Phys · renal
Electrolyte Disorders: Calcium, Magnesium and Phosphate
Also known as hypercalcaemia · hypocalcaemia · corrected calcium · ionised calcium · primary hyperparathyroidism · hypoparathyroidism · familial hypocalciuric hypercalcaemia · FHH · hypomagnesaemia · torsades de pointes · hyperphosphataemia · hypophosphataemia · CKD-MBD · tumor lysis syndrome · refeeding syndrome · hungry bone syndrome
Consultant-physician-depth guide to divalent ion disorders — corrected versus ionised calcium, the PTH-first hypercalcaemia workup and severe hypercalcaemia sequence, asymptomatic primary hyperparathyroidism decisions, post-surgical hypocalcaemia and acute tetany management, magnesium as the hidden blocker of potassium and calcium correction, torsades, CKD-MBD, tumor lysis and refeeding syndrome — structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Electrolyte Disorders: Calcium, Magnesium and Phosphate
The answer first
Calcium, magnesium and phosphate questions look like biochemistry and behave like physiology. Five rules carry you through almost every DWE vignette and every ward call [3]:
- Trust the right calcium. A total calcium without an albumin is half a result. Correct for albumin as a screen, but reach for ionised calcium whenever the correction assumptions break — critical illness, acid-base disturbance, paraproteinaemia, or when the story and the number disagree [1].
- PTH is the first test in hypercalcaemia. One number splits the differential in two: high or inappropriately normal PTH means parathyroid disease; suppressed PTH means malignancy, vitamin D or one of the rare mimics [2] [3].
- Severe hypercalcaemia is saline first. Volume repletion before any antiresorptive; calcitonin for speed within hours; a bisphosphonate — or denosumab in CKD — for durability over days [3] [7].
- Magnesium before potassium before calcium. Refractory hypokalaemia and refractory hypocalcaemia are both magnesium stories until proven otherwise — the kidney cannot hold potassium and the parathyroid cannot work without it [18] [19].
- Thiamine before calories. In the malnourished patient, feeding without thiamine and slow refeeding is how refeeding syndrome happens — phosphate, potassium and magnesium crash together [26].
The calcium you should trust: corrected and ionised
Total serum calcium is three pools: protein-bound (about 40%, mostly albumin), complexed to citrate and phosphate, and the free ionised fraction that actually does the biology. The albumin correction — corrected calcium equals measured calcium plus 0.02 mmol/L for every g/L the albumin sits below 40 — descends from Payne's 1973 regression against abnormal protein states, and it inherits every assumption of that dataset [1].
The correction misleads precisely in the patients you worry about most. It assumes a fixed, normal binding relationship, and that fails in critical illness (acidosis frees ionised calcium from albumin), in chronic kidney disease, in paraproteinaemias where immunoglobulin binds calcium, and at the extremes of albumin where the linear model falls apart. Multiple validation studies have shown corrected formulas misclassify true calcium status often enough that an abnormal or surprising result should trigger an ionised calcium rather than a debate about the formula [1] [3].
Hypercalcaemia: the PTH-first workup
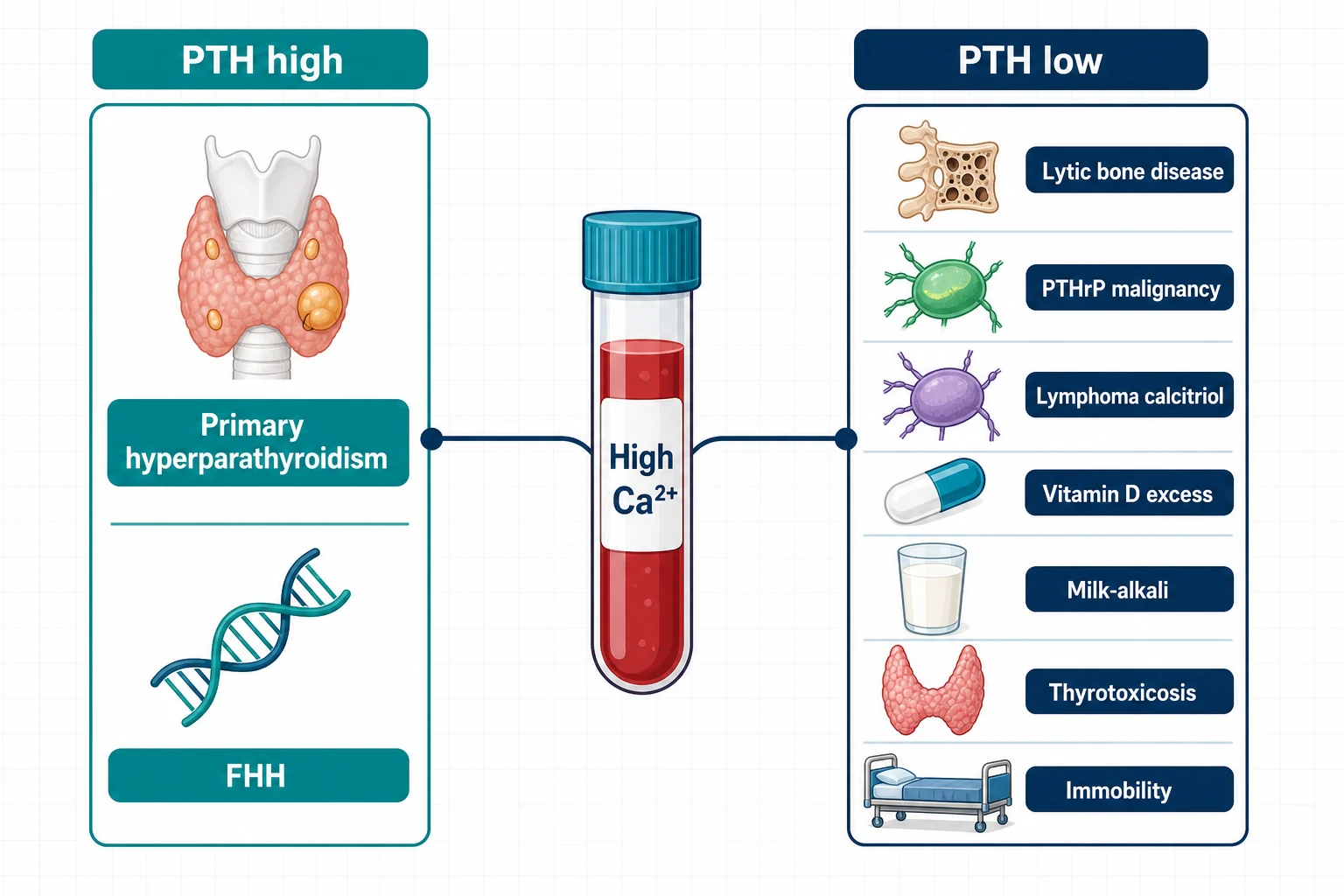
Two diseases account for the overwhelming majority of hypercalcaemia: primary hyperparathyroidism in the community and malignancy in the hospital. Everything else is a distant third, and the intact PTH — measured while the calcium is high — is the fork in the road [2] [3].
A high calcium with a high or inappropriately normal PTH is parathyroid-driven disease. The calcium-sensing receptor (CaSR) on parathyroid chief cells sets the point at which PTH secretion is suppressed; in primary hyperparathyroidism that setpoint is reset upward — usually by a single adenoma (about 85% of cases), less often by hyperplasia or, rarely, carcinoma — so the gland behaves as if a normal calcium is too low [3]. A PTH in the upper-normal range with a frankly high calcium is not "normal" — a properly suppressed gland should be undetectable at that calcium, so upper-normal is inappropriate and still means parathyroid disease [2].
| Pattern | Diagnoses | Next discriminating test |
|---|---|---|
| High Ca, high/inappropriately normal PTH | Primary hyperparathyroidism (adenoma, hyperplasia, carcinoma); familial hypocalciuric hypercalcaemia; lithium; thiazide-unmasked disease; tertiary hyperparathyroidism in CKD | 24-hour urine calcium with calcium:creatinine clearance ratio; drug history; family history [5] |
| High Ca, suppressed PTH | Malignancy (PTHrP humoral, lytic bone disease, calcitriol-producing lymphoma); vitamin D excess; granulomatous disease (sarcoid, TB); milk-alkali; thyrotoxicosis; immobility | PTHrP; 25-OH and 1,25-dihydroxy vitamin D; SPEP/free light chains; chest imaging; TSH [3] [10] |
Familial hypocalciuric hypercalcaemia (FHH) is the great mimic to name in the viva. An inactivating CaSR mutation raises the setpoint in both parathyroid and kidney, so the kidney reabsorbs calcium avidly despite hypercalcaemia — the signature is a low urine calcium. The calcium:creatinine clearance ratio (urine calcium times plasma creatinine, over plasma calcium times urine creatinine) is typically below 0.01 in FHH and above 0.02 in primary hyperparathyroidism, with a grey zone between [5] [6]. Two cautions the examiners love: vitamin D deficiency collapses urine calcium and can fake an FHH ratio, so replete first; and FHH needs no parathyroidectomy — surgery does not cure a receptor [5].
The suppressed-PTH branch is where malignancy lives, in three mechanisms. PTHrP humoral hypercalcaemia (squamous cancers, breast, renal) mimics PTH at its receptor — high calcium, low phosphate tendency, suppressed endogenous PTH, detectable PTHrP. Lytic bone disease (myeloma, breast metastases) is local osteoclast activation — check SPEP and free light chains. Calcitriol-mediated hypercalcaemia — lymphoma and granulomatous disease — is ectopic 1-alpha-hydroxylase converting 25-OH vitamin D to active calcitriol outside renal control, so the diagnostic signature is a high 1,25-dihydroxy vitamin D with a non-suppressed 25-OH level; in Donovan's series of 101 calcitriol-mediated cases, lymphoma and granulomatous disease dominated, and the calcium responded to glucocorticoids [10].
The remainder of the suppressed-PTH list is short and gettable from history: vitamin D excess (supplement overdose — high 25-OH, suppressed PTH) [12]; milk-alkali syndrome (calcium plus alkali intake, classically calcium carbonate for dyspepsia or osteoporosis — high calcium, suppressed PTH, metabolic alkalosis, renal impairment) [11]; thyrotoxicosis and immobility (increased bone turnover), and granulomatous disease on the calcitriol mechanism [2] [3]. Thiazides deserve their own line: they reduce urinary calcium excretion and can unmask underlying primary hyperparathyroidism — a thiazide-associated high calcium that persists after withdrawal is parathyroid disease until proven otherwise [2].
Severe hypercalcaemia: the management sequence
Severe hypercalcaemia — corrected calcium above 3.5 mmol/L, or any level with neurological, cardiac or renal features — is a same-hour problem. The pathophysiology is a vicious circle: hypercalcaemia causes nephrogenic diabetes insipidus and vomiting, dehydration collapses the GFR, and a falling GFR reduces calcium excretion further. Breaking that circle is why the sequence starts with salt [3].
The severe hypercalcaemia sequence
Isotonic saline
Restore volume first — typically 0.9% saline at 200-300 mL/h titrated to the patient, then maintain a diuresis; renal calcium excretion tracks sodium excretion. No antiresorptive works well in an empty tank
Calcitonin for speed
Calcitonin 4-8 IU/kg SC/IM works within hours by inhibiting osteoclasts and increasing renal calcium excretion — but tachyphylaxis limits it to 48-72 hours, so it buys time, it does not finish the job
Antiresorptive for durability
Zoledronate 4 mg IV over at least 15 minutes is the standard — onset 2-4 days, duration weeks; use with renal-dose caution when eGFR is low. Denosumab 120 mg SC is the answer when CKD precludes bisphosphonate
Treat the mechanism
Glucocorticoids (e.g. prednisone 40-60 mg daily or hydrocortisone) when calcitriol-mediated — lymphoma, sarcoid, vitamin D excess; withdraw thiazides, lithium, calcium and vitamin D; mobilise
Dialysis when refractory
Haemodialysis with a low-calcium bath for life-threatening hypercalcaemia with renal or cardiac failure that precludes saline loading — rare but examinable
The evidence behind the antiresorptive choice: the pooled phase III trials showed zoledronate 4 mg normalised calcium faster, in more patients, and for longer than pamidronate 90 mg in hypercalcaemia of malignancy — which is why zoledronate became the default bisphosphonate [7]. The renal caveat is real: bisphosphonates are cleared renally and can worsen renal failure, so in significant CKD the options are a reduced/cautious zoledronate dose or denosumab 120 mg subcutaneously, which is not renally cleared and was effective in bisphosphonate-refractory malignant hypercalcaemia [8] [9].
Two practical warnings earn marks. First, loop diuretics are not part of acute treatment — furosemide after repletion was historically taught, but it adds nothing to saline-driven calciuresis and causes dehydration and electrolyte harm; it survives only as treatment for fluid overload in the repleted cardiac patient [3]. Second, watch the magnesium and phosphate as the calcium falls — aggressive treatment can swing patients into hypocalcaemia, especially with denosumab in CKD [8].
Primary hyperparathyroidism: operate or watch
Most primary hyperparathyroidism now presents as asymptomatic hypercalcaemia found on routine bloods — which is why the decision, not the diagnosis, is the long-case centrepiece. Parathyroidectomy is the only cure; the question is who needs it [4].
The Fourth International Workshop criteria recommend surgery for asymptomatic patients with any of: corrected calcium 0.25 mmol/L or more above the upper limit; age below 50; reduced creatinine clearance (below 60 mL/min), a stone on imaging or hypercalciuria; or osteoporosis — a T-score at or below −2.5 at any site, or a fragility fracture [4].
| Trigger domain | Surgical criterion | Why it matters |
|---|---|---|
| Calcium | 0.25 mmol/L or more above the upper reference limit | Marks more severe biochemical disease |
| Age | Below 50 years | Decades of exposure ahead; surgery is curative |
| Kidney | Creatinine clearance below 60 mL/min, stone on imaging, or marked hypercalciuria | Silent renal damage is common and preventable [4] |
| Bone | T-score at or below −2.5, vertebral fracture on imaging, or fragility fracture | Cortical bone (distal radius) suffers first; bone density improves after cure [4] |
Patients meeting no criterion can be monitored: annual calcium and creatinine, bone density every 1–2 years, and imaging if symptoms develop — with the explicit understanding that roughly a quarter to a third will meet a criterion within a decade, and that monitoring is a decision, not a default [4].
Localisation is for the surgeon, not the diagnostician. The diagnosis of primary hyperparathyroidism is biochemical — high calcium, inappropriately normal or high PTH, normal vitamin D status, urine calcium checked for FHH. Sestamibi and 4D-CT are ordered after the decision to operate, to enable minimally invasive parathyroidectomy; a negative scan never excludes the disease and a positive scan never makes the diagnosis [4]. In the viva, the candidate who orders a sestamibi to "confirm" hyperparathyroidism has the logic backwards.
The elderly comorbid patient is the real exam discussion: an 80-year-old with a calcium of 2.75, a T-score of −2.8 and three comorbidities meets a bone criterion, but operative benefit must be weighed against frailty, anaesthetic risk and life expectancy. The defensible answer is individualised: osteoporosis that is partly parathyroid-driven tips toward referral for a surgical opinion, severe dementia with limited life expectancy tips toward monitoring — and either way, stop the thiazide and replete vitamin D before finalising the biochemistry [4].
Hypocalcaemia: think in PTH pairs
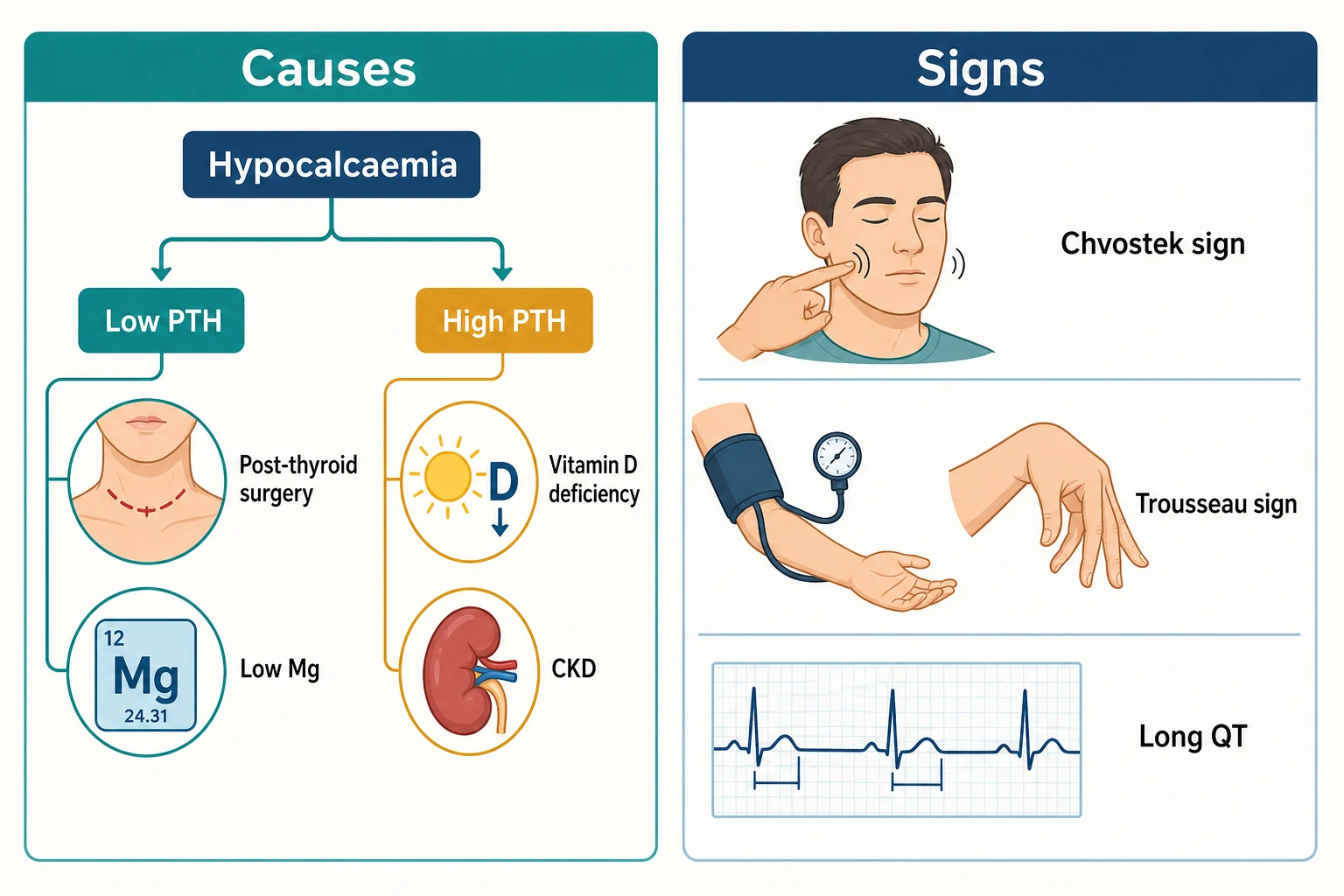
The same PTH-first logic runs the low-calcium workup. A low calcium with a low or inappropriately normal PTH means the gland has failed; a low calcium with a high PTH means the gland is working but something is resisting it [13].
| PTH | Cause | Clues |
|---|---|---|
| Low/inappropriately normal PTH | Post-surgical hypoparathyroidism (after total thyroidectomy or parathyroidectomy) | Neck scar; calcium falls over 24–72 h [13] |
| Low/inappropriately normal PTH | Autoimmune or congenital hypoparathyroidism; infiltrative (haemochromatosis, Wilson disease, metastasis); radiation | Young age, family history, other autoimmune disease [13] |
| Low/inappropriately normal PTH | Hypomagnesaemia | PPI, diuretic or alcohol history; refractory to calcium alone [18] |
| High PTH | Vitamin D deficiency | Low 25-OH vitamin D, high phosphate tendency reversed, myopathy [12] |
| High PTH | CKD-mineral bone disorder | High phosphate, high FGF23, low calcitriol, rising PTH [24] |
| High PTH | Pseudohypoparathyroidism (PTH resistance) | Albright hereditary osteodystrophy features; family history [15] |
| High PTH | Hungry bone syndrome (after parathyroidectomy) | Recent cure of severe hyperparathyroidism; calcium, phosphate and magnesium all falling [28] |
Post-surgical hypoparathyroidism is the long-stay trap. After total thyroidectomy or parathyroidectomy the calcium typically nadirs at 24–72 hours — after the patient has often gone home. The safe unit checks calcium (and PTH where available) the morning after surgery, treats a falling trajectory before symptoms, and discharges with explicit paraesthesia instructions. Permanent hypoparathyroidism (persisting beyond 6–12 months) commits the patient to calcium plus calcitriol for life, with its burden of hypercalciuria, renal calculi and basal ganglia calcification — which is why guidelines push to avoid overtreatment and keep calcium at the low-normal end [13] [14].
Hungry bone syndrome is the mirror image: after successful parathyroidectomy for severe disease, the suddenly PTH-free skeleton avidity takes up calcium, phosphate and magnesium, causing profound hypocalcaemia that can last weeks. Predictors are the severity of preoperative bone disease — high PTH, high alkaline phosphatase, older age, large adenoma — exactly the Brasier and Nussbaum observations from 1988 that still guide which post-parathyroidectomy patients get pre-emptive calcium and calcitriol [28].
Pseudohypoparathyroidism is PTH resistance, not PTH failure: low calcium, high phosphate, high PTH, classically with Albright hereditary osteodystrophy (short stature, round facies, brachydactyly, subcutaneous ossification) from GNAS imprinting defects. Pseudo-pseudohypoparathyroidism carries the same skeletal phenotype with normal biochemistry — the distinction is the calcium and PTH, and both sit within the 2018 international consensus classification of PTH-resistance disorders [15] [16]. One line suffices in the exam — the concept that matters clinically is checking whether PTH is appropriately elevated.
Magnesium is the hidden cause. Severe hypomagnesaemia (typically below about 0.4 mmol/L) both suppresses PTH secretion and induces end-organ PTH resistance, so the biochemistry can look exactly like hypoparathyroidism — low calcium, inappropriately low PTH — until you replace magnesium and watch the PTH normalise within days. Any hypocalcaemia that is refractory to calcium is a magnesium problem until proven otherwise [18] [19].
Acute symptomatic hypocalcaemia: tetany management
Symptomatic hypocalcaemia — perioral and digital paraesthesia, carpopedal spasm, tetany, laryngospasm, seizures, or a long QT — is treated intravenously, and the details matter [13] [14].
Acute symptomatic hypocalcaemia
Calcium gluconate IV, slowly
10-20 mL of 10% calcium gluconate (about 1-2 g) over 10-20 minutes with ECG monitoring — never a push. Calcium chloride contains three times the elemental calcium and is a vesicant: resuscitation bays and central lines only
Then an infusion
The bolus effect lasts hours, not days; follow with a calcium infusion (e.g. 10% calcium gluconate 60-100 mL over 4-12 hours titrated to 6-hourly calcium) until oral therapy holds the level
Fix the magnesium
Check and replace magnesium — without it, PTH secretion and action stay broken and the calcium will not hold
Transition to oral
Oral calcium carbonate (e.g. 1-2 g elemental calcium daily in divided doses) PLUS calcitriol 0.25-1 micrograms daily — active vitamin D, because without PTH the kidney cannot activate cholecalciferol efficiently
Aim low-normal, not high
Chronic hypoparathyroidism without renal PTH-driven calcium retention means every mmol of serum calcium is excreted: target the low-normal range, asymptomatic, with monitoring of urine calcium and renal imaging
The two chronic-therapy points examiners probe: use calcitriol (or alfacalcidol), not plain cholecalciferol, as the backbone — hypoparathyroid patients cannot upregulate 1-alpha-hydroxylase without PTH [13]; and the therapeutic target is symptom relief at low-normal calcium with a normal urine calcium, not biochemical perfection — overtreatment causes nephrocalcinosis in a kidney that has lost its PTH brake on calcium excretion [14].
Magnesium: the quiet gatekeeper
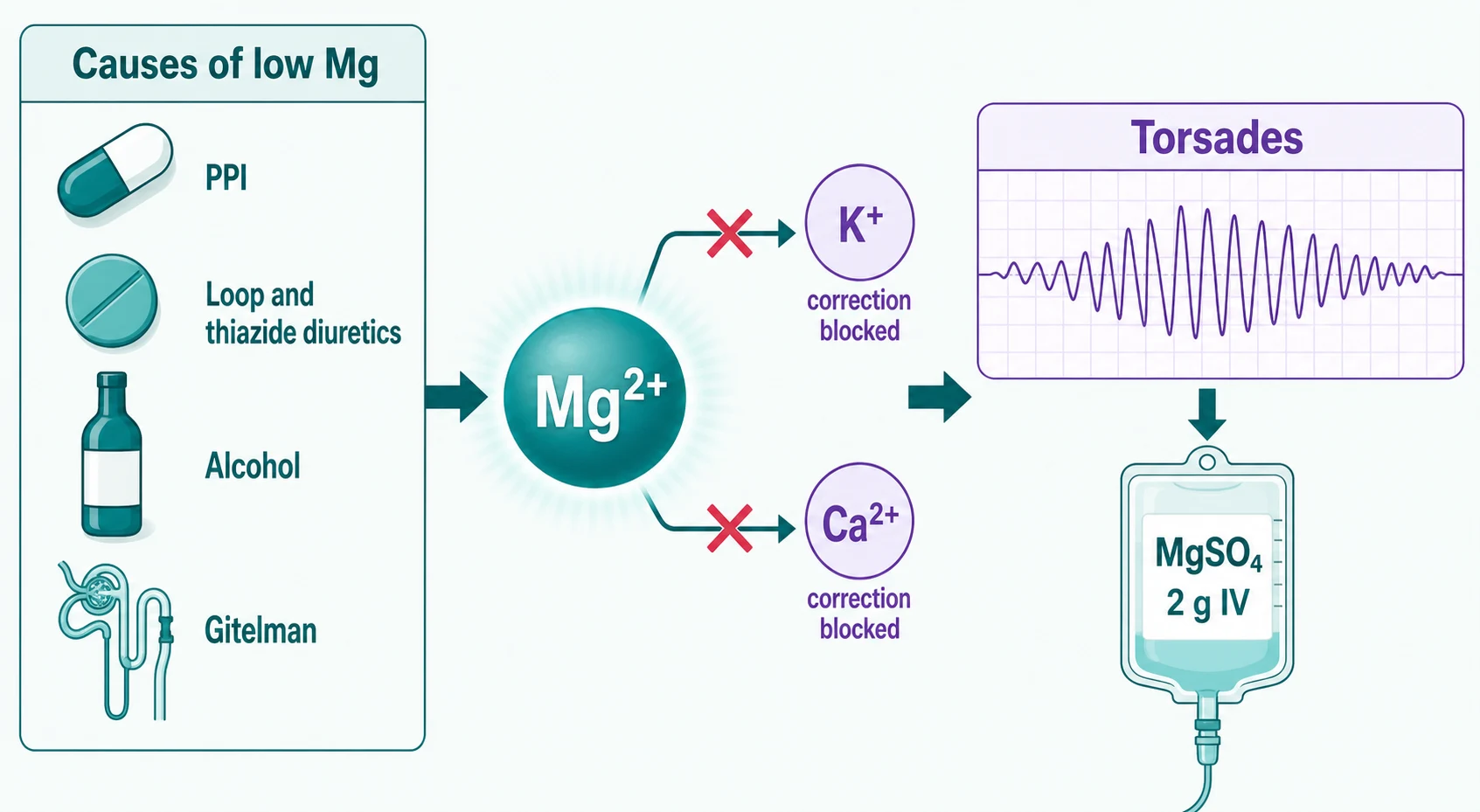
Magnesium is mostly intracellular and mostly unmeasured; the serum level carries about 1% of body stores, so a "normal" level does not exclude depletion and a low level always means significant depletion [19] [20].
The cause list is a ward list: proton pump inhibitors (chronic use impairs intestinal TRPM6-mediated absorption — the Danziger study linked PPI use to lower serum magnesium in a dose-related way, and the effect reverses on withdrawal) [21]; loop and thiazide diuretics (renal wasting); alcohol (poor intake plus renal leak plus phosphate shifts); diarrhoea and stomas; and the renal tubular leak states — Gitelman syndrome (thiazide-like: hypokalaemic metabolic alkalosis with hypomagnesaemia and low urine calcium) and Bartter syndrome (loop-like, with high urine calcium), distinguished partly by that urine calcium [22]. Nephrotoxic drugs — cisplatin, aminoglycosides, amphotericin, ciclosporin — complete the renal-wasting list [19].
The reason magnesium earns its own figure is the refractoriness rule. Magnesium deficiency opens ROMK channels in the collecting duct, so potassium is secreted regardless of how much you replace — the hypokalaemia of magnesium deficiency is unfixable until magnesium is repleted [18]. And as above, severe magnesium deficiency silences the parathyroid axis, producing hypocalcaemia that ignores calcium infusion. The exam sequence is magnesium, then potassium, then calcium — in that order — for any electrolyte that refuses to correct [18] [19].
Replacement route follows severity: severe or symptomatic depletion (arrhythmia, tetany, seizures) gets IV magnesium sulfate — typically 2 g over 10–15 minutes in emergencies, slower infusions otherwise — while chronic depletion gets oral organic salts (citrate, lactate, aspartate), which are better absorbed and less cathartic than oxide [20]. And stop the cause: deprescribing the PPI is the treatment, not an optional extra [21].
Phosphate: the forgotten electrolyte
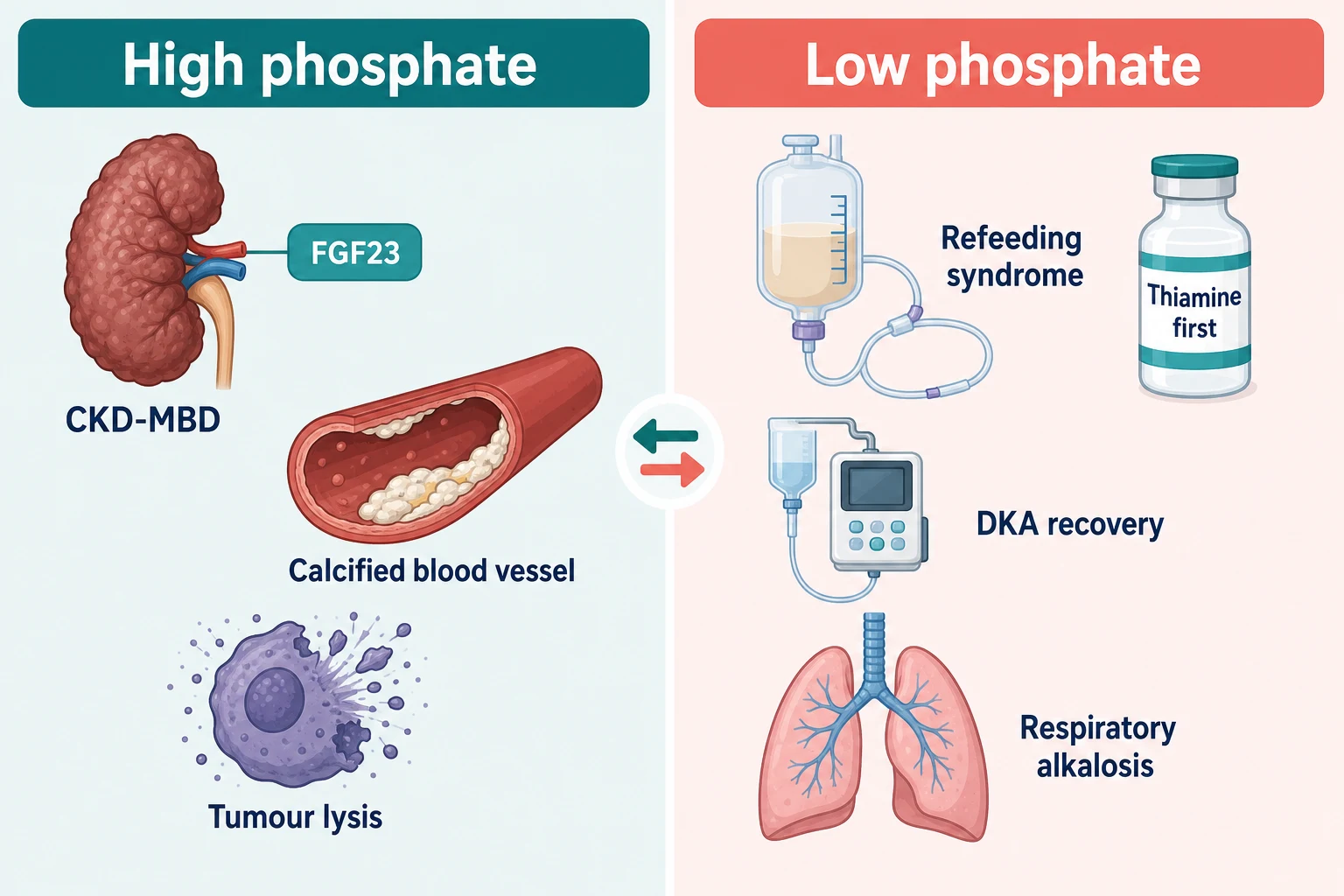
Phosphate questions come in two costumes: the chronic elevation of CKD-mineral bone disorder, and the acute crash of refeeding, DKA recovery and tumor lysis's mirror image [24] [25].
Hyperphosphataemia — CKD-MBD and tumor lysis
In CKD the phosphate story starts earlier than the phosphate level suggests: FGF23 rises first (with its co-receptor Klotho), driving phosphaturia and suppressing calcitriol synthesis, and PTH rises second — by the time serum phosphate is frankly high, the hormonal disturbance is years old. The KDIGO CKD-MBD update frames management around the whole axis — phosphate, calcium, PTH and FGF23 — rather than any single number, and ties it to the outcomes that matter: vascular calcification, renal osteodystrophy, fracture and cardiovascular death [24].
Management is dietary restriction of phosphate additives first, then phosphate binders with meals — calcium-based binders (cheap, but the calcium load feeds vascular calcification) versus non-calcium binders (sevelamer, lanthanum) when calcium load or calcification is the concern — and dialysis once end-stage, acknowledging binders work on gut phosphate and cannot outrun a poor diet [24].
Tumor lysis syndrome is the acute high-phosphate emergency: massive cell death after chemotherapy (or spontaneously in high-turnover lymphoma and leukaemia) releases potassium, phosphate and nucleic acids — the uric acid crystallises in tubules and the phosphate binds calcium, so the signature is high potassium, high phosphate, high uric acid, high LDH, low calcium and acute kidney injury [25]. Prevention is risk-stratified: hydration for all; allopurinol (a xanthine oxidase inhibitor — prevents new uric acid formation) for low-intermediate risk; rasburicase (recombinant urate oxidase — enzymatically degrades existing uric acid within hours) for high-risk disease, bulky burden, or established TLS, with the phase III data showing rasburicase controls uric acid faster and more completely than allopurinol [25] [27]. Two cautions: rasburicase is contraindicated in G6PD deficiency (haemolysis and methaemoglobinaemia), and it keeps working in the sample tube — uric acid after rasburicase must be sent on ice [25].
Hypophosphataemia — refeeding, DKA recovery and the shifts
Serious hypophosphataemia (below about 0.3–0.4 mmol/L) means cellular energy failure: ATP cannot be made, 2,3-BPG falls, and the presentation is proximal weakness, respiratory failure from diaphragmatic weakness, rhabdomyolysis, haemolysis and cardiac failure [26].
Refeeding syndrome is the exam favourite. After prolonged starvation, insulin falls and cells run on fat; restart carbohydrate and the insulin surge drives phosphate, potassium and magnesium into cells — serum levels crash within 2–5 days of feeding. The at-risk patient (chronic alcohol use, anorexia, malignancy, bariatric surgery, the elderly inpatient who has "not eaten for a week") is identified before the first meal: give thiamine first (the glucose load without thiamine precipitates Wernicke encephalopathy), start calories low and increase slowly, replace phosphate/potassium/magnesium proactively, and monitor daily for the first week [26].
The other two causes to name: DKA recovery — total-body phosphate is depleted through the osmotic diuresis, and insulin therapy plus fluids unmasks the deficit as phosphate shifts intracellularly, though routine replacement is reserved for symptomatic or severe depletion because over-replacement causes hypocalcaemia [25]; and respiratory alkalosis — intracellular alkalosis activates phosphofructokinase, consuming phosphate acutely, which is why the hyperventilating ICU patient can have a strikingly low phosphate with normal total-body stores. And do not forget hungry bone syndrome from the calcium section — phosphate, calcium and magnesium fall together into the healing skeleton [28].
Replacement is oral when mild and tolerated (phosphate is cathartic in large oral doses), IV when severe or symptomatic or the gut is unreliable, and always paired with potassium and magnesium attention — the three move together [26].
DCE angles: the long case and the short case
The long case is asymptomatic hypercalcaemia. Your patient is the 72-year-old referred with a corrected calcium of 2.95 found on routine bloods, a PTH at the top of the reference range, a T-score of −2.6 at the hip, on a thiazide for hypertension. The examiner's interest is not the diagnosis — it is the reasoning: confirm the biochemistry off the thiazide (withdraw for 2–4 weeks and repeat calcium and PTH — a thiazide-unmasked calcium that persists is parathyroid disease), replete vitamin D so the urine calcium for the FHH ratio is interpretable, image the kidneys for silent stones, and then have the operate-or-watch conversation as a genuine equipoise discussion — her hip T-score technically meets a surgical criterion, but her surgical fitness, fracture risk on other grounds, and preferences decide the referral [4] [5]. Structure the defence as: what is the diagnosis, what have I excluded (FHH, malignancy, vitamin D), what does she risk if watched (fracture, stone, renal decline), what does she risk if operated (anaesthesia, hungry bone is unlikely in mild disease, recurrent laryngeal injury), and who else decides with her [4].
The short case is the tetany examination. Asked to "examine for hypocalcaemia", the honours performance: inspect first (carpopedal spasm at rest, perioral paraesthesia story, neck scar from thyroid or parathyroid surgery, cataracts, band keratopathy at the slit lamp), then Chvostek sign — tap the facial nerve just anterior to the tragus and watch for ipsilateral facial twitching — then Trousseau sign — inflate the cuff above systolic pressure for up to 3 minutes and watch for carpopedal spasm (thumb adduction, metacarpophalangeal flexion, interphalangeal extension — the main d'accoucheur hand) [17]. Then say the honest sentence: Hoffman's clinical study found Chvostek positive in a substantial minority of people with normal calcium, and both signs can be absent in chronic hypocalcaemia — so the signs support, they never exclude [17]. Finish with the ECG (long QT) and the offer to check calcium, phosphate, magnesium and PTH [13].
Exam traps, collected
References
- [1]Payne RB, Little AJ, Williams RB, et al. Interpretation of serum calcium in patients with abnormal serum proteins Br Med J, 1973.PMID 4758544
- [2]Carroll MF, Schade DS. A practical approach to hypercalcemia Am Fam Physician, 2003.PMID 12751658
- [3]Minisola S, Pepe J, Piemonte S, et al. The diagnosis and management of hypercalcaemia BMJ, 2015.PMID 26037642
- [4]Bilezikian JP, Brandi ML, Eastell R, et al. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Fourth International Workshop J Clin Endocrinol Metab, 2014.PMID 25162665
- [5]Christensen SE, Nissen PH, Vestergaard P, et al. Familial hypocalciuric hypercalcaemia: a review Curr Opin Endocrinol Diabetes Obes, 2011.PMID 21986511
- [6]Marx SJ. Familial Hypocalciuric Hypercalcemia as an Atypical Form of Primary Hyperparathyroidism J Bone Miner Res, 2018.PMID 29115694
- [7]Major P, Lortholary A, Hon J, et al. Zoledronic acid is superior to pamidronate in the treatment of hypercalcemia of malignancy: a pooled analysis of two randomized, controlled clinical trials J Clin Oncol, 2001.PMID 11208851
- [8]Hu MI, Glezerman IG, Leboulleux S, et al. Denosumab for treatment of hypercalcemia of malignancy J Clin Endocrinol Metab, 2014.PMID 24915117
- [9]Hu MI, Glezerman I, Leboulleux S, et al. Denosumab for patients with persistent or relapsed hypercalcemia of malignancy despite recent bisphosphonate treatment J Natl Cancer Inst, 2013.PMID 23990665
- [10]Donovan PJ, Sundac L, Pretorius CJ, et al. Calcitriol-mediated hypercalcemia: causes and course in 101 patients J Clin Endocrinol Metab, 2013.PMID 23979953
- [11]Beall DP, Henslee HB, Webb HR, et al. Milk-alkali syndrome: a historical review and description of the modern version of the syndrome Am J Med Sci, 2006.PMID 16702792
- [12]Holick MF. Vitamin D deficiency N Engl J Med, 2007.PMID 17634462
- [13]Brandi ML, Bilezikian JP, Shoback D, et al. Management of Hypoparathyroidism: Summary Statement and Guidelines J Clin Endocrinol Metab, 2016.PMID 26943719
- [14]Bollerslev J, Rejnmark L, Marcocci C, et al. European Society of Endocrinology Clinical Guideline: Treatment of chronic hypoparathyroidism in adults Eur J Endocrinol, 2015.PMID 26160136
- [15]Mantovani G. Clinical review: Pseudohypoparathyroidism: diagnosis and treatment J Clin Endocrinol Metab, 2011.PMID 21816789
- [16]Mantovani G, Bastepe M, Monk D, et al. Diagnosis and management of pseudohypoparathyroidism and related disorders: first international Consensus Statement Nat Rev Endocrinol, 2018.PMID 29959430
- [17]Hoffman E. The Chvostek sign; a clinical study Am J Surg, 1958.PMID 13545482
- [18]Huang CL, Kuo E. Mechanism of hypokalemia in magnesium deficiency J Am Soc Nephrol, 2007.PMID 17804670
- [19]Ayuk J, Gittoes NJ. How should hypomagnesaemia be investigated and treated? Clin Endocrinol (Oxf), 2011.PMID 21569071
- [20]Ayuk J, Gittoes NJ. Treatment of hypomagnesemia Am J Kidney Dis, 2014.PMID 24100128
- [21]Danziger J, William JH, Scott DJ, et al. Proton-pump inhibitor use is associated with low serum magnesium concentrations Kidney Int, 2013.PMID 23325090
- [22]Blanchard A, Bockenhauer D, Bolignano D, et al. Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference Kidney Int, 2017.PMID 28003083
- [23]Panchal AR, Bartos JA, Cabañas JG, et al. Part 3: Adult Basic and Advanced Life Support: 2020 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care Circulation, 2020.PMID 33081529
- [24]Ketteler M, Block GA, Evenepoel P, et al. Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease-Mineral and Bone Disorder: Synopsis of the Kidney Disease: Improving Global Outcomes 2017 Clinical Practice Guideline Update Ann Intern Med, 2018.PMID 29459980
- [25]Howard SC, Jones DP, Pui CH. The tumor lysis syndrome N Engl J Med, 2011.PMID 21561350
- [26]Mehanna HM, Moledina J, Travis J. Refeeding syndrome: what it is, and how to prevent and treat it BMJ, 2008.PMID 18583681
- [27]Cortes J, Moore JO, Maziarz RT, et al. Control of plasma uric acid in adults at risk for tumor Lysis syndrome: efficacy and safety of rasburicase alone and rasburicase followed by allopurinol compared with allopurinol alone--results of a multicenter phase III study J Clin Oncol, 2010.PMID 20713865
- [28]Brasier AR, Nussbaum SR. Hungry bone syndrome: clinical and biochemical predictors of its occurrence after parathyroid surgery Am J Med, 1988.PMID 3400660