axis 3 — Time course
- Acute (days to 4 weeks) — GBS is the prototypical acute neuropathy. Acute onset also raises porphyria, tick paralysis, botulism, diphtheria, and toxic causes (organophosphates, thallium, arsenic).
- Subacute (weeks to months) — vasculitic neuropathy, nutritional neuropathy (B12, thiamine), paraneoplastic, drug-induced, and some toxin exposures.
- Chronic (months to years) — hereditary CMT, CIDP, diabetic DSPN, uraemic neuropathy, paraproteinaemic neuropathy. [1]
The time course immediately partitions the differential. A neuropathy that reaches its nadir in days demands urgent evaluation for GBS; one that has been present since childhood with foot deformity demands genetic testing for CMT. [1]
Axis 4 — Axonal versus demyelinating (the NCS discriminator)
This is the single most important paraclinical finding, because it partitions the cause into two largely non-overlapping lists [3]:
- Axonal neuropathy (the axon is damaged, the myelin intact) — reduced amplitudes of the compound muscle action potential and sensory nerve action potential, with relatively preserved conduction velocity. Needle EMG shows fibrillation potentials (denervation) and large, polyphasic motor unit potentials (reinnervation). Causes: diabetes, alcohol, B12, toxins, drugs, uraemia, vasculitis, critical illness — the vast majority of neuropathies are axonal.
- Demyelinating neuropathy (the myelin sheath is damaged, the axon intact) — markedly slowed conduction velocity (below 70 per cent of the lower limit of normal), prolonged distal latencies, prolonged or absent F-waves, conduction block (a marked drop in amplitude across a nerve segment), and temporal dispersion. Causes: GBS and CIDP, CMT type 1 (PMP22 duplication), anti-MAG neuropathy, POEMS, multifocal motor neuropathy with conduction block, and leprosy. [1]
Exam trap: Never generate a neuropathy differential before stating whether it is axonal or demyelinating. The differential for axonal neuropathy (diabetes, alcohol, B12) is completely different from the differential for demyelinating neuropathy (GBS, CIDP, CMT1, anti-MAG, POEMS). The NCS result is the gatekeeper of the differential. [1]
Distal symmetric polyneuropathy — the commonest pattern
Distal symmetric polyneuropathy (DSPN) is a length-dependent process: the longest nerves are affected first because they are most vulnerable to metabolic and toxic injury and most distant from the neuronal cell body's synthetic machinery. The result is a characteristic ascending pattern — symptoms begin in the toes, ascend to the knees, and only then appear in the fingertips (the stocking-glove distribution), because the fibres to the hands are the next longest. [1]
The clinical presentation is stereotyped: numbness and tingling in the toes, often with burning pain that is worse at night, loss of ankle reflexes early and knee reflexes later, loss of vibration sense (test at the great toe with a 128 Hz tuning fork) before loss of pinprick and temperature, and, in advanced disease, distal weakness (toe and ankle dorsiflexion, then foot drop) and a sensory ataxic gait with a positive Romberg sign. [1]
The approach to the patient with DSPN is structured and evidence-based [3]:
- Confirm it is a neuropathy — the symptoms and signs are usually sufficient, but nerve conduction studies confirm the neuropathy and determine axonal versus demyelinating.
- Screen for the common causes — fasting glucose and HbA1c, vitamin B12 (with methylmalonic acid or homocysteine), full blood count, urea and electrolytes, liver function, thyroid function, and serum protein electrophoresis with immunofixation and serum free light chains (the most cost-effective single test for a paraproteinaemic cause).
- Pursue targeted additional tests based on the clinical picture and risk factors — ANA, ANCA, rheumatoid factor, cryoglobulins, complements (vasculitic), HIV and hepatitis B and C (HIV neuropathy and cryoglobulinaemia), folate and thiamine (nutritional), vitamin B6 and vitamin E (toxicity and deficiency), heavy metals (lead, arsenic, mercury), and a hereditary neuropathy gene panel.
- Reserve nerve biopsy for specific scenarios — suspected vasculitic neuropathy or amyloidosis when less invasive tests are non-diagnostic [4].
- Accept that a cause is not found in a substantial minority — up to 25 per cent of DSPN remains idiopathic despite thorough evaluation; the management is then symptom control and surveillance.
The causes — by category
Diabetes mellitus — the most common cause
Diabetic peripheral neuropathy is the most common neuropathy in the developed world and the most common cause of non-traumatic amputation. It has several recognisable patterns [1]:
- Distal symmetric polyneuropathy (DSPN) — the most common pattern, as described above. It is a mixed sensorimotor and often autonomic neuropathy. Glycaemic control is the only disease-modifying intervention, and it is most effective in type 1 diabetes (where intensive therapy reduces neuropathy incidence by approximately 60 per cent over 5 years) and disappointingly less effective in type 2 diabetes (where multimodal cardiovascular risk factor management — blood pressure, lipids, smoking cessation, weight — is equally important).
- Diabetic lumbosacral radiculoplexus neuropathy (diabetic amyotrophy) — a painful, asymmetric, subacute weakness and wasting of the proximal thigh muscles (quadriceps, hip flexors), often with weight loss, that begins unilaterally and may spread. It is thought to be a microvasculitic process. It is largely self-limiting over months but may leave residual weakness; pain control and rehabilitation are the mainstays, with immunotherapy used in severe or progressive cases.
- Cranial mononeuropathy — acute, painful third nerve palsy (with pupillary sparing, because the parasympathetic fibres run in the periphery of the nerve and are spared by the central ischaemic lesion), or sixth nerve palsy, typically resolving over months. The pupil-sparing third nerve palsy in a diabetic is a classic DWE discriminator from a compressive (aneurysmal) third nerve palsy, which involves the pupil.
- Autonomic neuropathy — orthostatic hypotension, gastroparesis, diabetic diarrhoea, neurogenic bladder, erectile dysfunction, gustatory sweating, and resting tachycardia with loss of heart rate variability. Cardiac autonomic neuropathy is associated with increased mortality.
- Mononeuritis multiplex — diabetes is a common cause of this pattern, either from superimposed vasculitis or from a diabetic microvasculopathy. [1]
Painful diabetic neuropathy affects approximately 20 per cent of diabetics and is a major cause of reduced quality of life [2]. The pain is characteristically burning, worst at night, and may be accompanied by allodynia and hyperalgesia. First-line pharmacotherapy is a gabapentinoid (pregabalin, gabapentin), a serotonin-noradrenaline reuptake inhibitor (duloxetine), or a tricyclic antidepressant (amitriptyline), used as monotherapy and then in combination if inadequate [10].
DWE trap: A normal serum B12 does not exclude deficiency, particularly in metformin-treated patients. Metformin impairs B12 absorption in up to 30 per cent of long-term users. Check methylmalonic acid and homocysteine if the clinical picture fits — and consider empirical B12 replacement given its safety. [1]
Alcohol and nutritional neuropathy
Alcohol causes neuropathy by three mechanisms: direct neurotoxicity, thiamine (vitamin B1) deficiency (producing Wernicke encephalopathy and a neuropathy), and nutritional deficiency more broadly. The neuropathy is a distal, predominantly sensory and painful axonal neuropathy, often with autonomic features and calf tenderness. Management is abstinence, thiamine replacement (100 mg daily, parenteral initially if Wernicke is suspected), and a multivitamin including folate and B12. [1]
Vitamin B12 deficiency produces a mixed picture: a large fibre sensorimotor axonal neuropathy and subacute combined degeneration of the spinal cord (demyelination of the dorsal columns, producing loss of vibration and proprioception, and the lateral corticospinal tracts, producing upper motor neuron signs). The combined peripheral and central lesion is the explanation for the apparent paradox of brisk knee reflexes with absent ankle reflexes. Causes include pernicious anaemia, terminal ileal disease, gastric surgery, nitrous oxide abuse (which oxidises B12), and metformin. Treatment is parenteral hydroxocobalamin. Always check B12 in any neuropathy, and treat aggressively — the neurological deficit is partly reversible if caught early but becomes permanent. [1]
Other nutritional neuropathies include thiamine deficiency (beriberi) — wet beriberi (cardiac failure and oedema) and dry beriberi (sensorimotor neuropathy); folate deficiency; vitamin B6 (pyridoxine) toxicity — an important and under-recognised cause of a pure sensory neuropathy or neuronopathy from high-dose supplementation (typically more than 100 mg per day chronically); and vitamin E deficiency (malabsorption, abetalipoproteinaemia) — a large fibre ataxic sensory neuropathy mimicking Friedreich ataxia. [1]
Drug-induced and toxic neuropathies
A careful drug and toxin history is mandatory — the list of causative agents is long, and removal of the agent is often the only treatment required. [1]
| Agent | Pattern | Key feature |
|---|---|---|
| Vincristine | Sensorimotor axonal | Dose-dependent; autonomic (constipation, ileus); occurs early in treatment |
| Cisplatin, oxaliplatin | Sensory neuronopathy (dorsal root ganglion) | Oxaliplatin also causes acute cold-induced neuropathy |
| Isoniazid | Sensorimotor axonal | Prevent with pyridoxine 25 to 50 mg daily |
| Metronidazole | Predominantly sensory axonal | Often with prolonged or repeated courses |
| Phenytoin | Mild sensorimotor axonal | Long-term therapy; also causes cerebellar signs |
| Statins | Mild axonal, or worsens existing neuropathy | Controversial; usually mild and reversible |
| Taxanes (paclitaxel) | Sensory predominant axonal | Cumulative dose-related |
| Bortezomib | Sensory predominant, painful | Often with worsening on treatment |
| Lead | Motor predominant, upper limb (wrist drop) | Occupational; abdominal pain and microcytic anaemia |
| Arsenic | Painful sensorimotor | Acute or chronic; Mees lines on nails |
| Mercury | Sensory neuropathy, tremor, cognitive change | Organic mercury (fish, industrial) |
| Organophosphates | Acute cholinergic crisis then delayed neuropathy | Agricultural and industrial exposure |
| Thallium | Painful sensorimotor, alopecia | Rat poison; mimic of GBS acutely |
| Nitrofurantoin | Acute or chronic sensorimotor | Renal impairment is a risk factor |
Lead neuropathy deserves emphasis as a high-yield exam topic. It is a purely motor neuropathy affecting the upper limbs — classically wrist and finger extensor weakness (wrist drop) — in a worker with occupational exposure (battery manufacture, lead smelting, old paint). It is accompanied by abdominal pain, constipation, microcytic anaemia with basophilic stippling, and a gum lead line. The mechanism is direct toxicity to the anterior horn cell and the motor axon. [1]
Hereditary neuropathies
Charcot-Marie-Tooth disease (CMT) is the most common inherited neuropathy, with a prevalence of approximately 1 in 2500. It is a slowly progressive distal motor and sensory neuropathy beginning in childhood or adolescence. The clinical hallmark is the combination of distal muscle wasting producing the inverted champagne-bottle leg (wasted below the knee), foot deformity (pes cavus, hammer toes), stepping gait from foot drop, loss of ankle reflexes, and distal sensory loss that is often asymptomatic [7]. The hands become involved later (intrinsic muscle wasting). NCS partition the subtypes: CMT type 1 (demyelinating) from PMP22 duplication on chromosome 17 (the most common cause, 70 per cent of CMT1), and CMT type 2 (axonal). Genetic testing (PMP22 first, then a hereditary neuropathy gene panel) confirms the diagnosis. Management is supportive — podiatry, orthotics (ankle-foot orthoses for foot drop), physiotherapy, and avoidance of neurotoxic drugs. Life expectancy is normal in CMT1 and CMT2.
Clinical pearl: The combination of long-standing symptoms, foot deformity (pes cavus, hammer toes), a family history, and the absence of positive sensory symptoms (despite sensory signs) points to hereditary CMT — send a PMP22 test and a gene panel rather than an exhaustive autoimmune and paraneoplastic workup. [1]
Hereditary neuropathy with liability to pressure palsy (HNPP) is caused by a PMP22 deletion (the reciprocal of the CMT1 duplication). It presents with recurrent, painless mononeuropathies at common compression sites (peroneal at the fibular head, ulnar at the elbow, radial in the spiral groove, brachial plexus) that recover over weeks. NCS shows a generalised demyelinating neuropathy with tomacula (focal myelin thickenings) on nerve biopsy. Management is avoidance of pressure and compression. [1]
Autoimmune and inflammatory neuropathies
Guillain-Barre syndrome (GBS) is an acute, monophasic, immune-mediated demyelinating polyradiculoneuropathy, usually triggered by an infection (Campylobacter jejuni, cytomegalovirus, Epstein-Barr virus, Mycoplasma, influenza, Zika). It presents with progressive, symmetrical, ascending weakness with areflexia, reaching nadir within 4 weeks, and may involve respiratory muscles (20 to 30 per cent require mechanical ventilation) and cranial nerves (facial diplegia, ophthalmoplegia in the Miller Fisher variant). The diagnosis is clinical, supported by cytoalbuminologic dissociation in the CSF (raised protein with normal cell count) and demyelination on NCS. Treatment is intravenous immunoglobulin (0.4 g per kg per day for 5 days) or plasma exchange, which are equally effective and must be given within 2 to 4 weeks of onset [5]. Corticosteroids are not effective in GBS. Respiratory and autonomic monitoring (forced vital capacity, single breath count, heart rate variability, blood pressure) is critical, as is prophylactic anticoagulation. GBS is covered in depth in its own topic.
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is the chronic counterpart of GBS — the same immune-mediated demyelinating process but progressing or relapsing over more than 8 weeks. It presents with symmetrical proximal and distal weakness, sensory loss, and diffuse areflexia, often with large fibre sensory involvement and a relapsing or progressive course. The 2021 EAN/PNS guideline simplifies the diagnosis to CIDP or possible CIDP, based on clinical phenotype (typical versus atypical variants such as multifocal acquired demyelinating sensory and motor neuropathy, distal acquired demyelinating symmetric neuropathy, and pure motor or pure sensory forms) and electrophysiological evidence of demyelination [6]. CSF shows cytoalbuminologic dissociation. First-line treatment is intravenous immunoglobulin, corticosteroids, or plasma exchange (all effective); many patients need maintenance IV immunoglobulin infusions, and steroid-sparing agents (azathioprine, mycophenolate, rituximab) are used for refractory or steroid-dependent disease.
Vasculitic neuropathy is a neurological emergency. It is an immune-mediated inflammation of the vasa nervorum producing ischaemic infarction of individual nerve fascicles, which explains the clinical signature: mononeuritis multiplex — painful, asymmetric, stepwise deficits in the distribution of individual peripheral nerves (peroneal, ulnar, radial, sciatic), evolving over days to weeks [9]. Causes include systemic vasculitis (polyarteritis nodosa, ANCA-associated vasculitis, cryoglobulinaemia, rheumatoid vasculitis) and nonsystemic vasculitic neuropathy (confined to the peripheral nerves, without systemic involvement). Diagnosis rests on sural nerve biopsy (showing vasculitis of the vasa nervorum with asymmetric fibre loss) combined with NCS (axonal, often asymmetric) and a systemic vasculitis screen (ANA, ANCA, rheumatoid factor, cryoglobulins, complements, hepatitis B and C). Treatment is urgent high-dose corticosteroids (oral prednisolone 1 mg per kg, or intravenous methylprednisolone for severe disease) with cyclophosphamide for severe or rapidly progressive disease, followed by maintenance immunosuppression (azathioprine or methotrexate). Delay leads to cumulative, irreversible nerve infarction and disability.
Red flag: Painful, asymmetric, stepwise multifocal neuropathy is mononeuritis multiplex from vasculitis until proven otherwise. This is a neurological emergency — urgent sural nerve biopsy and high-dose immunosuppression to prevent irreversible disability. [1]
Paraproteinaemic neuropathy
A serum paraprotein is found in approximately 10 per cent of neuropathies, and every unexplained neuropathy warrants serum protein electrophoresis with immunofixation and serum free light chains [3]. The associations include:
- MGUS (monoclonal gammopathy of undetermined significance) — the most common; the neuropathy may be coincidental or causally related. Anti-MAG IgM paraproteins produce a distal demyelinating neuropathy with large fibre sensory loss, sensory ataxia, and tremor, with marked prolongation of distal latencies on NCS. Anti-MAG neuropathy is typically refractory to standard CIDP therapies.
- Multiple myeloma — usually an axonal neuropathy, but amyloid deposition may produce a painful sensorimotor and autonomic neuropathy.
- POEMS syndrome (Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal plasma cell disorder, Skin changes) — a demyelinating and axonal neuropathy with a lambda light chain paraprotein, Castleman disease, and characteristic features (hepatosplenomegaly, endocrine dysfunction, sclerotic bone lesions, hyperpigmentation, papilloedema, and very high VEGF levels). Treatment targets the plasma cell clone (radiotherapy for a sclerotic lesion, melphalan and autologous stem cell transplant for diffuse disease).
- Waldenstrom macroglobulinaemia — IgM paraprotein, anti-MAG neuropathy, hyperviscosity.
- Amyloidosis (AL amyloid) — a progressive, painful sensorimotor and prominent autonomic neuropathy, often with carpal tunnel syndrome as an early feature. Congo red staining of a biopsy (fat pad, rectum, or nerve) with apple-green birefringence confirms the diagnosis. [1]
Uraemic and critical illness neuropathy
Uraemic neuropathy is a distal, symmetric, sensorimotor axonal neuropathy of chronic kidney disease, correlating with the severity of renal impairment and improving with dialysis and kidney transplantation. It is often asymptomatic in early stages, detected by reduced ankle reflexes and vibration sense. [1]
Critical illness polyneuropathy and myopathy (CIP and CIM) are major causes of ICU-acquired weakness, presenting as flaccid quadriparesis and failure to wean from mechanical ventilation in a patient who has survived the acute critical illness [8]. The pathophysiology involves microvascular dysfunction, hyperglycaemia, catabolism, and an acquired sodium channelopathy, with axonal degeneration (CIP) and muscle necrosis with myosin loss (CIM). Diagnosis is by electrophysiology and clinical assessment; biopsy is rarely needed. Management is prevention — intensive insulin glycaemic control (modest effect), minimising sedation and neuromuscular blocking agents, early mobilisation, and rehabilitation. Recovery is slow and often incomplete, with persistent weakness and disability at 1 to 2 years in a substantial proportion of survivors.
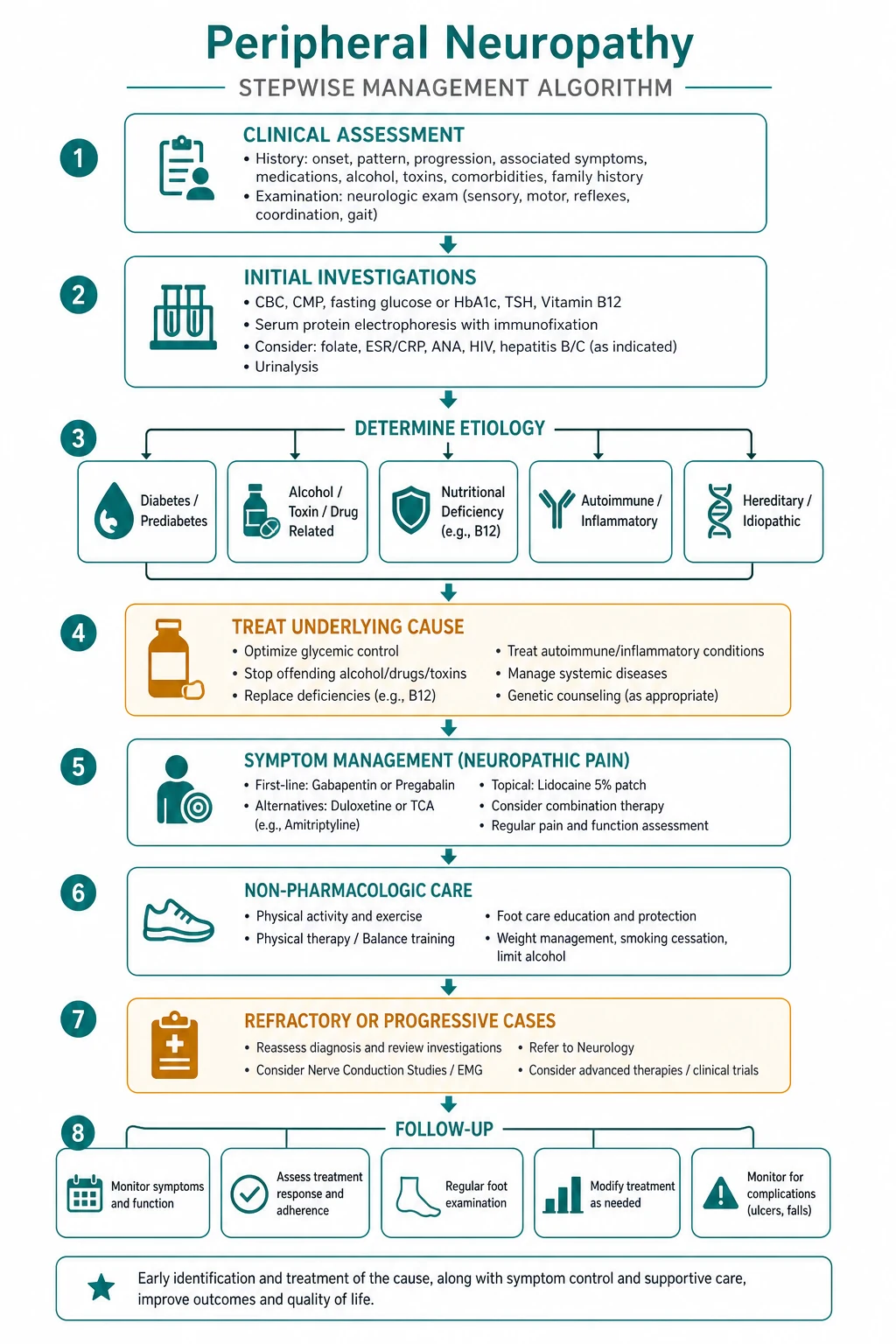
Investigation — the structured pathway
The investigation of an unexplained neuropathy follows a logical, cost-effective sequence [3] [4].
Step 1 — Nerve conduction studies and EMG
The NCS confirms the neuropathy, determines axonal versus demyelinating, grades severity, and clarifies the distribution. The key parameters: [1]
- Conduction velocity — markedly slowed (below 70 per cent of the lower limit) indicates demyelination; relatively preserved despite reduced amplitude indicates axonal loss.
- Distal latency — prolonged in demyelination.
- F-wave — assesses the proximal nerve segment and root; prolonged or absent in demyelinating neuropathy (GBS, CIDP) and proximal lesions.
- Conduction block and temporal dispersion — hallmarks of acquired demyelination (CIDP, multifocal motor neuropathy); their absence in hereditary demyelination (CMT1) is because the slowing is uniform.
- Needle EMG — fibrillation potentials and positive sharp waves (denervation), and large polyphasic motor unit potentials (reinnervation) in axonal neuropathy. [1]
Step 2 — First-line blood panel (every patient)
- Fasting glucose and HbA1c (impaired glucose tolerance is a common cause of small fibre neuropathy)
- Vitamin B12 with methylmalonic acid or homocysteine
- Full blood count, urea and electrolytes, liver function, thyroid function
- Serum protein electrophoresis with immunofixation and serum free light chains (the most cost-effective test for a paraproteinaemic cause — the AAN specifically recommends immunofixation over standard electrophoresis because it detects small monoclonal bands) [1]
Step 3 — Targeted additional tests (guided by the clinical picture)
- Vasculitic screen: ANA, extractable nuclear antigens, ANCA, rheumatoid factor, complements, cryoglobulins
- Infectious: HIV, hepatitis B and C (cryoglobulinaemia), Lyme serology where appropriate
- Nutritional and toxic: folate, thiamine, vitamin B6, vitamin E; heavy metals (lead, arsenic, mercury) if exposure is suspected; porphyrins
- Antibodies: anti-MAG, anti-GQ1b (Miller Fisher), anti-GM1 (multifocal motor neuropathy), anti-Hu (paraneoplastic sensory neuronopathy) [1]
Step 4 — CSF, genetics, and biopsy
- Lumbar puncture and CSF — for suspected GBS and CIDP (cytoalbuminologic dissociation: high protein, normal cell count), and suspected leptomeningeal disease.
- Genetic testing — a hereditary neuropathy gene panel for suspected CMT (PMP22 duplication or deletion first, then sequencing of MPZ, GJB1, MFN2, and others) guided by the phenotype and inheritance pattern.
- Sural nerve biopsy — reserved for suspected vasculitic neuropathy or amyloidosis when less invasive tests are non-diagnostic [4]. It carries a risk of permanent sensory loss in the biopsied nerve distribution, wound infection, and delayed healing, and should be reserved for when the result will change management.
- Skin biopsy — a 3 mm punch biopsy with reduced intraepidermal nerve fibre density confirms small fibre neuropathy when nerve conduction studies are normal [4].
Management — the three pillars
Pillar 1 — Treat the underlying cause
| Cause | Disease-modifying treatment |
|---|---|
| Diabetes | Glycaemic control (especially type 1); multimodal cardiovascular risk factor management |
| B12 deficiency | Parenteral hydroxocobalamin |
| Alcohol | Abstinence, thiamine, multivitamin |
| Drug or toxin | Cessation or avoidance |
| Uraemia | Dialysis, kidney transplantation |
| Hypothyroidism | Thyroxine replacement |
| GBS | IV immunoglobulin or plasma exchange (acute) |
| CIDP | IV immunoglobulin, corticosteroids, or plasma exchange (ongoing) |
| Vasculitic neuropathy | Corticosteroids with cyclophosphamide, then maintenance immunosuppression |
| POEMS | Radiotherapy for a sclerotic lesion; melphalan and autologous stem cell transplant |
| Anti-MAG | Rituximab (variable response) |
Pillar 2 — Control neuropathic pain
Neuropathic pain is often the most distressing symptom and the main driver of reduced quality of life in diabetic neuropathy [2]. The EFNS guidelines recommend a stepped approach [10]:
- First-line monotherapy — a gabapentinoid (gabapentin 300 to 3600 mg per day in three divided doses, or pregabalin 150 to 600 mg per day), a serotonin-noradrenaline reuptake inhibitor (duloxetine 30 to 120 mg daily, venlafaxine), or a tricyclic antidepressant (amitriptyline 10 to 75 mg at night, nortriptyline). Choose based on comorbidity (duloxetine for depression, amitriptyline for insomnia, a gabapentinoid for anxiety), and titrate to the maximum tolerated dose.
- Combination therapy — if monotherapy is inadequate, combine agents with different mechanisms (a gabapentinoid plus an SNRI or TCA).
- Second-line — topical lidocaine 5 per cent patches or capsaicin 8 per cent patches for focal pain, and tramadol for refractory pain.
- Avoid — strong opioids long-term where possible (limited efficacy, tolerance, dependence, and adverse effects); NICE specifically advises against initiating strong opioids for chronic neuropathic pain except in acute crises. [1]
Tricyclics require caution in the elderly (anticholinergic effects, falls, confusion) and in patients with cardiac conduction disease, glaucoma, and urinary retention. Gabapentinoids require dose reduction in renal impairment and carry a risk of sedation, dizziness, and weight gain. Duloxetine is contraindicated in uncontrolled hypertension and hepatic impairment. [1]
Pillar 3 — Prevent complications
Foot care is the cornerstone of preventing amputation in diabetic neuropathy. The neuropathic foot loses protective sensation (painless injury), develops deformity (claw toes, prominent metatarsal heads, Charcot changes) that creates pressure points, and, combined with peripheral arterial disease, is vulnerable to ulceration and infection. Prevention requires: [1]
- Daily foot inspection — by the patient or carer, looking for cuts, blisters, redness, and callus.
- Well-fitting footwear — accommodative shoes, insoles, and orthotics to offload pressure points; custom footwear for deformity.
- Regular podiatry — callus debridement, nail care, and pressure offloading.
- Prompt attention to any break in the skin — early treatment of ulcers prevents deep infection and amputation.
- Glycaemic control and smoking cessation. [1]
Charcot neuroarthropathy is a destructive arthritis of the foot or ankle in a neuropathic patient, presenting with warmth, swelling, and erythema with relatively little pain (because of the loss of protective sensation). It progresses to fracture, dislocation, and the rocker-bottom foot deformity if untreated. It is often misdiagnosed as cellulitis, gout, or osteomyelitis. Management is immediate offloading (total contact cast), and bisphosphonates and referral to a specialist foot clinic. [1]
Falls and burns prevention — the neuropathic patient has sensory ataxia (loss of proprioception), distal weakness (foot drop), and orthostatic hypotension, all of which increase fall risk. Prevention includes home safety assessment (remove rugs and clutter, adequate lighting, handrails), lower the hot water thermostat (to prevent painless scalds from loss of temperature sensation), protective footwear, and referral to occupational therapy and physiotherapy. Orthostatic hypotension is managed with adequate hydration, gradual mobilisation, compression stockings, fludrocortisone, or midodrine. [1]
High-yield DWE points and exam traps
- Always classify before generating a differential. State the distribution (distal symmetric, mononeuropathy, mononeuritis multiplex, polyradiculoneuropathy) and the axonal-versus-demyelinating status from the NCS. This is the single discipline that structures the answer.
- Diabetes is the most common cause of distal symmetric polyneuropathy; CMT is the most common hereditary neuropathy.
- Mononeuritis multiplex (painful, asymmetric, stepwise) is vasculitis until proven otherwise — a neurological emergency.
- GBS reaches nadir within 4 weeks; CIDP progresses beyond 8 weeks. Both are demyelinating with cytoalbuminologic dissociation. Corticosteroids do not work in GBS but do in CIDP.
- A normal serum B12 does not exclude deficiency — check methylmalonic acid and homocysteine, particularly in metformin-treated patients.
- Immunofixation and free light chains, not standard electrophoresis, for the paraprotein screen — standard electrophoresis misses small monoclonal bands.
- Lead neuropathy is purely motor and affects the upper limbs (wrist drop) — a classic discriminator.
- B6 toxicity causes a sensory neuronopathy from high-dose supplementation — the one vitamin that causes neuropathy from overdose rather than deficiency.
- Pupil-sparing third nerve palsy in a diabetic is microvascular ischaemia; a pupil-involving third nerve palsy is compressive (posterior communicating artery aneurysm) until proven otherwise.
- The neuropathic foot ulcer — combined neuropathy, ischaemia, and deformity; 85 per cent of diabetes-related amputations are preceded by ulceration, and most are preventable.
- Charcot neuroarthropathy presents as a warm, swollen, relatively painless foot — do not misdiagnose it as cellulitis or osteomyelitis; offload immediately. [1]
Key references
The foundational evidence for this topic includes the Toronto consensus on diabetic neuropathy definitions and diagnostic criteria [1], the review of painful diabetic neuropathy and quality of life [2], the AAN practice parameters on the evaluation of distal symmetric polyneuropathy covering laboratory and genetic testing [3] and autonomic testing, nerve biopsy, and skin biopsy [4], the comprehensive review of Guillain-Barre syndrome [5], the 2021 EAN/PNS guideline on CIDP [6], the review of Charcot-Marie-Tooth disease [7], the review of critical illness polyneuropathy and myopathy [8], the Peripheral Nerve Society guideline on nonsystemic vasculitic neuropathy [9], and the EFNS guidelines on the pharmacological treatment of neuropathic pain [10].
References
- [1]Tesfaye S, Boulton AJ, Dyck PJ, Freeman R, Horowitz M, Kempler P, Lauria G, Malik RA, Spallone V, Vinik A, Bernardi L, Valensi P, Toronto Diabetic Neuropathy Expert Group Diabetic neuropathies: update on definitions, diagnostic criteria, estimation of severity, and treatments Diabetes Care, 2010.PMID 20876709
- [2]Tesfaye S, Boulton AJ, Dickenson AH. Mechanisms and management of diabetic painful distal symmetrical polyneuropathy Diabetes Care, 2013.PMID 23970715
- [3]England JD, Gronseth GS, Franklin G, Miller RG, Dubinsky RM, French JA, Gray J, Herrmann DN, Hughes R, Kasckisow J, Latov N, Lewis RA, Low PA, Pfeifer MA, American Academy of Neurology, American Association of Neuromuscular and Electrodiagnostic Medicine, American Academy of Physical Medicine and Rehabilitation Practice Parameter: evaluation of distal symmetric polyneuropathy: role of laboratory and genetic testing (an evidence-based review). Report of the American Academy of Neurology, American Association of Neuromuscular and Electrodiagnostic Medicine, and American Academy of Physical Medicine and Rehabilitation Neurology, 2009.PMID 19056666
- [4]England JD, Gronseth GS, Franklin G, Miller RG, Dubinsky RM, French JA, Gray J, Herrmann DN, Hughes R, Kasckisow J, Latov N, Lewis RA, Low PA, Pfeifer MA, American Academy of Neurology, American Association of Neuromuscular and Electrodiagnostic Medicine, American Academy of Physical Medicine and Rehabilitation Practice Parameter: evaluation of distal symmetric polyneuropathy: role of autonomic testing, nerve biopsy, and skin biopsy (an evidence-based review) [RETIRED]. Report of the American Academy of Neurology, American Association of Neuromuscular and Electrodiagnostic Medicine, and American Academy of Physical Medicine and Rehabilitation Neurology, 2009.PMID 19056667
- [5]Willison HJ, Jacobs BC, van Doorn PA Guillain-Barré syndrome Lancet, 2016.PMID 26948435
- [6]Van den Bergh PYK, van Doorn PA, Hadden RDM, Avau B, Veltkamp R, Cornblath DR, Gosselin S, Leger JM, Nobile-Orazio E, van Schaik IN, Antonini G, Attarian S, Bruno M, Cruciatti B, Faggioli R, Harbo T, Hughes RAC, Kuitwaard K, Rajabally YA, Umapathi T, Wang Y, Yuki N, Bergh PV, Braathen G, Collongues N, Eftimov F, Kiplelis E, Kwavic M, Lissens V, Loncarevic D, Mandarakas G, Ozkara B, Petrou P, Rao M, Razali S, Sadig S, Saifee TA, Savic-Pavicevic D, Wieser T, EAN/PNS Guideline Task Force European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint Task Force-Second revision J Peripher Nerv Syst, 2021.PMID 34085743
- [7]Pareyson D, Marchesi C Diagnosis, natural history, and management of Charcot-Marie-Tooth disease Lancet Neurol, 2009.PMID 19539237
- [8]Latronico N, Bolton CF Critical illness polyneuropathy and myopathy: a major cause of muscle weakness and paralysis Lancet Neurol, 2011.PMID 21939902
- [9]Collins MP, Dyck PJ, Gronseth GS, Guilcher JR, Jann S, Leger JM, Lewis RA, Lozeron P, Mellgren SI, Merkies IS, Nobile-Orazio E, Sommer CL, Torbergsen T, Vrancken AFNE, Gorson KC, Peripheral Nerve Society Guideline Peripheral Nerve Society Guideline on the classification, diagnosis, investigation, and immunosuppressive therapy of non-systemic vasculitic neuropathy: executive summary J Peripher Nerv Syst, 2010.PMID 21040139
- [10]Attal N, Cruccu G, Baron R, Haanpaa M, Hansson P, Jensen TS, Nurmikko T, EFNS Task Force EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision Eur J Neurol, 2010.PMID 20402746