Phys · renal
Acid-Base Disorders
Also known as acid-base disturbance · metabolic acidosis · metabolic alkalosis · respiratory acidosis · respiratory alkalosis · high anion gap metabolic acidosis · HAGMA · normal anion gap acidosis · hyperchloraemic metabolic acidosis · renal tubular acidosis · RTA · distal RTA · proximal RTA · type 4 RTA · lactic acidosis · diabetic ketoacidosis · DKA · Winter's formula · delta-delta · anion gap · osmolar gap · MUDPILES · GOLD MARK
Consultant-physician-depth guide to systematic acid-base interpretation and management — the six-step algorithm (pH, primary disorder, compensation, anion gap, delta-delta, osmolar gap), high and normal anion gap metabolic acidoses (MUDPILES/GOLD MARK and renal tubular acidoses), metabolic alkalosis, respiratory acid-base disorders, and treatment principles. Structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Acid-Base Disorders
The answer first
Acid-base interpretation is a process, not a diagnosis. The diagnosis falls out of a reproducible, six-step algorithm applied to every blood gas — arterial or venous. The registrar who can recite the algorithm under viva pressure and reach a complete answer (primary disorder, compensation, mixed disorders, and aetiology) will pass the acid-base station; the one who eyeballs the gas and guesses will miss mixed disorders and fail. [1]
The mandate is simple: [1]
- Read the pH first — acidaemia or alkalaemia.
- Name the primary disorder — respiratory or metabolic, using PaCO2 and bicarbonate.
- Assess compensation — is it appropriate? If not, a second primary disorder is present (a mixed disorder).
- Calculate the anion gap (AG) — every metabolic acidosis is high-AG or normal-AG.
- Apply the delta-delta — does the AG explain the bicarbonate? This unmasks a hidden metabolic alkalosis or a second acidosis.
- Check the osmolar gap — when a toxic alcohol is possible. [1]
The single most important principle for the exam: never stop at the primary disorder. A septic patient with a lactic acidosis and an apparent "appropriate" PaCO2 may still have a hidden metabolic alkalosis from vomiting — only the delta-delta reveals it. Mixed disorders are the discriminator that separates a pass from a distinction [1].
The six-step algorithm
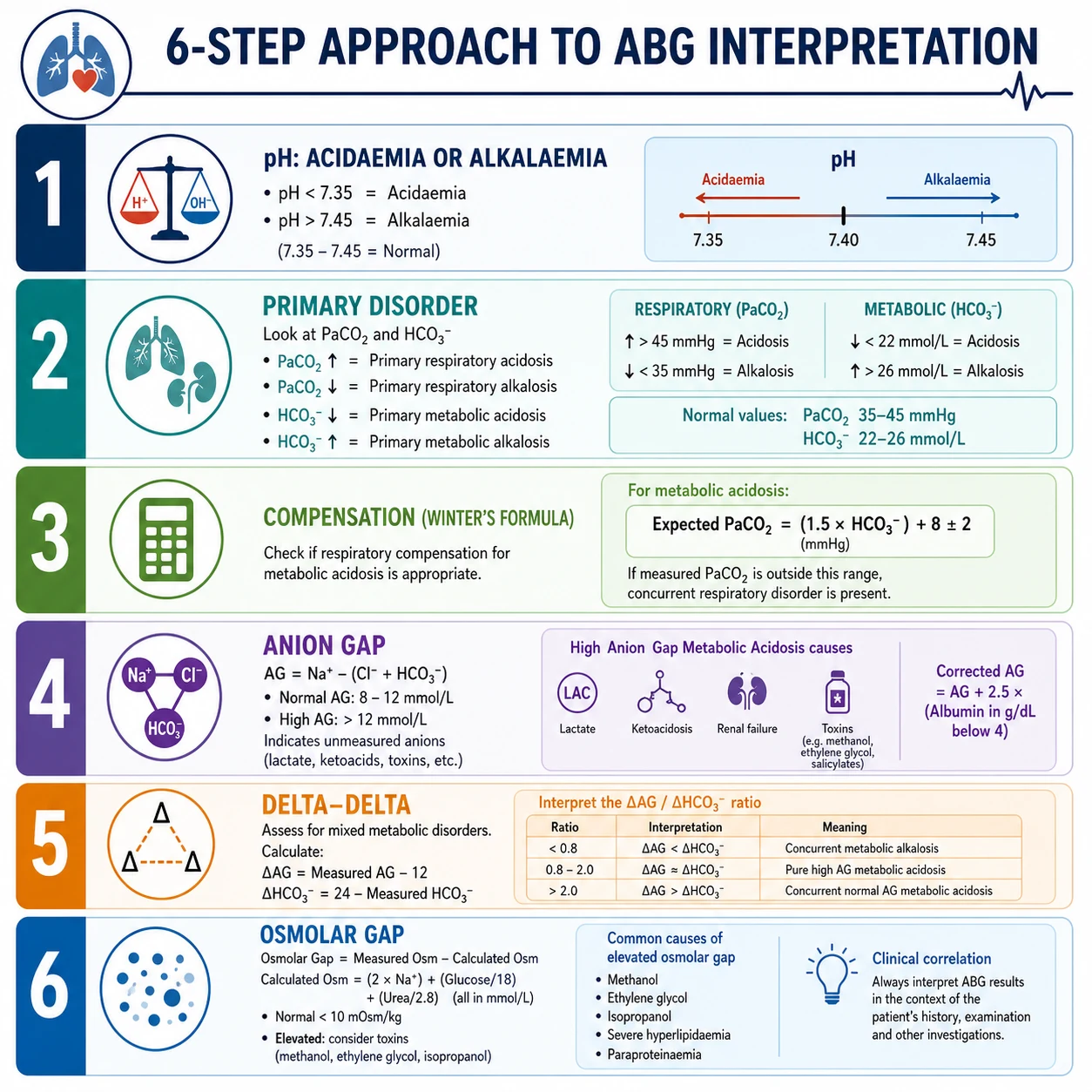
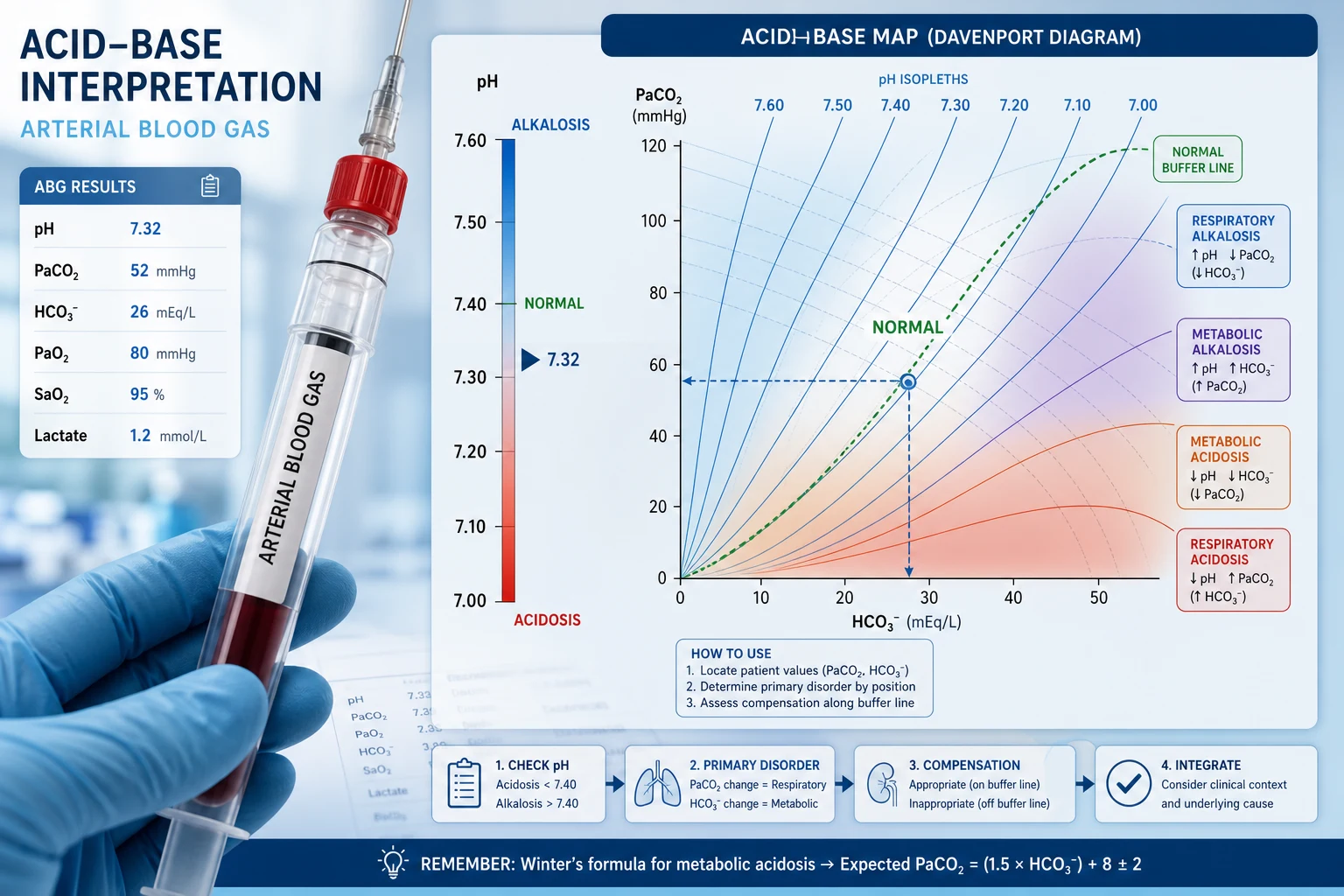
Step 1 — The pH: acidaemia or alkalaemia
pH is the dependent variable. Normal arterial pH is 7.35–7.45. [1]
- pH < 7.35 → acidaemia (the blood is acidotic).
- pH > 7.45 → alkalaemia (the blood is alkalotic).
- pH 7.35–7.45 → normal, but a mixed disorder can hide here (two opposing processes cancelling out). [1]
Always state whether the patient is acidaemic or alkalaemic before anything else. This anchors the whole interpretation. A patient with pH 7.30 has acidaemia; the next question is whether the cause is respiratory (high PaCO2) or metabolic (low bicarbonate). [1]
Viva trap: "Acidosis" and "alkalosis" are processes; "acidaemia" and "alkalaemia" describe the blood. A patient can have a metabolic acidosis (a process) with a normal pH if a second disorder alkalinises them. Reserve the "-aemia" suffix for the pH. [1]
Step 2 — The primary disorder
Compare PaCO2 and bicarbonate against normal and against the direction of the pH change. [1]
| Arterial value | Normal range |
|---|---|
| pH | 7.35–7.45 |
| PaCO2 | 35–45 mmHg (4.7–6.0 kPa) |
| Bicarbonate (HCO3) | 22–26 mmol/L |
The rule: the abnormal variable that moves in the same direction as the pH change is not the cause. The variable that moves in the direction predicted by the pH is the primary process. [1]
- Metabolic acidosis: low bicarbonate AND low pH (PaCO2 will also be low as compensation).
- Metabolic alkalosis: high bicarbonate AND high pH (PaCO2 rises as compensation).
- Respiratory acidosis: high PaCO2 AND low pH.
- Respiratory alkalosis: low PaCO2 AND high pH. [1]
If the pH is low and the PaCO2 is also low, the PaCO2 cannot be driving the acidaemia — a high PaCO2 would cause acidaemia, so a low PaCO2 in an acidaemic patient is compensation, and the primary disorder is metabolic. State this logic aloud in the viva. [1]
DCE examiner question: "Is this a respiratory or metabolic process?" Lead with the answer: "The pH is 7.20 (acidaemia). The PaCO2 is 28, which is low — a low PaCO2 would alkalinise, so it is not the cause. The bicarbonate is 12, also low, and a low bicarbonate causes acidaemia. This is a primary metabolic acidosis with respiratory compensation." [1]
Step 3 — Compensation
Once the primary disorder is named, calculate the expected compensatory response. If the measured value falls outside the predicted range, a second primary disorder is present — a mixed disturbance [1].

Metabolic acidosis — Winter's formula
The expected PaCO2 for a metabolic acidosis is: [1]
Expected PaCO2 = 1.5 × HCO3 + 8 ± 2 mmHg [1]
If the measured PaCO2 is within this range, compensation is appropriate. If it is higher than expected, there is a concurrent respiratory acidosis (the patient is under-ventilating). If it is lower, there is a concurrent respiratory alkalosis (over-ventilating — common in sepsis, salicylate toxicity, hypoxia, pregnancy). [1]
DWE high-yield: Winter's formula is the single most commonly tested compensation rule. A worked example: HCO3 10 → expected PaCO2 = (1.5 × 10) + 8 = 23 ± 2, i.e. 21–25 mmHg. If the measured PaCO2 is 35, the patient has a metabolic acidosis plus a respiratory acidosis — they are failing to compensate and may need ventilatory support. [1]
Metabolic alkalosis
Expected PaCO2 rises to compensate. The PaCO2 rises by approximately 0.6–0.7 mmHg for every 1 mmol/L rise in bicarbonate, up to a ceiling around 55–60 mmHg (hypoxaemia eventually limits hypoventilation). A practical rule: expected PaCO2 ≈ 0.7 × HCO3 + 20 ± 5 mmHg. [1]
If the measured PaCO2 is lower than expected, a respiratory alkalosis is also present; if higher, a respiratory acidosis. [1]
Respiratory acidosis — acute vs chronic
The kidney takes 2–5 days to amplify bicarbonate reabsorption. So compensation differs by chronicity. [1]
| Change per 10 mmHg rise in PaCO2 | pH change per 10 mmHg | |
|---|---|---|
| Acute respiratory acidosis | HCO3 rises 1 mmol/L | pH falls 0.08 |
| Chronic respiratory acidosis | HCO3 rises 3.5–4 mmol/L | pH falls 0.03 |
The clinical implication: in a COPD patient with chronic CO2 retention, a "normal" bicarbonate of 24 would be abnormal — chronic compensation should have raised it. A bicarbonate that is lower than expected signals a concurrent metabolic acidosis. [1]
Respiratory alkalosis — acute vs chronic
| Change per 10 mmHg fall in PaCO2 | pH change per 10 mmHg | |
|---|---|---|
| Acute respiratory alkalosis | HCO3 falls 2 mmol/L | pH rises 0.08 |
| Chronic respiratory alkalosis | HCO3 falls 4–5 mmol/L | pH rises 0.03 |
Chronic respiratory alkalosis is the rule in pregnancy, high altitude, and chronic liver disease (the latter from portopulmonary stimulation of the respiratory centre). [1]
Step 4 — The anion gap (AG)
For every metabolic acidosis, calculate the anion gap: [1]
Anion gap = Na − Cl − HCO3 [1]
Normal range 8–12 mmol/L (some references use 8–16; pick one and apply it consistently). The gap exists because plasma has more unmeasured anions (proteins, phosphate, organic anions) than unmeasured cations, and it widens when an acid adds an unmeasured anion (lactate, ketones, sulphate, oxalate, salicylate). [1]
This step bifurcates the metabolic acidosis into two families with entirely different differentials: [1]
- High anion gap metabolic acidosis (HAGMA): AG > 12 (or > 16 by stricter cutoff). An unmeasured acid is present — think MUDPILES / GOLD MARK.
- Normal anion gap (hyperchloraemic) metabolic acidosis: AG 8–12. Bicarbonate has been lost or a chloride-containing acid gained — think diarrhoea or renal tubular acidosis. [1]
Correct for albumin
Albumin is the dominant unmeasured anion. In hypoalbuminaemia the baseline AG falls, so a "normal" AG can mask a high-AG acidosis. Correct the gap: [1]
Corrected AG = observed AG + 2.5 × (4.0 − albumin in g/dL) [1]
In practice: for every 10 g/L (1 g/dL) the albumin falls below 40 g/L, the lower limit of the AG rises by about 2.5–3 mmol/L. A patient with albumin 20 g/L (2 g/dL) and a measured AG of 14 actually has a corrected AG of 14 + 2.5 × 2 = 19 — that is a high anion gap. In the critically ill, never interpret an AG without checking the albumin. [1]
DWE exam trap: The classic discriminator is "what is the anion gap in a patient with albumin 18 g/L and AG 12?" The uncorrected AG looks normal; the corrected AG (about 22) is high. Always correct for albumin in the ICU patient. [1]
Step 5 — The delta-delta (ΔAG / ΔHCO3)
This step asks: does the rise in the anion gap fully account for the fall in bicarbonate? It unmasks a second, hidden metabolic disorder that the AG step alone cannot see. [1]
Calculate two deltas: [1]
- ΔAG = observed AG − 12 (the rise in the gap above normal).
- ΔHCO3 = 24 − observed HCO3 (the fall in bicarbonate below normal). [1]
Then the ratio ΔAG / ΔHCO3: [1]
| Ratio | Interpretation |
|---|---|
| 1–2 | Pure high-AG metabolic acidosis (the AG rise matches the bicarbonate fall) |
| < 1 | Concurrent normal anion gap acidosis (bicarbonate falls more than the gap rises — e.g. HAGMA from sepsis plus diarrhoea or RTA) |
| > 2 | Concurrent metabolic alkalosis (bicarbonate is higher than the gap rise predicts — e.g. HAGMA from uraemia plus vomiting, or diuretic-induced alkalosis) |
The clinical payoff is large. A patient with sepsis and a lactic acidosis who has been vomiting has a delta-delta > 2 — the metabolic alkalosis from vomiting is "protecting" the bicarbonate and partially the pH, and you will miss it without this step. [1]
Viva question: "Why is the bicarbonate only mildly low when the anion gap is markedly raised?" Model answer: "Because there is a concurrent metabolic alkalosis elevating the bicarbonate. The delta-delta ratio is greater than two. I would look for vomiting, diuretic use, or nasogastric losses as the cause of the alkalosis." [1]
Step 6 — The osmolar gap
When a high-AG acidosis has no immediately obvious cause, calculate the osmolar gap, especially if altered consciousness or a history of ingestion is present. [1]
Calculated osmolality = 2 × Na + glucose + urea (all in mmol/L) [1]
Osmolar gap = measured osmolality − calculated osmolality. Normal < 10 mOsm/kg. [1]
A high osmolar gap with a high-AG acidosis points to a toxic alcohol (methanol, ethylene glycol, diethylene glycol, propylene glycol) [2][8]. The osmolar gap is highest early, when the parent alcohol is circulating; as hepatic metabolism converts it to organic acids, the osmolar gap falls and the anion gap rises. A high gap with a normal AG and a high anion gap may simply reflect the natural history — the two gaps are moving in opposite directions over time.
Critical trap: Isopropanol (isopropyl alcohol) raises the osmolar gap and causes ketosis without anion-gap acidosis (it is metabolised to acetone, not an organic acid). A high osmolar gap, no acidosis, and ketonuria without glucosuria is isopropanol until proven otherwise. [1]
High anion gap metabolic acidosis — MUDPILES and GOLD MARK
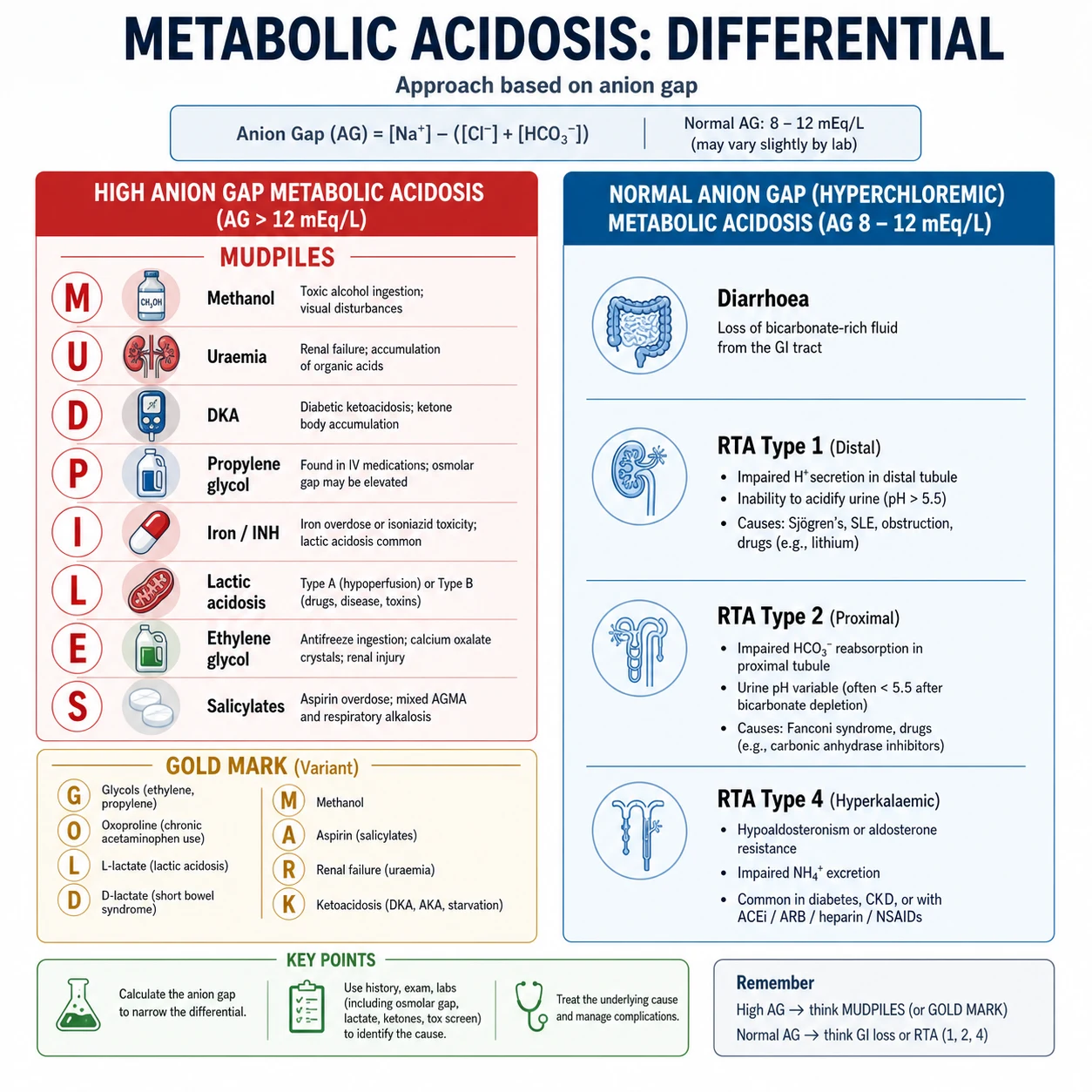
The traditional mnemonic is MUDPILES: Methanol, Uraemia, Diabetic ketoacidosis (and other ketoacidoses), Propylene glycol, Iron/INH, Lactic acidosis, Ethylene glycol, Salicylates. [1]
The modern alternative, GOLD MARK, updates the list for the 21st century and drops outdated causes (paraldehyde): Glycols (ethylene and propylene), Oxoproline (5-oxoproline/pyroglutamic acid, from chronic paracetamol), L-lactate, D-lactate (from bacterial overgrowth), Methanol, Aspirin, Renal failure, Ketoacidosis [5]. Both mnemonics are acceptable in the exam; know both, because examiners may ask you to justify one over the other.
Lactic acidosis — the most common HAGMA
Lactic acidosis is the single commonest cause of a high-AG acidosis in hospital. Use the Cohen and Woods classification: [1]
- Type A — tissue hypoperfusion / hypoxia: shock (septic, cardiogenic, hypovolaemic, haemorrhagic), severe hypoxia, mesenteric ischaemia, seizures, strenuous exercise. This is the form that responds to restoring perfusion, not to bicarbonate.
- Type B — impaired metabolism without overt hypoperfusion: malignancy (lymphoma, leukaemia), liver failure (impaired lactate clearance), thiamine deficiency, metformin accumulation, mitochondrial toxins (antiretrovirals, linezolid, propofol infusion syndrome), toxins (cyanide, carbon monoxide). [1]
Management principle (Surviving Sepsis Campaign): In septic shock with a lactate > 2 mmol/L, the response is fluid resuscitation, early antibiotics, source control, and vasopressors to restore perfusion — not sodium bicarbonate. Lactate is a marker of the illness; treating the number with bicarbonate treats the marker, not the disease [7].
Metformin-associated lactic acidosis (MALA) is a Type B acidosis: metformin inhibits mitochondrial complex I, shifting metabolism toward anaerobic glycolysis. Risk is highest in CKD, dehydration, hypoxia, and acute illness — hence the "sick-day rule" to hold metformin during intercurrent illness. The acidosis can be profound (pH < 7.1, lactate > 10) and may require haemodialysis both for the acidosis and for metformin removal. [1]
Ketoacidosis
- Diabetic ketoacidosis (DKA): absolute insulin deficiency drives ketogenesis (beta-hydroxybutyrate, acetoacetate). Classic triad: hyperglycaemia, ketosis, high-AG acidosis. Treat with insulin infusion, intravenous fluids, and potassium replacement [4].
- Alcoholic ketoacidosis: chronically malnourished, binge-drinking patient, often with vomiting and a low or normal glucose. High-AG acidosis, ketonuria. Treat with dextrose-containing saline and thiamine — dextrose stimulates insulin and stops ketogenesis; thiamine prevents Wernicke.
- Starvation ketosis: mild, usually a mixed disturbance, bicarbonate rarely below 18.
DWE high-yield — euglycaemic DKA: DKA with a near-normal glucose (< 11 mmol/L) occurs in type 1 diabetes with reduced oral intake, pregnancy, and SGLT2 inhibitor use. The glucosuria from the SGLT2i keeps glucose low while insulin deficiency drives ketogenesis. If a patient on an SGLT2i presents unwell with a high-AG acidosis and a "normal" sugar, check ketones — the diagnosis will be missed otherwise. [1]
Toxic alcohols — methanol and ethylene glycol
Both cause a high-AG acidosis with a high osmolar gap early, and characteristic end-organ toxicity: [1]
- Methanol → metabolised to formaldehyde then formic acid, causing visual toxicity ("snowfield vision", optic disc oedema, ultimately blindness) and basal ganglia infarcts.
- Ethylene glycol → metabolised to glycolate then oxalate, causing acute kidney injury (calcium oxalate crystals in urine) and hypocalcaemia. [1]
The management of both is the same and is time-critical [2]:
- Fomepizole (15 mg/kg IV loading) or ethanol infusion to inhibit alcohol dehydrogenase and halt conversion to toxic metabolites.
- Haemodialysis to remove the parent alcohol and metabolites — indicated for severe acidosis, renal failure, visual symptoms, or high levels.
- Cofactors: folate and thiamine/pyridoxine to divert metabolism away from toxic intermediates.
- Sodium bicarbonate for the acidosis while awaiting definitive removal. [1]
Exam trap: The "lactate gap" — glycolate (an ethylene glycol metabolite) is falsely read as lactate by some point-of-care analysers, producing an apparent lactate of 10–15 that the laboratory assay does not confirm. A discordant high point-of-care lactate with a high-AG acidosis should prompt measurement of an ethylene glycol level and the osmolar gap. [1]
Salicylate toxicity
Salicylates cause a mixed disorder: initially a respiratory alkalosis (direct stimulation of the medullary respiratory centre) followed by a high-AG metabolic acidosis (uncoupling of oxidative phosphorylation drives lactate and ketoacid production). A patient with a respiratory alkalosis and a metabolic acidosis simultaneously — a mixed disorder on Step 3 — is salicylate toxicity until excluded. Urine alkalinisation (intravenous bicarbonate to keep urine pH > 7.5) enhances salicylate elimination; haemodialysis is indicated for severe toxicity (level > 700 mg/L, or lower with end-organ damage). [1]
5-Oxoproline (pyroglutamic acid)
An under-recognised cause of high-AG acidosis in the chronically ill: chronic paracetamol (acetaminophen) use depletes glutathione and disrupts the gamma-glutamyl cycle, accumulating 5-oxoproline. Suspect it in a septic or malnourished patient on regular paracetamol with an unexplained high-AG acidosis. Stop the paracetamol and give N-acetylcysteine. [1]
Normal anion gap (hyperchloraemic) metabolic acidosis
When the AG is normal and the patient is acidotic, bicarbonate has been lost or a chloride-containing acid has been gained. The kidney normally responds by raising the chloride to maintain electroneutrality as bicarbonate falls — hence "hyperchloraemic". The discriminator between renal and GI causes is the urinary anion gap and the serum potassium. [1]
Urinary anion gap (UAG) = urine Na + urine K − urine Cl. [1]
- Negative UAG (e.g. −20): the kidney is appropriately excreting ammonium (NH4+) — the GI tract is the source of bicarbonate loss (diarrhoea).
- Positive UAG (e.g. +30): the kidney is failing to excrete acid — a renal tubular acidosis. [1]
GI bicarbonate loss — diarrhoea
Diarrhoea is the commonest cause of a normal-AG acidosis. Stool is rich in bicarbonate; loss of bicarbonate-rich fluid drives a hyperchloraemic acidosis with a negative UAG (the kidney compensates by excreting acid). The serum potassium is usually low (total body potassium depletion from secondary hyperaldosteronism). Other GI causes: villous adenoma, ureterosigmoidostomy, pancreatic fistula. [1]
Renal tubular acidosis
RTA is a syndrome of impaired renal acid excretion (distal) or bicarbonate reclamation (proximal), or aldosterone failure (type 4), producing a normal-AG acidosis with a positive UAG. The serum potassium distinguishes the three types [6].
| Feature | Type 1 (distal) | Type 2 (proximal) | Type 4 (hyperkalaemic) |
|---|---|---|---|
| Defect | Impaired H+ secretion (alpha-intercalated cell) | Impaired HCO3 reabsorption (proximal tubule) | Aldosterone deficiency or resistance |
| Serum K+ | Low | Low | High |
| Bicarbonate | Often very low (10–20) | Moderately low (12–20) | Mildly low (usually > 17) |
| Urine pH | > 5.5 (cannot acidify) | Can fall < 5.5 after equilibrium | Can fall < 5.5 |
| Complications | Nephrocalcinosis, nephrolithiasis (calcium phosphate), osteomalacia | Rickets/osteomalacia, Fanconi syndrome features | Usually benign, often incidental |
| Treatment | Oral bicarbonate / citrate (1–2 mmol/kg/day) | High-dose bicarbonate (10–15 mmol/kg/day), thiazide | Fludrocortisone, or treat the cause |
Type 1 — distal RTA
The alpha-intercalated cell of the collecting duct cannot secrete hydrogen ion (via the H+-ATPase pump) or cannot reclaim the secreted H+ via the Cl/HCO3 exchanger (AE1). Causes: autoimmune (Sjogren's — the classic association, rheumatoid arthritis, SLE), hereditary, amphotericin B, lithium, toluene, obstructive uropathy. The inability to acidify urine below pH 5.5, even in the face of systemic acidosis, is the hallmark. Chronic metabolic acidosis mobilises bone buffer, causing hypercalciuria; the alkaline urine precipitates calcium phosphate stones and nephrocalcinosis. Most patients present with nephrolithiasis or osteomalacia and a hypokalaemic, normal-AG acidosis. [1]
Type 2 — proximal RTA
The proximal tubule reclaims 85% of filtered bicarbonate; failure causes bicarbonaturia until the plasma bicarbonate falls to a new lower threshold (typically 12–20 mmol/L), at which point the reduced filtered load can be reclaimed by the distal nephron and urine pH falls. Usually part of a generalised proximal tubulopathy (Fanconi syndrome): glycosuria with normal glucose, aminoaciduria, phosphaturia, uricosuria. Causes: myeloma (the classic association in older adults), ifosfamide, tenofovir, aminoglycosides, valproate, hereditary (cystinosis in children, Wilson's disease). Treatment requires high-dose bicarbonate (the proximal tubule will keep wasting it) — often 10–15 mmol/kg/day — plus a thiazide diuretic to reduce extracellular volume and lower the filtered load. [1]
Type 4 — hyperkalaemic RTA
The commonest RTA in adults. Hypoaldosteronism (or aldosterone resistance) impairs distal Na+ reabsorption, K+ secretion, and H+ secretion — so the patient develops hyperkalaemia with a mild normal-AG acidosis. Causes: diabetic nephropathy (hyporeninaemic hypoaldosteronism — the single most common cause), adrenal insufficiency, ACE inhibitors/ARBs, K-sparing diuretics (spironolactone, eplerenone, amiloride, triamterene), NSAIDs, heparin, calcineurin inhibitors. Unlike types 1 and 2, the urine can still be acidified (pH < 5.5) because the H+-ATPase is intact; the problem is reduced distal NH3 buffering from hyperkalaemia (hyperkalaemia suppresses ammoniagenesis) [6]. Treatment is to address the cause; fludrocortisone (0.1 mg/day) is effective in true hypoaldosteronism, but beware of worsening oedema or hypertension in the cardiac patient.
DWE discriminator: The serum potassium sorts the RTA instantly. Hypokalaemia with a normal-AG acidosis is type 1 or type 2; hyperkalaemia with a normal-AG acidosis is type 4. If an MCQ gives a diabetic patient on an ACE inhibitor with a high potassium and a mild acidosis, the answer is type 4 RTA. [1]
Metabolic alkalosis — chloride-responsive vs chloride-resistant
Metabolic alkalosis is a high bicarbonate with a high pH. The discriminator that drives both diagnosis and management is the urinary chloride, which separates chloride-responsive from chloride-resistant causes. [1]
Chloride-responsive (urine Cl < 10–20 mmol/L) [1]
The kidney is chloride-depleted and retains bicarbonate in compensation. These alkaloses correct with saline and chloride. [1]
- Vomiting and nasogastric suction: loss of gastric HCl leaves bicarbonate behind; volume contraction drives aldosterone-mediated bicarbonate retention. The urine chloride is low (the kidney avidly retains chloride).
- Diuretics (loop and thiazide): chronic use causes contraction alkalosis; the urine chloride may be high during active diuresis but becomes low after the drug wears off.
- Post-hypercapnia: the chronically CO2-retaining patient whose ventilation is suddenly improved (intubation) retains their high bicarbonate, producing an alkalosis until chloride is repleted.
- Cystic fibrosis (sweat chloride loss), villous adenoma (rarely). [1]
The treatment is isotonic saline with potassium chloride. The alkalosis is "saline-responsive" — it resolves as chloride is restored. [1]
DWE high-yield: The urine chloride is the discriminator. A vomiting patient with a metabolic alkalosis has a urine chloride < 10; the patient with hyperaldosteronism has a urine chloride > 20. This single test distinguishes the two. [1]
Chloride-resistant (urine Cl > 20 mmol/L) [1]
The kidney is not chloride-depleted; mineralocorticoid excess drives hydrogen and potassium secretion while retaining bicarbonate. These alkaloses do not correct with saline. [1]
- Primary hyperaldosteronism (Conn's syndrome) — the classic cause. Hypertension, hypokalaemia, metabolic alkalosis, and a high urine chloride and aldosterone with a suppressed renin.
- Cushing's syndrome, exogenous mineralocorticoids (liquorice — inhibits 11-beta-hydroxysteroid dehydrogenase, exposing the mineralocorticoid receptor to cortisol), congenital adrenal hyperplasia.
- Bartter syndrome (loop-like defect, normotensive) and Gitelman syndrome (thiazide-like defect, hypomagnesaemia, normotensive) — inherited tubulopathies presenting with hypokalaemic alkalosis and a normal or low blood pressure.
- Severe potassium depletion itself perpetuates alkalosis (increases renal ammoniagenesis and bicarbonate reabsorption). [1]
Treatment is to address the cause (adrenal adenoma resection, stop the exogenous source) and potassium repletion, which alone can partially correct the alkalosis. [1]
Respiratory acidosis and alkalosis
Respiratory acidosis
High PaCO2 with acidaemia — alveolar hypoventilation. Causes divide into CNS depression and neuromuscular or airway disease: [1]
- CNS: opioids, sedatives, stroke, brainstem lesions, obesity hypoventilation syndrome.
- Airway/parenchyma: COPD exacerbation, severe asthma (the tired, tiring patient), upper airway obstruction, obstructive sleep apnoea.
- Neuromuscular: Guillain-Barre, myasthenia gravis, severe hypokalaemia/hypophosphataemia, neuromuscular blocking agents.
- Chest wall/mechanics: kyphoscoliosis, flail chest, massive obesity. [1]
Management is to treat the cause and support ventilation — oxygen for hypoxaemia, non-invasive ventilation (BiPAP) for COPD with respiratory acidosis, intubation for the patient who cannot protect their airway or is tiring. Oxygen alone will not lower the CO2. In the opioid-overdose patient, naloxone reverses the cause; in COPD, bronchodilators, steroids, antibiotics and BiPAP address the exacerbation. [1]
Critical trap: Giving high-flow oxygen to a COPD patient with chronic CO2 retention can worsen hypercapnia (reduced hypoxic drive, worsened V/Q matching, Haldane effect). Titrate oxygen to a target SpO2 of 88–92% in COPD and use arterial blood gases to monitor. [1]
Respiratory alkalosis
Low PaCO2 with alkalaemia — alveolar hyperventilation. Causes: [1]
- Hypoxaemia-driven: high altitude, pulmonary embolism, pneumonia, pulmonary oedema, early asthma.
- CNS/drive: anxiety (the classic cause — look for carpopedal spasm from hypocalcaemia), pain, fever, sepsis, salicylate toxicity (early), pregnancy (progesterone-driven), hepatic failure.
- Iatrogenic: excessive mechanical ventilation. [1]
Management is to treat the cause (analgesia, anxiolysis, oxygen for hypoxaemia, treat sepsis). Acute alkalaemia causes a leftward shift of the oxyhaemoglobin dissociation curve and can provoke paraesthesiae, carpopedal spasm, and seizures (from decreased ionised calcium). Rebreathing into a bag is a temporising manoeuvre only. [1]
Treatment principles
Sodium bicarbonate — when and when not
Bicarbonate is one of the most overused drugs in acid-base medicine. The evidence is clear: bicarbonate does not improve outcomes in lactic acidosis. The body metabolises lactate to bicarbonate once perfusion is restored; giving exogenous bicarbonate generates CO2 (which diffuses into cells and paradoxically worsens intracellular acidosis), causes hypernatraemia and volume overload, and shifts potassium. [1]
The BICAR-ICU trial (PMID 29910040) randomised critically ill patients with severe metabolic acidaemia (pH < 7.20) to sodium bicarbonate or control: there was no significant difference in the primary composite outcome of death or organ failure at day 7, though a pre-specified subgroup with severe AKI showed reduced need for renal replacement therapy [3].
Legitimate indications for bicarbonate: [1]
- Hyperkalaemia with ECG changes (as part of the shifting strategy).
- Severe acidaemia (pH < 7.1–7.15) with haemodynamic instability where acidosis itself is impairing catecholamine responsiveness — a bridge to definitive treatment.
- Tricyclic antidepressant toxicity (alkalinises the blood and the myocardium).
- Toxic alcohol and salicylate poisoning (urine alkalinisation and correction of acidosis).
- Specific renal tubular acidoses (the proximal and distal tubule cannot reclaim bicarbonate — supplementation is the treatment). [1]
For the garden-variety lactic acidosis of sepsis or shock, the answer is resuscitation, source control, and time — not bicarbonate. [1]
The general approach to any metabolic acidosis
- Resuscitate — ABCDE, oxygen, IV access, treat shock. The acidosis is a marker; the patient comes first.
- Identify and treat the cause — the six-step algorithm names the disorder; the differentials (MUDPILES, RTA, alkalosis) name the cause.
- Correct potassium — acidosis shifts K+ out of cells; correction of acidosis shifts it back. Monitor closely.
- Reserve bicarbonate for specific indications — as above.
- Renal replacement therapy for refractory acidosis, refractory hyperkalaemia, or volume overload — the definitive removal strategy. [1]
DCE long-case approach
Opening statement (SASPOP)
"Mr Tran is a 64-year-old Vietnamese-Australian man, a retired plumber, who presents to the emergency department with two days of vomiting, abdominal pain, and confusion. He has a background of type 2 diabetes for 20 years, chronic kidney disease stage 3b (baseline eGFR 38), hypertension, and ischaemic heart disease. He takes metformin, empagliflozin, perindopril, frusemide, and atorvastatin." [1]
"His main problems are:
- Severe high anion gap metabolic acidosis — likely a combination of metformin-associated lactic acidosis and euglycaemic ketoacidosis precipitated by an SGLT2 inhibitor and acute illness, on a background of chronic kidney disease
- Acute kidney injury, KDIGO stage 2, on chronic CKD — pre-renal from vomiting and hypovolaemia compounded by nephrotoxic medications
- Hyperkalaemia, K+ 6.4, with peaked T waves — a medical emergency
- Possible intra-abdominal sepsis as the precipitant — abdominal pain and a high lactate demand investigation
- Chronic multimorbidity — diabetes, CKD, ischaemic heart disease — dictating drug choices and prognosis." [1]
Integrated management plan
Present the plan in domains: [1]
- Resuscitate: ABCDE. Oxygen. Two large-bore cannulae. Treat the hyperkalaemia immediately — calcium gluconate 10 mL of 10% IV to stabilise the myocardium, then insulin-dextrose and a beta-agonist to shift potassium. Continuous cardiac monitoring.
- Interpret and confirm the acid-base disorder: arterial blood gas, apply the six-step algorithm. Confirm HAGMA, calculate the delta-delta to exclude a concurrent metabolic alkalosis from vomiting, check the osmolar gap.
- Treat the cause: stop metformin and empagliflozin immediately. Give IV fluids (balanced crystalloid) for volume depletion. Investigate for sepsis — blood cultures, lactate, urinalysis, abdominal imaging. Empirical broad-spectrum antibiotics within one hour if sepsis is suspected.
- Definitive removal: this patient has severe acidaemia with AKI and is on metformin — discuss early with nephrology and ICU for haemodialysis, which corrects the acidosis, removes metformin, and addresses the potassium.
- Longitudinal care: review the medication list on recovery — metformin may be contraindicated if CKD progresses; the SGLT2 inhibitor should be held during acute illness (sick-day rule) but is renal- and cardioprotective long-term. Diabetes and CKD follow-up. Patient education on sick-day rules. [1]
DCE examiner probing questions:
- "Would you give bicarbonate?" → "Not routinely. His pH is 7.08, so I would discuss bicarbonate as a bridge to haemodialysis in conjunction with ICU, but I would not give it as a standalone treatment — the cause is metformin and hypoperfusion, and dialysis is the definitive removal strategy."
- "How does the delta-delta change your management?" → "He has been vomiting, so I expect a concurrent metabolic alkalosis. If the delta-delta is greater than two, the bicarbonate is being artificially supported and his true acid load is worse than the number suggests — I would move to dialysis sooner." [1]
DCE short-case discussion: ABG interpretation
Instruction: "Interpret this arterial blood gas and discuss your management." [1]
Systematic presentation template
"I will interpret this gas in six steps. First, the pH is 7.20 — that is acidaemia. Second, the PaCO2 is 24 and the bicarbonate is 9 — both are low. A low PaCO2 would alkalinise, so it cannot be the cause of the acidaemia; the low bicarbonate is the primary process. This is a primary metabolic acidosis with respiratory compensation." [1]
"Third, I apply Winter's formula: the expected PaCO2 is 1.5 times the bicarbonate plus 8, which is 1.5 times 9 plus 8, equals 21.5, plus or minus 2 — so 19.5 to 23.5. The measured PaCO2 is 24, which is at the upper limit, so compensation is borderline appropriate with a very mild concurrent respiratory acidosis." [1]
"Fourth, the anion gap is sodium 138 minus chloride 100 minus bicarbonate 9, which is 29 — that is a high anion gap metabolic acidosis. Fifth, the delta-delta: the delta AG is 29 minus 12, which is 17; the delta bicarbonate is 24 minus 9, which is 15; the ratio is 17 over 15, about 1.1 — consistent with a pure high-AG acidosis. Sixth, the osmolar gap is 8, which is normal, making a toxic alcohol less likely." [1]
"In summary, this is a severe high anion gap metabolic acidosis, most likely lactic acidosis or ketoacidosis. My immediate management is ABCDE resuscitation, treat the hyperkalaemia, identify and treat the cause — I would check the lactate, ketones, glucose, and renal function now — and involve ICU early given the severity." [1]
Discussion questions
Examiner: "What is the most dangerous cause to miss?" [1]
"A toxic alcohol. Although the osmolar gap is normal here, the gap falls as the parent alcohol is metabolised, so a late-presenting ingestion can have a normal osmolar gap with a high anion gap. I would explicitly take a history of possible ingestion, check the visual system for methanol toxicity, examine the urine for crystals in ethylene glycol, and if there is any doubt, give fomepizole — it is safe and time-critical." [1]
Examiner: "When would you start renal replacement therapy?" [1]
"For absolute indications — refractory hyperkalaemia, refractory severe acidaemia with haemodynamic instability, pulmonary oedema, uraemic complications — or for a specific toxin amenable to dialysis such as metformin or a toxic alcohol. In this patient with metformin-associated lactic acidosis, severe acidaemia, and AKI, I would start haemodialysis early." [1]
Key DWE MCQ patterns
- Apply Winter's formula and identify a mixed disorder. The stem gives a metabolic acidosis with a PaCO2 that is too high (concurrent respiratory acidosis) or too low (concurrent respiratory alkalosis from sepsis or salicylates). Calculate, do not guess.
- Delta-delta to find a hidden alkalosis. A patient with sepsis and vomiting has a HAGMA but a "normal" bicarbonate — the ratio is > 2; the answer is concurrent metabolic alkalosis.
- Correct the anion gap for albumin. A hypoalbuminaemic ICU patient with a measured AG of 14 has a corrected AG that is high — the answer is HAGMA, not normal-AG.
- Identify the toxic alcohol from the clinical clue. Visual symptoms → methanol; renal failure and calcium oxalate crystals → ethylene glycol; high osmolar gap, ketosis, no acidosis → isopropanol.
- Distinguish the RTA by potassium. Hypokalaemic normal-AG acidosis with nephrocalcinosis → type 1; with Fanconi features → type 2; hyperkalaemic normal-AG acidosis in a diabetic on an ACE inhibitor → type 4.
- Distinguish chloride-responsive from resistant alkalosis by urine chloride. Vomiting (low urine Cl, saline-responsive) vs Conn's (high urine Cl, saline-resistant).
- Recognise euglycaemic DKA. Unwell patient on an SGLT2 inhibitor with high-AG acidosis and a near-normal glucose — check ketones.
- Respiratory compensation rules — acute vs chronic. A COPD patient with PaCO2 70 and bicarbonate 36 has chronic respiratory acidosis (appropriate compensation); a bicarbonate of 24 would signal an additional metabolic acidosis. [1]
References
[1] Adrogue HJ, Madias NE. Management of life-threatening acid-base disorders (Part 1). NEJM 1998;338:26–34. The canonical two-part review of acid-base physiology, compensation rules, and treatment — the source for Winter's formula and the acute/chronic respiratory compensation tables. [2] Kraut JA, Mullins ME. Toxic alcohols. NEJM 2018. The definitive modern review of methanol, ethylene glycol, diethylene glycol, propylene glycol and isopropanol — pathophysiology, the anion and osmolal gaps, fomepizole and haemodialysis. [3] Jaber S, et al. BICAR-ICU. Lancet 2018. Sodium bicarbonate for severe metabolic acidaemia in the ICU: no overall mortality benefit, with a signal toward reduced RRT need in the severe-AKI subgroup. Underpins the modern cautious use of bicarbonate. [4] Kitabchi AE, et al. Hyperglycemic crises in adult patients with diabetes. Diabetes Care 2009. The consensus statement defining DKA and HHS diagnostic criteria and the insulin-fluid-potassium management paradigm. [5] Mehta AN, Emmett JB, Emmett M. GOLD MARK: an anion gap mnemonic for the 21st century. Lancet 2008. The modern alternative to MUDPILES, adding 5-oxoproline and D-lactate and dropping obsolete causes. [6] Karet FE. Mechanisms in hyperkalemic renal tubular acidosis. JASN 2009. The molecular basis of type 4 RTA — hypoaldosteronism, distal tubule defects, and the role of hyperkalaemia in suppressing ammoniagenesis. [7] Evans L, et al. Surviving Sepsis Campaign 2021. Crit Care Med 2021. Lactate-guided resuscitation and the principle that lactic acidosis is treated by restoring perfusion, not by administering bicarbonate. [8] Kraut JA, Kurtz I. Toxic alcohol ingestions: clinical features, diagnosis, and management. CJASN 2008. The foundational nephrology review of toxic alcohol diagnosis using the combined anion and osmolal gap approach.
Adrogue & Madias, NEJM 1998; Surviving Sepsis Campaign 2021; ADA Standards of Care; KDIGO CKD Guideline (acidosis section). [1]
References
- [1]Adrogue HJ, Madias NE Management of life-threatening acid-base disorders. First of two parts N Engl J Med, 1998.PMID 9414329
- [2]Kraut JA, Mullins ME Toxic Alcohols N Engl J Med, 2018.PMID 29342392
- [3]Jaber S, Paugam C, Futier E, et al. Sodium bicarbonate therapy for patients with severe metabolic acidaemia in the intensive care unit (BICAR-ICU): a multicentre, open-label, randomised controlled, phase 3 trial Lancet, 2018.PMID 29910040
- [4]Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN Hyperglycemic crises in adult patients with diabetes Diabetes Care, 2009.PMID 19564476
- [5]Mehta AN, Emmett JB, Emmett M GOLD MARK: an anion gap mnemonic for the 21st century Lancet, 2008.PMID 18790311
- [6]Karet FE Mechanisms in hyperkalemic renal tubular acidosis J Am Soc Nephrol, 2009.PMID 19193780
- [7]Evans L, Rhodes A, Alhazzani W, et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock 2021 Crit Care Med, 2021.PMID 34605781
- [8]Kraut JA, Kurtz I Toxic alcohol ingestions: clinical features, diagnosis, and management Clin J Am Soc Nephrol, 2008.PMID 18045860