Phys · renal
Diabetic Kidney Disease
Also known as DKD · diabetic nephropathy · diabetic kidney disease · Kimmelstiel-Wilson disease · nodular glomerulosclerosis · diabetic glomerulosclerosis · microalbuminuria · diabetic renal disease · diabetic kidney failure
Consultant-physician-depth guide to diabetic kidney disease (DKD) — the leading cause of end-stage kidney disease worldwide. Covers the hyperglycaemia to AGE to fibrosis pathophysiology, Mogensen five-stage natural history, screening with annual albumin-to-creatinine ratio, evidence-based management (SGLT2 inhibitors per CREDENCE/DAPA-CKD/EMPA-KIDNEY, ACEi/ARB per RENAAL/IDNT, finerenone per FIDELIO-DKD, multifactorial intervention per Steno-2), normoalbuminuric DKD, and renal biopsy indications. Structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Diabetic Kidney Disease
The answer first
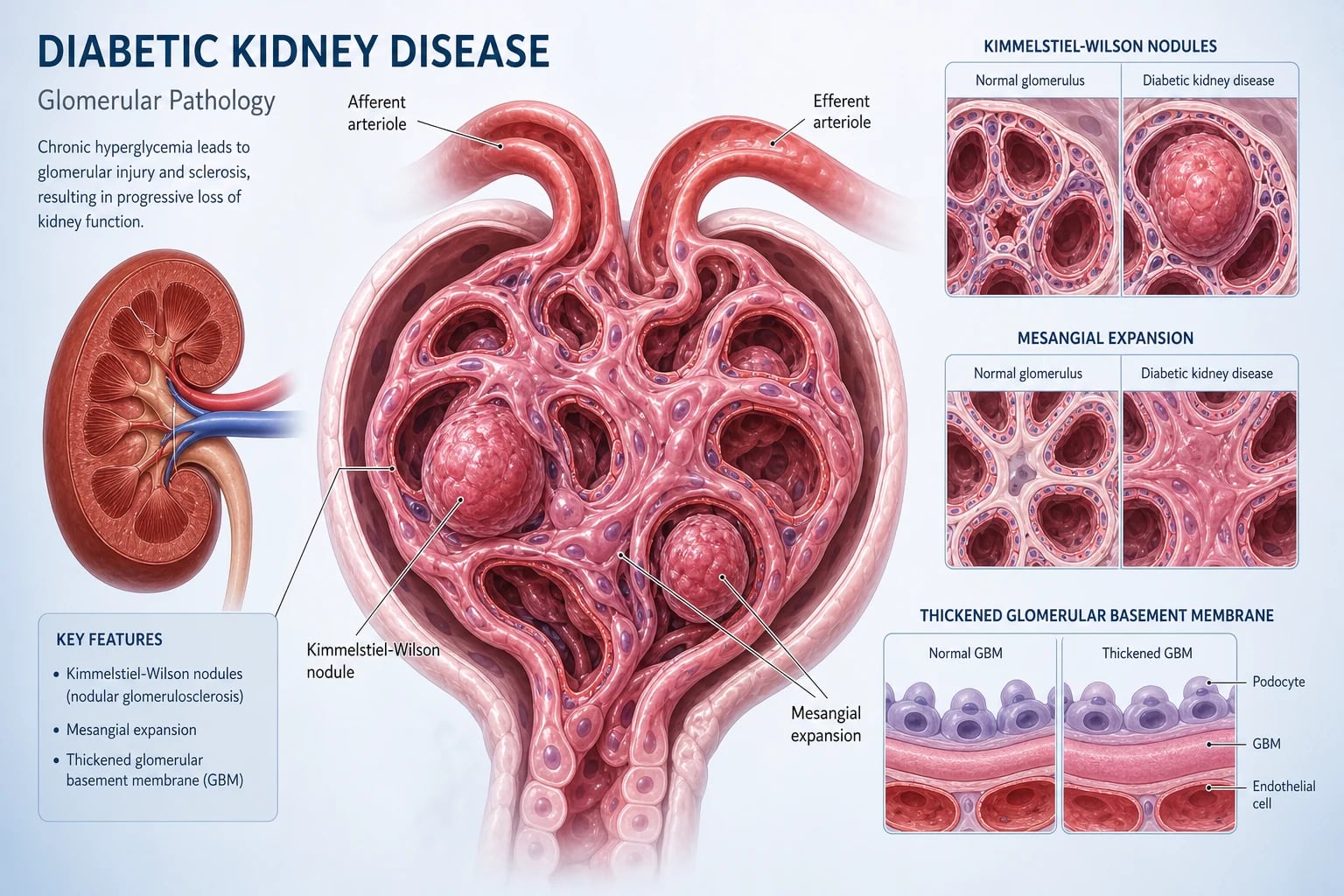
Diabetic kidney disease (DKD) is the chronic kidney injury caused by diabetes — structurally a combination of glomerular basement membrane thickening, mesangial expansion with Kimmelstiel-Wilson nodules, afferent and efferent arteriolar hyalinosis, and tubulointerstitial fibrosis. Clinically it is the leading single cause of end-stage kidney disease (ESKD) worldwide, responsible for around 40 to 50% of new dialysis patients in Australia, the UK and the US [2].
The two questions that dominate DKD management: [1]
- Is this really DKD, or is there a superimposed or alternative process? Classic DKD in a patient with long-standing diabetes and retinopathy needs no biopsy. Atypical features — rapid decline, haematuria, active sediment, short diabetes duration, no retinopathy in T1DM — demand a renal biopsy to exclude a treatable glomerulonephritis.
- How do I slow progression and reduce the cardiovascular death that kills most of these patients before they reach dialysis? [1]
The management mandate, in priority order: [1]
- Start an SGLT2 inhibitor in every patient with T2DM and eGFR greater than or equal to 20 with albuminuria or eGFR-based CKD, irrespective of glycaemia — CREDENCE, DAPA-CKD and EMPA-KIDNEY are the three pillars of this mandate [3][4][5].
- Maximise RAAS blockade — ACE inhibitor or ARB (never both) at maximally tolerated dose for any patient with albuminuria, the foundation laid by RENAAL, IDNT and ADVANCE [7][8][9].
- Add finerenone in T2DM CKD with albuminuria on top of maximal RAAS blockade — FIDELIO-DKD [6].
- Control blood pressure, glycaemia and lipids — the multifactorial Steno-2 model halves cardiovascular events over the long term [10].
- Prevent and manage CKD complications — anaemia, CKD-MBD, acidosis, hyperkalaemia, volume overload.
- Prepare for kidney replacement therapy early and offer a conservative care pathway where dialysis will not benefit.
The single most important exam principle: cardiovascular disease kills more DKD patients than kidney failure does. A stage 3 DKD patient is far more likely to die of an MI than reach dialysis. Statin and BP control are non-negotiable, and a DKD patient without a statin is a long-case fail. [1]
Epidemiology — why DKD dominates the renal landscape
Diabetes affects roughly 1 in 11 adults globally, and DKD develops in 30 to 40% of them over their lifetime [2]. The cumulative result is that diabetes is the single largest contributor to ESKD in every developed health system — around 45% of incident dialysis in Australia (ANZDATA), around 38% in the UK, and nearly 50% in the US (USRDS).
Three epidemiological facts every physician must carry: [1]
- DKD is a coronary risk equivalent. A patient with T2DM and CKD has a 10-year cardiovascular risk comparable to a patient with established coronary disease. Mortality in DKD stage 3 is dominated by cardiovascular events, not ESKD.
- Most patients die with their kidneys, not of them. For every DKD patient who reaches dialysis, several die first of cardiovascular disease. This reframes management: slowing renal decline is also cardiovascular risk reduction.
- The epidemic is not slowing in absolute terms. Despite SGLT2 inhibitors, the absolute number of patients with DKD is rising with the global diabetes epidemic, particularly in Asia and Indigenous populations (high-risk groups include Aboriginal and Torres Strait Islander peoples, Māori/Pacific peoples, South Asian and African ancestry with APOL1-mediated risk in the latter). [1]
DWE exam trap: A common MCQ stem states diabetes is "the second leading cause of ESKD." This is now wrong — diabetes is the leading cause in most developed nations. Hypertensive nephrosclerosis, glomerulonephritis, polycystic disease and reflux nephropathy follow. [1]
Pathophysiology — from glucose to fibrosis
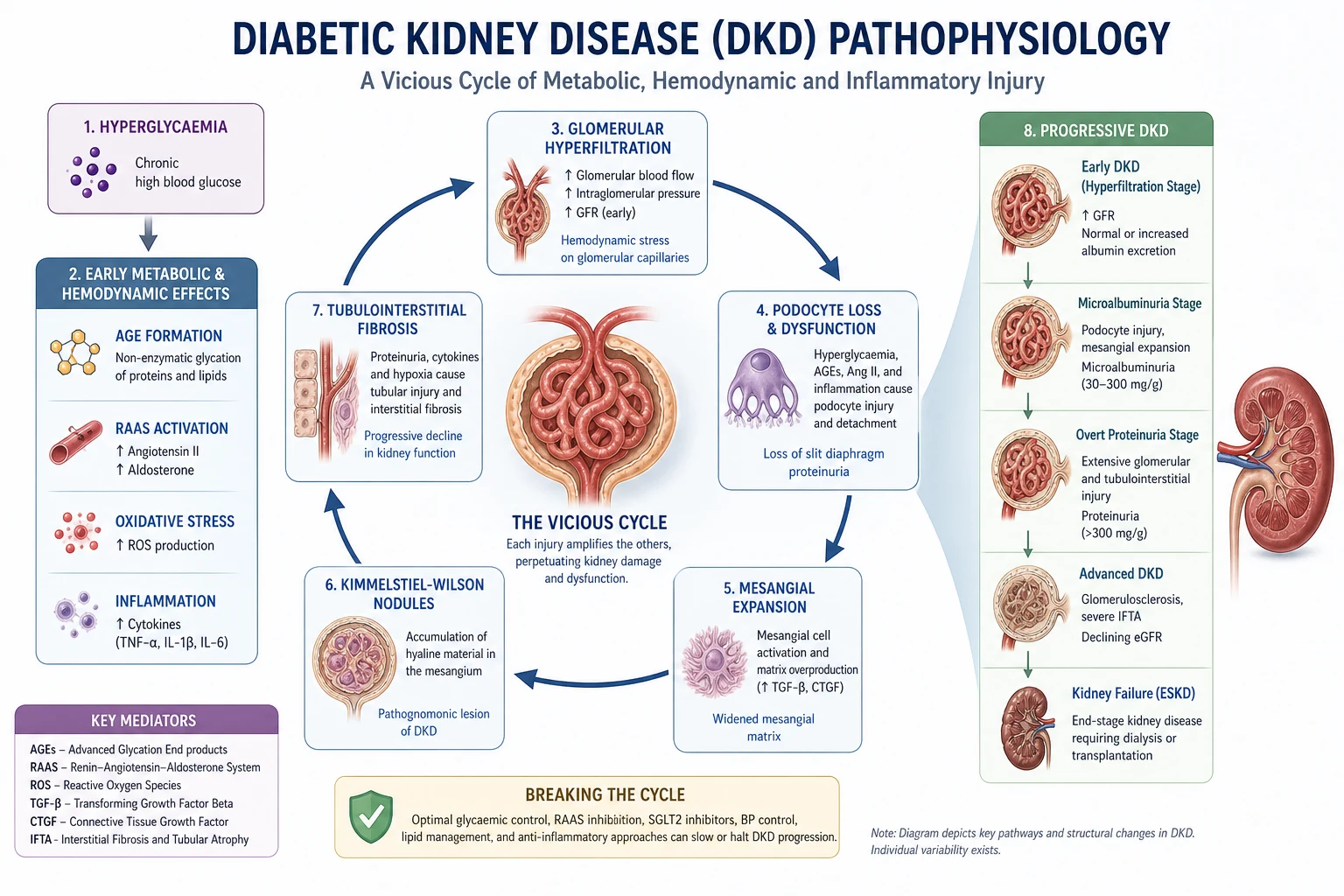
The biochemical injury: hyperglycaemia as a toxic exposure
Sustained hyperglycaemia saturates the glycolytic pathway of cells that cannot down-regulate glucose uptake — particularly mesangial cells, podocytes and proximal tubular cells. Excess intracellular glucose is shunted into four damaging pathways [2]:
- Polyol pathway — aldose reductase converts glucose to sorbitol, consuming NADPH and generating oxidative stress.
- Hexosamine pathway — generates UDP-GlcNAc that pathologically O-GlcNAcylates transcription factors (notably Sp1, driving TGF-beta and PAI-1).
- Protein kinase C (PKC) activation — via diacylglycerol; drives TGF-beta, VEGF, NF-kappaB, fibronectin and endothelial dysfunction.
- Advanced glycation end-product (AGE) formation — non-enzymatic glycation of proteins and lipids; AGEs cross-link matrix, generate oxidative stress, and bind RAGE receptors that amplify inflammation and fibrosis. [1]
The common downstream effect is TGF-beta activation — the master profibrotic cytokine that drives mesangial matrix expansion, fibroblast activation and epithelial-to-mesenchymal transition. TGF-beta is the molecular signature of DKD, and targeting it (indirectly via glycaemic control, RAAS blockade, SGLT2i and finerenone) is the mechanistic basis of every disease-modifying therapy we have. [1]
Glomerular haemodynamic injury: hyperfiltration
Early in diabetes, eGFR is often elevated (hyperfiltration). This is the Mogensen stage 1 finding and reflects two forces: [1]
- Afferent arteriolar vasodilatation — mediated by insulin, nitric oxide, and proximal tubular hypertrophy
- Efferent arteriolar vasoconstriction — mediated by angiotensin II [1]
The result is raised glomerular capillary pressure (intraglomerular hypertension) and raised single-nephron GFR. Hyperfiltration is adaptive in the short term but maladaptive in the long term: the raised pressure drives mechanical strain on podocytes, glomerular hypertrophy, and ultimately glomerulosclerosis. [1]
This is the mechanism that SGLT2 inhibitors reverse. By blocking proximal sodium reabsorption, more sodium reaches the macula densa, restoring tubuloglomerular feedback and causing afferent arteriolar constriction — lowering intraglomerular pressure and albuminuria independent of glycaemia. This is why SGLT2i protect the kidney even when HbA1c has barely moved. [1]
Structural lesions — what the biopsy shows
The histological signature of DKD (when biopsied) is a constellation seen in no other disease [2]:
| Lesion | Description | Significance |
|---|---|---|
| Glomerular basement membrane thickening | Earliest change, detectable within 2 years of diabetes | Functional barrier loss |
| Mesangial expansion | Diffuse increase in matrix; the dominant early lesion | Drives albuminuria |
| Kimmelstiel-Wilson nodules | Acellular PAS-positive nodules of matrix at the periphery of glomerular lobules | Pathognomonic of advanced DKD |
| Afferent AND efferent arteriolar hyalinosis | Both arterioles affected (hypertension affects only afferent) | Vascular injury, distinctive |
| Podocyte detachment and loss | Foot-process effacement; podocyte number falls | Albumin leak; podocytes do not regenerate |
| Tubulointerstitial fibrosis and tubular atrophy | Strongest histological predictor of progression | Final common pathway |
| Arteriolar thickening and rarefaction | Microvascular loss | Ischaemia |
The Kimmelstiel-Wilson nodule is the eponymous histological finding (described by Paul Kimmelstiel and Clifford Wilson in 1936) and is the answer to any "nodular glomerulosclerosis" question. [1]
Albuminuria as both marker and mechanism
Albumin in the urine is not a passive readout. Filtered albumin and other proteins are endocytosed by proximal tubular cells, which become inflamed and fibrogenic — secreting TGF-beta, MCP-1 and RANTES, recruiting inflammatory cells and activating fibroblasts. The end-point is tubulointerstitial fibrosis. [1]
This is why albuminuria reduction is a therapeutic target, not just a marker. Lowering ACR with an ACE inhibitor or SGLT2 inhibitor reduces tubular injury and slows fibrosis, independent of blood pressure. [1]
Normoalbuminuric DKD — the modern phenotype
A substantial and growing proportion of patients with DKD (20 to 50% in modern cohorts) develop a progressive decline in eGFR without ever developing albuminuria. This "normoalbuminuric DKD" phenotype reflects predominant tubulointerstitial, ischaemic and vascular injury with relative preservation of the glomerular filtration barrier. [1]
Clinically: a falling eGFR with a bland urine is still DKD until proven otherwise in a patient with diabetes — but it requires the atypical-presentation workup (see below) to exclude a superimposed or alternative process before being attributed purely to diabetes. [1]
Natural history — the Mogensen five stages
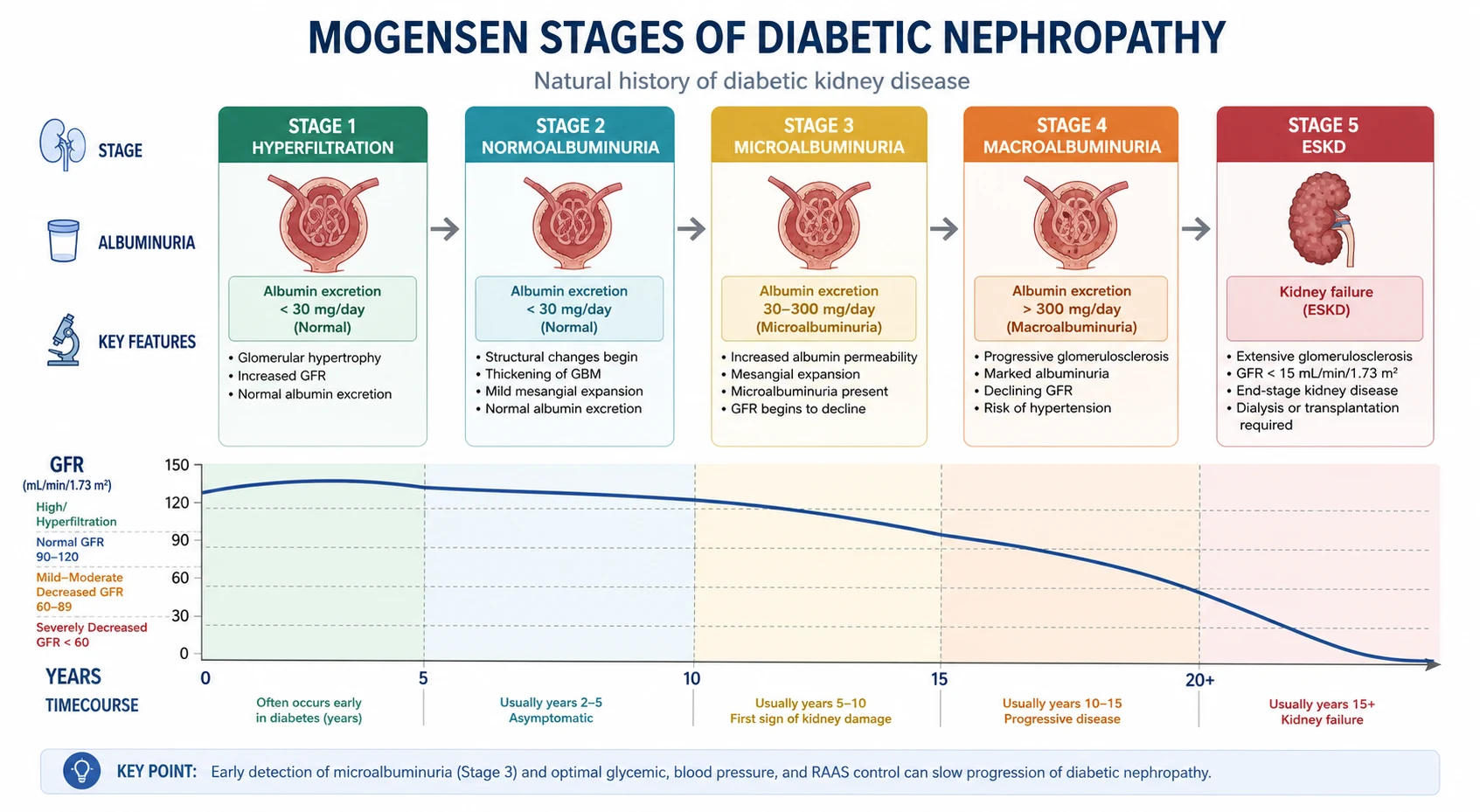
The Mogensen classification (originally described for T1DM, applied with modification to T2DM) frames DKD as a continuum of structural and functional change over 15 to 25 years [2]:
| Stage | Finding | eGFR | ACR | Reversibility |
|---|---|---|---|---|
| 1. Hyperfiltration | Glomerular hyperfiltration, renal hypertrophy | Elevated (120 to 140 mL/min) | Normal (A1) | Fully reversible with glycaemic control |
| 3. Microalbuminuria / early nephropathy | The first clinically detectable sign — persistent microalbuminuria | Normal or slightly reduced | A2: 3 to 30 mg/mmol (20 to 200 microg/min; 30 to 300 mg/g) | Reversible with RAAS blockade and glycaemic control; the critical intervention window |
| 4. Macroalbuminuria / overt nephropathy | Declining eGFR, often nephrotic-range proteinuria, hypertension, oedema | Progressive decline | A3: greater than 30 mg/mmol | Slowed but not reversed by therapy |
| 5. End-stage kidney disease | Requirement for dialysis or transplantation | Less than 15 mL/min/1.73 m² (or on KRT) | N/A | Requires kidney replacement therapy |
Why the Mogensen stages matter at the bedside
- Stages 1 and 2 are silent. The patient is unaware, the eGFR may be normal or high, and only sensitive testing (and eventually histology) shows disease. This is why annual screening is essential — see below.
- Stage 3 (microalbuminuria) is the therapeutic window. Intervention here with RAAS blockade and strict glycaemic control can reverse albuminuria and prevent progression. Once macroalbuminuria (stage 4) is established, the disease is largely irreversible.
- In T2DM, the timeline is compressed and less predictable. Many T2DM patients present with established DKD (or normoalbuminuric decline) because diabetes was present for years before diagnosis. The classic linear progression is less reliable in T2DM than T1DM.
- The rate of eGFR decline is the key monitoring metric. A typical untreated DKD patient loses 4 to 12 mL/min/year. Effective therapy (SGLT2i plus RAAS) can reduce this to under 2 mL/min/year. [1]
DWE exam trap: Microalbuminuria is defined as ACR 3 to 30 mg/mmol (or 30 to 300 mg/g, or 20 to 200 microg/min on a timed collection). Macroalbuminuria (overt nephropathy) is ACR greater than 30 mg/mmol. Nephrotic-range proteinuria is PCR greater than 300 mg/mmol or ACR greater than 220 mg/mmol. These thresholds are commonly tested. [1]
Screening — when, who, how
The screening algorithm is the single most exam-testable public-health intervention in DKD [1]:
| Population | Test | Frequency |
|---|---|---|
| Type 2 diabetes | eGFR AND urinary ACR (spot morning sample) | Annually from diagnosis |
| Type 1 diabetes | eGFR AND urinary ACR | Annually from 5 years after diagnosis (or puberty, whichever is later) |
| Pre-diabetes / high-risk (metabolic syndrome, family history) | eGFR and ACR | Clinician's discretion, typically every 1 to 2 years |
Why screening is annual from diagnosis in T2DM
T2DM is often present for 5 to 10 years before clinical diagnosis. Up to 7% of patients have DKD at the time their T2DM is first diagnosed. Waiting 5 years (as we do in T1DM, where the onset is acute and well-dated) would miss early disease in T2DM. [1]
Confirming the result
A single elevated ACR is not enough. ACR is influenced by exercise, fever, heart failure, UTI, uncontrolled hypertension, menstruation and posture. The KDIGO rule: confirm on at least two of three samples over 3 to 6 months before labelling a patient as having albuminuria. [1]
Persistent albuminuria (confirmed) plus diabetes is the clinical diagnosis of DKD in most cases — no biopsy required if the presentation is classic. [1]
Diagnosis — when to accept the clinical label, when to biopsy
DKD is a clinical diagnosis in the patient with:
- Long-standing diabetes (especially with retinopathy)
- Progressive albuminuria and/or declining eGFR
- Bland urine (no haematuria, no casts)
- Normal or enlarged kidneys on ultrasound [1]
In this classic picture, biopsy adds risk without changing management. But atypical features mandate biopsy — because the alternative diagnoses (IgA nephropathy, membranous nephropathy, FSGS, ANCA vasculitis, anti-GBM, lupus nephritis, amyloid, myeloma cast nephropathy) are treatable, and treating them as if they were DKD is harmful. [1]
Renal biopsy indications in suspected DKD
| Atypical feature | What it suggests |
|---|---|
| Rapid decline in eGFR (greater than expected for DKD) | Superimposed RPGN, acute interstitial nephritis, obstruction |
| Haematuria — dysmorphic RBCs or red cell casts | Glomerulonephritis (DKD has bland urine) |
| Active urinary sediment | Glomerulonephritis or vasculitis |
| Nephrotic-range proteinuria without retinopathy | Membranous nephropathy, FSGS, amyloid, minimal change |
| Short diabetes duration (less than 5 years in T1DM, or no retinopathy) | Alternative diagnosis more likely |
| Absence of retinopathy in T1DM with proteinuria | Strongly against classic DKD; biopsy |
| Signs of systemic disease | Lupus, vasculitis, myeloma — biopsy for diagnosis |
| Falling complement levels, ANA, ANCA, anti-GBM positive | Specific autoimmune GN |
Conversely, biopsy is generally not performed when kidneys are too small (high bleeding risk, low diagnostic yield) or when the diagnosis is classic and tissue would not change management. [1]
DCE long-case trap: The examiner will test whether you would biopsy. State the principle: "Classic DKD is a clinical diagnosis; I would biopsy if there were atypical features — rapid decline, haematuria, active sediment, short duration, no retinopathy, or signs of systemic disease." [1]
Management — the integrated, evidence-based plan
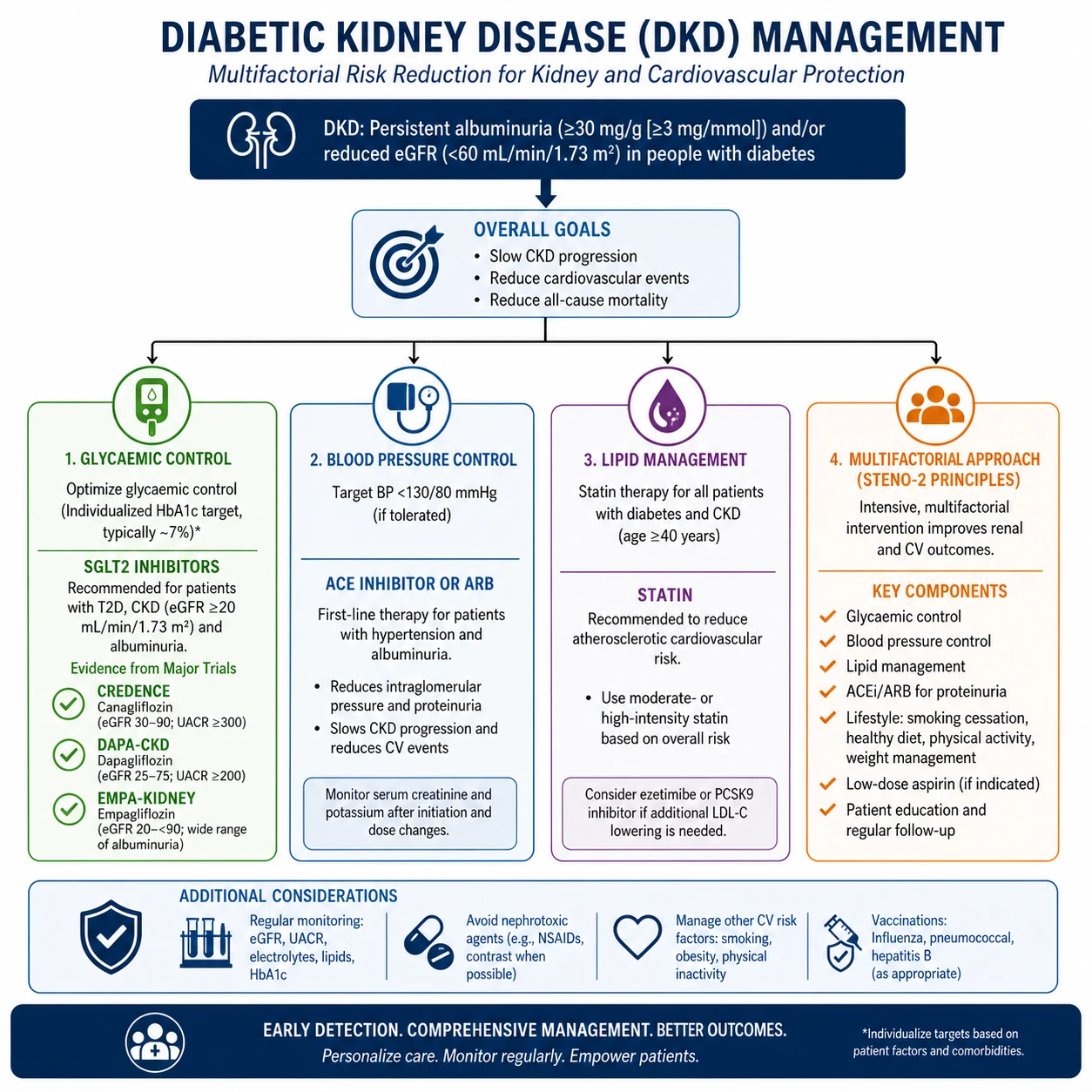
Pillar 1 — SGLT2 inhibitors (the foundation)
SGLT2 inhibitors are now standard of care for any patient with T2DM and CKD. Three landmark trials define the evidence: [1]
| Trial | Drug | Population | Primary outcome | Result |
|---|---|---|---|---|
| CREDENCE [3] | Canagliflozin 100 mg | 4,401 with T2DM, eGFR 30 to 90, ACR greater than 33.9 mg/mmol | ESKD, doubling creatinine, renal or CV death | 30% relative risk reduction; stopped early for efficacy |
| DAPA-CKD [4] | Dapagliflozin 10 mg | 4,304 with CKD (about 67% diabetic), eGFR 25 to 75, ACR 22.6 to 565 mg/mmol | eGFR decline 50%, ESKD, renal or CV death | 39% RRR |
| EMPA-KIDNEY [5] | Empagliflozin 10 mg | 6,609 with CKD (about 54% diabetic), eGFR 20 to 45 (or 45 to 90 with ACR 22.6 mg/mmol), broader albuminuria | Progressive CKD or CV death | 28% RRR |
The key practical points: [1]
- Start an SGLT2i in essentially every T2DM CKD patient with eGFR 20 or above — the benefit is renoprotective and cardiovascular, independent of glycaemia.
- Continue even when HbA1c is at target — the renal protection does not depend on glucose lowering.
- Continue even as eGFR falls (licensed down to eGFR 20) — do not stop solely because glycaemic efficacy has gone.
- Hold during intercurrent illness (volume depletion, infection, surgery, DKA risk) — sick-day rule.
- Counsel on genital mycotic infection, volume depletion, and (rarely) euglycaemic DKA. The amputation signal from CANVAS was not reproduced in CREDENCE. [1]
Pillar 2 — RAAS blockade (ACE inhibitor or ARB)
For patients with albuminuria and hypertension, an ACE inhibitor or ARB at the maximally tolerated dose is first-line. The evidence is from RENAAL, IDNT and ADVANCE [7][8][9]:
| Trial | Drug | Population | Key result |
|---|---|---|---|
| RENAAL [7] | Losartan 50 to 100 mg vs placebo | 1,513 with T2DM and nephropathy | 16% RRR in primary composite (doubling creatinine, ESKD, death); 28% RRR in ESKD; 35% reduction in proteinuria |
| IDNT [8] | Irbesartan vs amlodipine vs placebo | 1,715 with T2DM and overt nephropathy | 20% RRR vs placebo, 23% vs amlodipine; benefit independent of BP |
| ADVANCE [9] | Perindopril + indapamide vs placebo | 11,140 with T2DM | 21% RRR in renal events (mostly new-onset macroalbuminuria); 14% RRR all-cause mortality |
Practical rules: [1]
- Choose one of ACEi or ARB — never combine. Combination increases AKI, hyperkalaemia and mortality (ONTARGET). Do not add a direct renin inhibitor (aliskiren) on top — harm.
- Titrate to maximally tolerated dose — the antiproteinuric effect is dose-dependent.
- Expect a creatinine rise of up to 30% on initiation — this is functional (efferent vasodilatation) and acceptable; reassess at 2 to 4 weeks. A larger rise or hyperkalaemia prompts a search for volume depletion, bilateral renal artery stenosis, or intercurrent NSAID use.
- Do NOT start if bilateral renal artery stenosis is known or suspected — acute kidney injury risk.
- Continue even when eGFR falls — the renoprotective benefit persists; do not stop RAAS blockade solely for a number on the eGFR. Stop only for uncontrolled hyperkalaemia, bilateral renal artery stenosis, or a fall in eGFR beyond 30%. [1]
Pillar 3 — Finerenone (non-steroidal MRA)
Finerenone is a non-steroidal mineralocorticoid receptor antagonist that blocks the profibrotic and proinflammatory effects of aldosterone at the mineralocorticoid receptor with less hyperkalaemia than spironolactone. FIDELIO-DKD showed, in 5,734 patients with T2DM, CKD and ACR greater than 30 mg/mmol on maximal RAAS blockade [6]:
- 18% RRR in the primary composite (kidney failure, sustained doubling creatinine, renal death)
- 14% RRR in cardiovascular events
- Mean potassium rise of 0.2 to 0.5 mmol/L; hyperkalaemia-related discontinuation about 2.3% vs 0.9% placebo [1]
Indication: add finerenone to maximal ACEi/ARB in T2DM CKD with eGFR 25 or above and ACR greater than 30 mg/mmol. Monitor potassium at 4 weeks. Do not use with classic MRAs (spironolactone, eplerenone) or with potassium supplements. [1]
Pillar 4 — Glycaemic control
Tight glycaemic control in early DKD reduces the risk of developing albuminuria and slows progression (DCCT in T1DM, UKPDS in T2DM). But targets must be individualised: [1]
| Patient profile | HbA1c target | Rationale |
|---|---|---|
| Young, few comorbidities, early DKD | 53 mmol/mol (7.0%) or lower if safe | Maximum prevention of microvascular progression |
| Standard adult T2DM with DKD | 53 to 58 mmol/mol (7.0 to 7.5%) | Balance microvascular vs hypoglycaemia |
| Older, frail, limited life expectancy, high CV risk | 58 to 64 mmol/mol (7.5 to 8.0%) or higher | Avoid hypoglycaemia — ACCORD showed harm from very tight control in high-risk patients |
Drug selection in DKD: [1]
- Metformin remains first-line — dose-adjusted to 1 g daily at eGFR 30 to 45, stopped below 30. Lactic acidosis is rare but real in hypoperfusion.
- SGLT2i — preferred second-line for any T2DM CKD patient (renoprotection).
- GLP-1 receptor agonist (semaglutide, dulaglutide) — preferred if SGLT2i unsuitable; reduces albuminuria and CV events; useful for weight.
- Sulfonylureas — risk of hypoglycaemia in CKD; gliclazide preferred (least renal clearance).
- Insulin — often needed; clearance falls as eGFR drops — reduce dose by 25 to 50% at eGFR less than 30.
- DPP-4 inhibitors — neutral; linagliptin preferred (hepatic clearance, no dose adjustment).
- Avoid pioglitazone in heart failure (fluid retention) and in fracture risk. [1]
Pillar 5 — Blood pressure control
Target BP is individualised: [1]
- Younger patients with albuminuria: less than 130/80 mmHg (SPRINT supports intensive targets in non-diabetic CKD; the evidence for less than 130/80 in DKD is strong but watch orthostatic hypotension in autonomic neuropathy).
- Older, frail: less than 140/90 mmHg as a minimum floor; avoid falls. [1]
Drug hierarchy:
- ACEi or ARB first-line (mandatory if albuminuria)
- Calcium channel blocker (amlodipine for BP; diltiazem/verapamil add antiproteinuric effect)
- Thiazide/thiazide-like diuretic (chlorthalidone, indapamide) at eGFR greater than 30; loop diuretic at eGFR less than 30 for volume and potassium
- Beta-blocker if IHD or heart failure
- Alpha-blocker (doxazosin) as fifth-line; watch orthostatic drop [1]
Pillar 6 — Lipid management
Every DKD patient aged 40 years or older should be on a high-intensity statin (atorvastatin 40 to 80 mg). Atorvastatin is preferred as it is hepatically cleared and not affected by CKD. At eGFR less than 30 in a dialysis-naive patient, add ezetimibe if LDL remains elevated. Statins are less effective once on dialysis (4D, AURORA) — start them before dialysis. [1]
Pillar 7 — Multifactorial intervention (the Steno-2 model)
The Steno-2 study randomised 160 patients with T2DM and microalbuminuria to conventional versus targeted multifactorial intervention (behaviour modification, low-dose aspirin, RAAS blockade, BP less than 130/80, HbA1c less than 53 mmol/mol, total cholesterol less than 4.1, fasting cholesterol less than 1.7, lifestyle) [10]. After a mean 7.8 years:
- 53% RRR in cardiovascular events
- 61% RRR in nephropathy (progression to macroalbuminuria, doubling creatinine, ESKD)
- Long-term 21-year follow-up showed median 7.9 years of additional life [1]
The Steno-2 message is the central principle of DKD management: single-pill thinking fails; coordinated multifactorial care transforms outcomes. A DKD patient on SGLT2i but with BP 155/92, no statin and HbA1c 75 mmol/mol is not being managed. [1]
Preventing acute kidney injury on DKD
Patients with DKD have a lower threshold for AKI. The common precipitants: [1]
- Volume depletion — illness, GI losses, diuretics, fasting
- Nephrotoxins — NSAIDs (including over-the-counter), iodinated contrast, certain antibiotics (aminoglycosides)
- RAAS blockade — particularly in volume depletion or renal artery stenosis
- SGLT2i — in volume depletion or sepsis (rarely euglycaemic DKA)
- Contrast — for imaging or angiography [1]
Sick-day rule education is essential. Provide written instructions: during vomiting, diarrhoea or febrile illness, temporarily hold:
- ACE inhibitors and ARBs
- SGLT2 inhibitors
- Diuretics
- Metformin
- NSAIDs (avoid altogether in DKD) [1]
Resume when eating and drinking normally for 24 to 48 hours. [1]
Complications — the long-case checklist
As eGFR falls, the CKD complications accumulate earlier and more severely in DKD than in many other CKD aetiologies: [1]
| Complication | Onset | Management |
|---|---|---|
| Anaemia | Often at eGFR less than 45; earlier than non-diabetic CKD | Iron studies; IV iron if TSAT less than 30% or ferritin less than 100; ESA if Hb less than 100 g/L after iron repletion; target 100 to 120 g/L (TREAT showed harm above 115) |
| Metabolic acidosis | Bicarbonate less than 22 mmol/L | Sodium bicarbonate 600 mg TDS (titrate); slows progression |
| Hyperkalaemia | Amplified by RAAS, SGLT2i, acidosis | Dietary counselling; Patiromer/sodium zirconium if persistent; review RAAS dose |
| Volume overload | With hypertension, heart failure | Loop diuretic; salt restriction; treat heart failure |
| Cardiovascular disease | The dominant cause of death | Statin, antiplatelet, BP control, SGLT2i, GLP-1RA, consider coronary screening |
| Neuropathy and foot disease | Synergistic with vascular disease | Podiatry, foot examination every visit, glycaemic control |
| Retinopathy | Coexists with DKD in classic disease | Annual retinal screening; ophthalmology referral |
Kidney replacement therapy — when and what
Referral to a nephrologist for KRT planning should occur by eGFR less than 30. The milestones: [1]
| eGFR (mL/min) | Action |
|---|---|
| 30 | Refer to nephrology for KRT education, modality counselling, vascular access planning, transplant workup |
| 20 to 25 | Consider AV fistula creation (allow 6 to 12 months to mature); pre-emptive transplant listing if donor available |
| 15 to 20 | Initiate dialysis if symptomatic (uraemia, fluid overload refractory to diuretics); peritoneal or haemodialysis based on patient factors |
| Less than 10 | Dialysis or conservative care |
Pre-emptive transplant (before dialysis) is the best survival option for suitable candidates. Living donor transplant doubles graft survival compared to deceased donor. [1]
Conservative (non-dialysis) care is a legitimate pathway for elderly, frail or highly comorbid patients; median survival on dialysis in the over-80 diabetic is often under 2 years, and symptom burden is high. A values-based conversation, ideally before crisis, is essential. [1]
The DCE long case — putting it together
The DKD long case is a cardiorenal-metabolic patient — multi-morbid, multi-medicated, and the examiner will test your ability to integrate rather than treat each disease in isolation. [1]
Opening statement (SASPOP): "This is a 68-year-old retired truck driver with 22 years of type 2 diabetes presenting to the renal clinic with progressive nephropathy, on a background of hypertension, ischaemic heart disease, peripheral vascular disease and diabetic retinopathy. His main problems are progressive chronic kidney disease with macroalbuminuria, suboptimal blood pressure and glycaemic control, and high cardiovascular risk." [1]
Structured problem list:
- Progressive diabetic kidney disease (CKD G3b A3, eGFR 33, ACR 85 mg/mmol) — not yet on SGLT2i or finerenone [1]2. Suboptimal blood pressure (146/88 seated) on perindopril 10 mg and amlodipine 10 mg
- Suboptimal glycaemic control (HbA1c 62 mmol/mol) on metformin 1g BD and gliclazide 80 mg BD
- High cardiovascular risk — prior NSTEMI, on aspirin, atorvastatin and metoprolol
- CKD complications — anaemia (Hb 98, iron deficient), CKD-MBD (PTH 18), vitamin D deficiency
- Polypharmacy and sick-day risk [1]
Integrated management plan:
- Add dapagliflozin 10 mg daily — renoprotection independent of glycaemia; counsel on genital hygiene, sick-day rules, volume depletion [1]2. Titrate perindopril or add a loop diuretic for BP and volume; consider changing to or adding a thiazide-like diuretic (indapamide) — mirroring ADVANCE; recheck K+ and creatinine at 2 weeks
- Add finerenone 10 mg daily — for T2DM CKD with albuminuria on top of RAAS; recheck K+ at 4 weeks
- Switch glycaemic strategy — continue metformin (eGFR permits), add a GLP-1 receptor agonist (semaglutide) for both glycaemia and weight, consider stopping gliclazide
- Replete iron (IV iron), check 25-OH vitamin D, start cholecalciferol, review phosphate
- Confirm statin is high-intensity (atorvastatin 80 mg); consider ezetimibe if LDL elevated
- Referral to diabetes educator, dietitian, podiatry; sick-day rule education in writing
- Surveillance — repeat eGFR and ACR at 3 months; aim to reduce ACR and slow eGFR decline [1]
Insight — discuss the patient's perspective: medication cost, fear of dialysis, the desire to remain independent; align targets with what matters to him. [1]
The DCE short case — examination in DKD
When the short-case instruction is "Examine this patient's abdomen" or "Examine this patient with diabetes," structure your routine around end-organ damage and renal replacement therapy: [1]
- End of bed — insulin injection sites, body habitus, Cushingoid features if on steroids (post-transplant), dialysis alertness band
- Hands — AV fistula (thrill, bruit, scar, radio-radial delay, steal), peripheral neuropathy (glove-and-stocking), diabetic cheiroarthropathy, trigger finger
- Pulse and BP — lying and standing (autonomic neuropathy), atrial fibrillation, peripheral vascular disease
- Face — xanthelasma, anaemia, glossy tongue, gum hypertrophy (cyclosporin)
- Eyes — cataracts, retinopathy (background, pre-proliferative, proliferative, laser scars), hypertensive changes
- Neck — JVP (volume status), carotid bruits, thyroid (goitre)
- Chest — heart (LVH, murmurs, S3 heart failure), lungs (pulmonary oedema, effusion)
- Abdomen — renal transplant (iliac fossa, scar), AVF, PD catheter, hepatomegaly (NAFLD), abdominal obesity, aortic bruit, bladder
- Legs — peripheral pulses, foot examination (ulcers, callus, Charcot, infection), peripheral oedema, neuropathy [1]
Presentation template: "My peripheral and abdominal findings in this patient with diabetes include evidence of end-organ damage — background diabetic retinopathy on fundoscopy, a glove-and-stocking peripheral neuropathy, and a bruitable AV fistula at the left radiocephalic region indicating preparation for haemodialysis. There is no evidence of peripheral oedema or transplant. These findings, combined with a long history of type 2 diabetes, are consistent with advanced diabetic kidney disease with multi-organ involvement." [1]
High-yield exam discriminators
| Discriminator | Reasoning |
|---|---|
| SGLT2i reduces intraglomerular pressure via tubuloglomerular feedback | Mechanistic answer to "how does an SGLT2i protect the kidney?" |
| ACEi/ARB for albuminuria — choose one, never combine | The single most tested DKD prescribing rule |
| Microalbuminuria = ACR 3 to 30 mg/mmol; macroalbuminuria greater than 30 | Threshold question, frequently tested |
| Biopsy for atypical DKD — rapid decline, haematuria, no retinopathy in T1DM | Discriminator for the thoughtful candidate |
| Normoalbuminuric DKD exists — falling eGFR with bland urine can still be DKD | Modern phenotype, tests conceptual depth |
| Cardiovascular disease is the leading cause of death in DKD | The single most important prognostic fact |
| Steno-2 — multifactorial intervention halves CV events | The evidence base for integrated care |
| CREDENCE — 30% RRR; DAPA-CKD — 39% RRR; EMPA-KIDNEY — 28% RRR | Trial numbers worth memorising |
| Kimmelstiel-Wilson nodules = nodular glomerulosclerosis, pathognomonic | Eponymous histology question |
References and guidelines
KDIGO 2012 CKD Guideline [1]; KDIGO 2024 CKD update; KDIGO 2022 Diabetes in CKD Guideline; ADA Standards of Care; NICE NG203 (2021); CARI Guidelines; Kidney Health Australia; Diabetic kidney disease Primer (Alicic) [2]; CREDENCE [3]; DAPA-CKD [4]; EMPA-KIDNEY [5]; FIDELIO-DKD [6]; RENAAL [7]; IDNT [8]; ADVANCE [9]; Steno-2 [10]; Kidney Failure Risk Equation (Tangri) [11].
References
- [1]Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group Evaluation and management of chronic kidney disease: synopsis of the kidney disease: improving global outcomes 2012 clinical practice guideline Ann Intern Med, 2013.PMID 23732715
- [2]Alicic RZ, Rooney MT, Tuttle KR Diabetic Kidney Disease: Challenges, Progress, and Possibilities Clin J Am Soc Nephrol, 2017.PMID 28522654
- [3]Perkovic V, Jardine MJ, Neal B, et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy N Engl J Med, 2019.PMID 30990260
- [4]Heerspink HJL, Stefánsson BV, Correa-Rotter R, et al. Dapagliflozin in Patients with Chronic Kidney Disease N Engl J Med, 2020.PMID 32970396
- [5]The EMPA-KIDNEY Collaborative Group, Herrington WG, Staplin N, et al. Empagliflozin in Patients with Chronic Kidney Disease N Engl J Med, 2023.PMID 36331190
- [6]Bakris GL, Agarwal R, Anker SD, et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes N Engl J Med, 2020.PMID 33264825
- [7]Brenner BM, Cooper ME, de Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy N Engl J Med, 2001.PMID 11565518
- [8]Lewis EJ, Hunsicker LG, Clarke WR, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes N Engl J Med, 2001.PMID 11565517
- [9]ADVANCE Collaborative Group, Patel A, MacMahon S, et al. Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus (the ADVANCE trial): a randomised controlled trial Lancet, 2007.PMID 17765963
- [10]Gaede P, Vedel P, Larsen N, et al. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes N Engl J Med, 2003.PMID 12556541
- [11]Tangri N, Stevens LA, Griffith J, et al. A predictive model for progression of chronic kidney disease to kidney failure JAMA, 2011.PMID 21482743