Phys · renal
Potassium Disorders — Hyperkalaemia and Hypokalaemia
Also known as hyperkalaemia · hyperkalemia · hypokalaemia · hypokalemia · potassium · peaked T waves · sine wave · calcium gluconate · insulin-dextrose · insulin glucose · patiromer · sodium zirconium cyclosilicate · SZC · sodium polystyrene sulfonate · resonium · Bartter syndrome · Gitelman syndrome · primary aldosteronism · Conn syndrome · pseudohyperkalaemia · tumour lysis syndrome · rhabdomyolysis
Consultant-physician-depth guide to potassium homeostasis and its two emergencies. Covers hyperkalaemia (K+ above 5.5): the renal and pharmacological causes (AKI, CKD, RAAS inhibitors, K-sparing diuretics, Addison's, rhabdomyolysis, tumour lysis, acidosis), the ECG progression from peaked T waves through PR prolongation and QRS widening to sine wave and cardiac arrest, and emergency management with calcium gluconate for membrane stabilisation, insulin-dextrose and salbutamol to shift K+ intracellularly, bicarbonate in acidosis, the potassium binders (patiromer, sodium zirconium cyclosilicate, resonium) and dialysis. Covers hypokalaemia (K+ below 3.5): the GI and renal causes, diuretics, Bartter and Gitelman syndromes, Cushing and Conn syndromes, alkalosis, insulin, and refractory hypomagnesaemia; the clinical features of weakness, cramps, and U waves with QT prolongation; oral versus IV replacement with the maximum 10 to 20 mmol/h central-line rate; and the 24h urine potassium, renin/aldosterone and acid-base workup. Structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Potassium Disorders — Hyperkalaemia and Hypokalaemia
The answer first
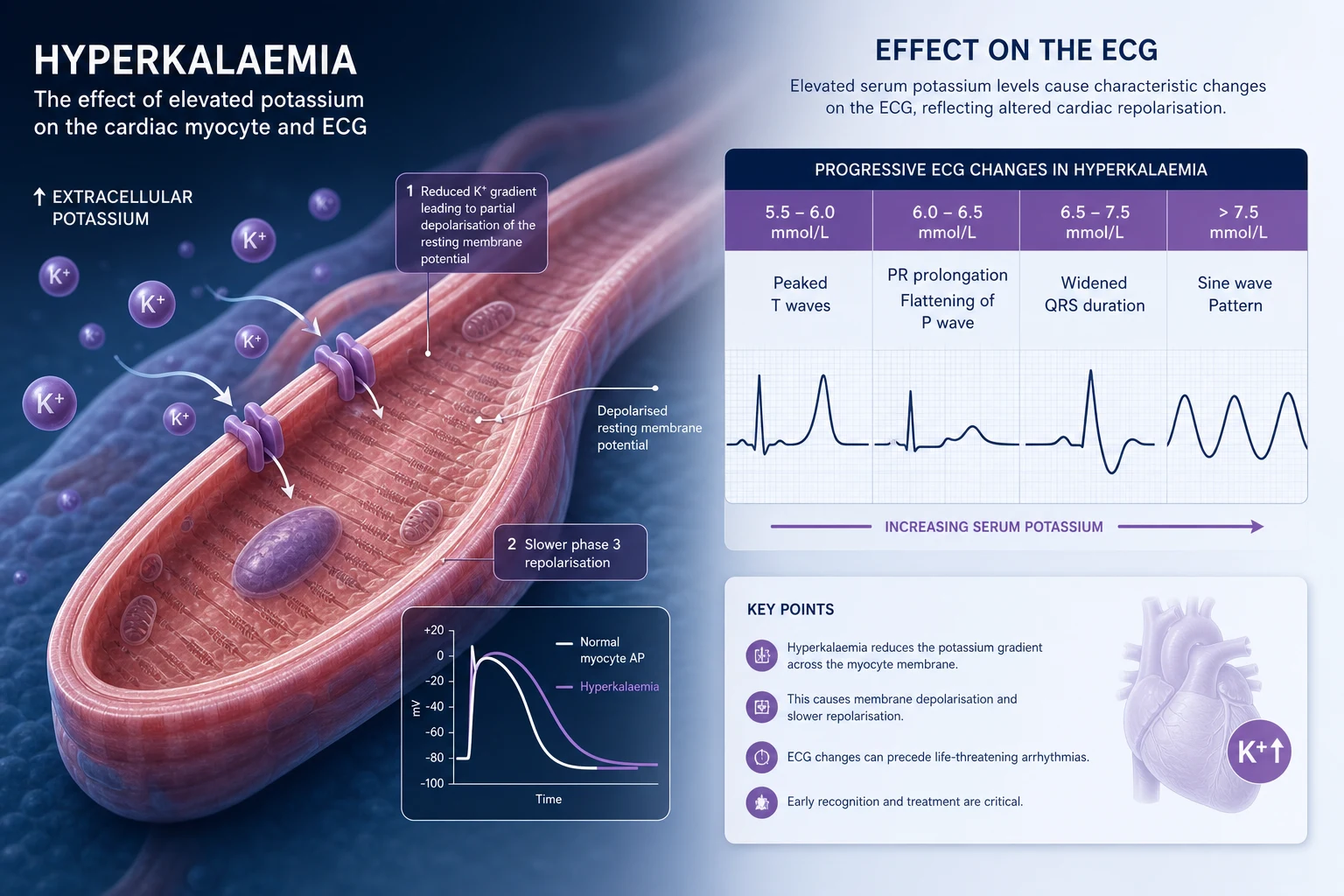
Potassium is an intracellular ion. Only 2 percent of total body potassium is in the extracellular fluid, yet that small fraction is what the serum potassium measures and what determines membrane excitability — in the heart most acutely. The serum potassium is therefore a poor index of total body potassium; it reflects the balance between intake, excretion, and the distribution of potassium across the cell membrane. A clinician faced with an abnormal potassium must answer three questions in parallel: is this real, what is the threat to the heart, and what is driving the disturbance [1][2].
The two emergencies are mirror images. Hyperkalaemia (K+ above 5.5 mmol/L) threatens the heart by depolarising the resting membrane potential: the myocyte cannot repolarise, conduction slows, and the rhythm degenerates from peaked T waves through QRS widening to a sine wave and ventricular fibrillation or asystole. Hypokalaemia (K+ below 3.5 mmol/L) hyperpolarises the membrane, prolongs repolarisation, and produces arrhythmias — classically QT prolongation with torsades, atrial ectopy, and re-entrant ventricular arrhythmias — as well as skeletal and smooth muscle weakness. [1]
In both, the ECG determines urgency more than the absolute number, but the ECG is imperfect: severe hyperkalaemia can have a near-normal ECG, and the first manifestation of an arrhythmia may be the arrest itself. Treat the patient and the trend, not the single value [13].
Potassium physiology — what a registrar must carry into every case
Total body potassium is approximately 50 mmol per kilogram, or 3500 mmol in a 70-kilogram adult. Of this, 98 percent is intracellular. The serum concentration is held tightly between 3.5 and 5.0 mmol/L by the interplay of four systems. [1]
1. The Na+/K+-ATPase pump. This moves three sodium ions out of the cell and two potassium ions in, consuming ATP. It is stimulated by insulin and beta-2 adrenergic agonists, and inhibited by alpha agonists and acidosis (organic acidosis less than mineral acidosis). This pump is the mechanism behind insulin-dextrose and salbutamol as acute hyperkalaemia therapies. [1]
2. Aldosterone. Aldosterone acts on the principal cell of the collecting duct to increase the number and activity of epithelial sodium channels (ENaC) at the apical membrane. Sodium reabsorption creates a lumen-negative gradient that drives potassium secretion through the ROMK channel. Anything that reduces aldosterone (Addison's, ACE inhibitors, ARBs, renin inhibitors, K-sparing diuretics, heparin) reduces potassium excretion and predisposes to hyperkalaemia. Anything that increases aldosterone or mimics it (Conn's, Cushing's, secondary hyperaldosteronism, liquorice, exogenous mineralocorticoid) increases potassium excretion and causes hypokalaemia [2].
3. Distal sodium delivery and urine flow. Potassium secretion tracks distal sodium delivery and tubular flow. Diuretics that act proximal to the collecting duct (loop, thiazide) increase distal sodium delivery and flow, enhancing potassium secretion — the mechanism of diuretic-induced hypokalaemia. Reduced distal sodium delivery (volume depletion, advanced CKD, ACE inhibition) limits potassium secretion and predisposes to hyperkalaemia. [1]
4. Acid-base balance. Metabolic acidosis shifts potassium out of cells (the exchange of intracellular K+ for extracellular H+ to buffer the acid load), raising the serum potassium by approximately 0.6 mmol/L for every 0.1 unit fall in pH — though this rule is loose, and the effect is greater with mineral (hyperchloraemic) acidosis than with organic (lactic, keto) acidosis. Alkalosis shifts potassium into cells, lowering the serum potassium [1].
The kidney is responsible for 90 to 95 percent of potassium excretion; the gut handles the remainder. In advanced CKD, colonic potassium excretion is upregulated and becomes clinically important — this is part of why constipation and certain drugs (resonium) that bind potassium in the gut have a role in chronic hyperkalaemia management. [1]
Hyperkalaemia
Definition and severity
Hyperkalaemia is a serum potassium above 5.5 mmol/L (some units define 5.0 to 5.5 as mild, above 5.5 as significant, above 6.5 as severe). A practical severity framework: [1]
| Severity | Serum K+ (mmol/L) | Action |
|---|---|---|
| Mild | 5.5 to 5.9 | Confirm real (repeat, exclude pseudohyperkalaemia); identify and address cause; review medications; ECG |
| Moderate | 6.0 to 6.4 | As above plus admit; consider shift therapy and binder; 6-hourly K+; urgent nephrology review if CKD |
| Severe | Above 6.5, or any K+ with ECG changes | Emergency: calcium, shift therapy, removal, and a plan for dialysis; cardiac monitoring [6][13] |
Pseudohyperkalaemia must be excluded before any aggressive treatment. It is a falsely elevated serum potassium due to release of potassium from cells during or after venesection. Causes: fist clenching during collection, prolonged tourniquet time, haemolysed sample, delayed transport, severe leucocytosis (WCC above 100) or thrombocytosis (platelets above 700) from cell lysis in the tube, and ex vivo haematological malignancy cell lysis. A point-of-care arterial or venous blood gas with a free-flowing sample, run immediately, is the fastest way to confirm the true value. A serum-to-plasma potassium difference greater than 0.4 mmol/L on paired samples confirms pseudohyperkalaemia. Never treat a pseudohyperkalaemia with calcium and insulin. [1]
Causes — organise by the mechanism of impaired excretion versus transcellular shift
Hyperkalaemia arises from one of three mechanisms (often combined): impaired renal excretion, a transcellular potassium shift out of cells, or increased intake — and in practice, the first two dominate. [1]
Impaired renal excretion (the commonest mechanism): [1]
| Category | Specific causes |
|---|---|
| Reduced glomerular filtration | Acute kidney injury, advanced chronic kidney disease (especially G4/G5), end-stage kidney disease — fewer functioning nephrons to secrete potassium |
| Reduced aldosterone effect | ACE inhibitors, angiotensin receptor blockers, direct renin inhibitors; K-sparing diuretics (spironolactone, eplerenone, amiloride, triamterene); Addison's disease / primary adrenal insufficiency; heparin (inhibits aldosterone synthesis); calcineurin inhibitors (cyclosporin, tacrolimus); congenital adrenal hyperplasia |
| Reduced distal sodium delivery / flow | Volume depletion, severe heart failure, effective circulating volume depletion (cirrhosis, nephrosis) |
| Tubulointerstitial disease | Obstructive uropathy, interstitial nephritis, lupus nephritis — the distal nephron is the secretory site |
| Type 4 renal tubular acidosis | Hyporeninaemic hypoaldosteronism (classic in diabetic nephropathy), the prototype of hyperkalaemic RTA |
Transcellular shift out of cells (potassium redistributed, not increased in total): [1]
| Category | Specific causes |
|---|---|
| Cell lysis | Rhabdomyolysis (crush, statins, prolonged immobilisation, exertional), tumour lysis syndrome (especially high-grade lymphoma and leukaemia after chemotherapy), massive haemolysis, severe burns |
| Acidosis | Mineral (hyperchloraemic) metabolic acidosis shifts potassium out of cells more than organic acidosis |
| Insulin deficiency | DKA and HHS — lack of insulin removes the principal push of potassium into cells; the total body potassium is in fact depleted |
| Hyperosmolarity | HHS, mannitol therapy — solvent drag pulls potassium out of cells |
| Drugs | Beta-blockers (especially non-selective, in overdose), digoxin toxicity (inhibits the Na+/K+-ATPase directly — a classic cause), suxamethonium (in denervation, burns, prolonged immobility — suxamethonium-induced hyperkalaemia) |
| Hyperkalaemic periodic paralysis | Rare channelopathy with intermittent attacks; distinct from the more common hypokalaemic form |
Increased intake is rarely the sole cause in a patient with normal renal function — the kidney can excrete hundreds of mmol of potassium per day. It becomes relevant in CKD: salt substitutes (high in potassium chloride), potassium-containing penicillins, large dietary loads (bananas, tomatoes, nuts, dried fruit), and packed red cell transfusions (each unit leaks potassium, especially irradiated and older units). [1]
The clinical presentation
Most patients with hyperkalaemia are asymptomatic and the finding is on bloods taken for another reason. Symptoms, when present, are non-specific: muscle weakness (the classic ascending pattern mimicking Guillain-Barre in severe cases), paraesthesiae, and palpitations. The emergency presentations are cardiac: the arrhythmia, the arrest, or the ECG change that precedes them. A patient with hyperkalaemia on the monitor can deteriorate to asystole in minutes — this is why the ECG and cardiac monitoring are the first responses to any significant K+ result [7][9].
ECG changes — the progression that determines urgency
The ECG is the single most important clinical tool in hyperkalaemia. The changes track the progressive impairment of myocardial conduction and repolarisation as the serum potassium rises. The classic progression is taught as a sequence, but individual patients do not always follow it, and the ECG may be near-normal even at lethal potassium levels — never be reassured by a normal-looking ECG in a severe hyperkalaemia [6].
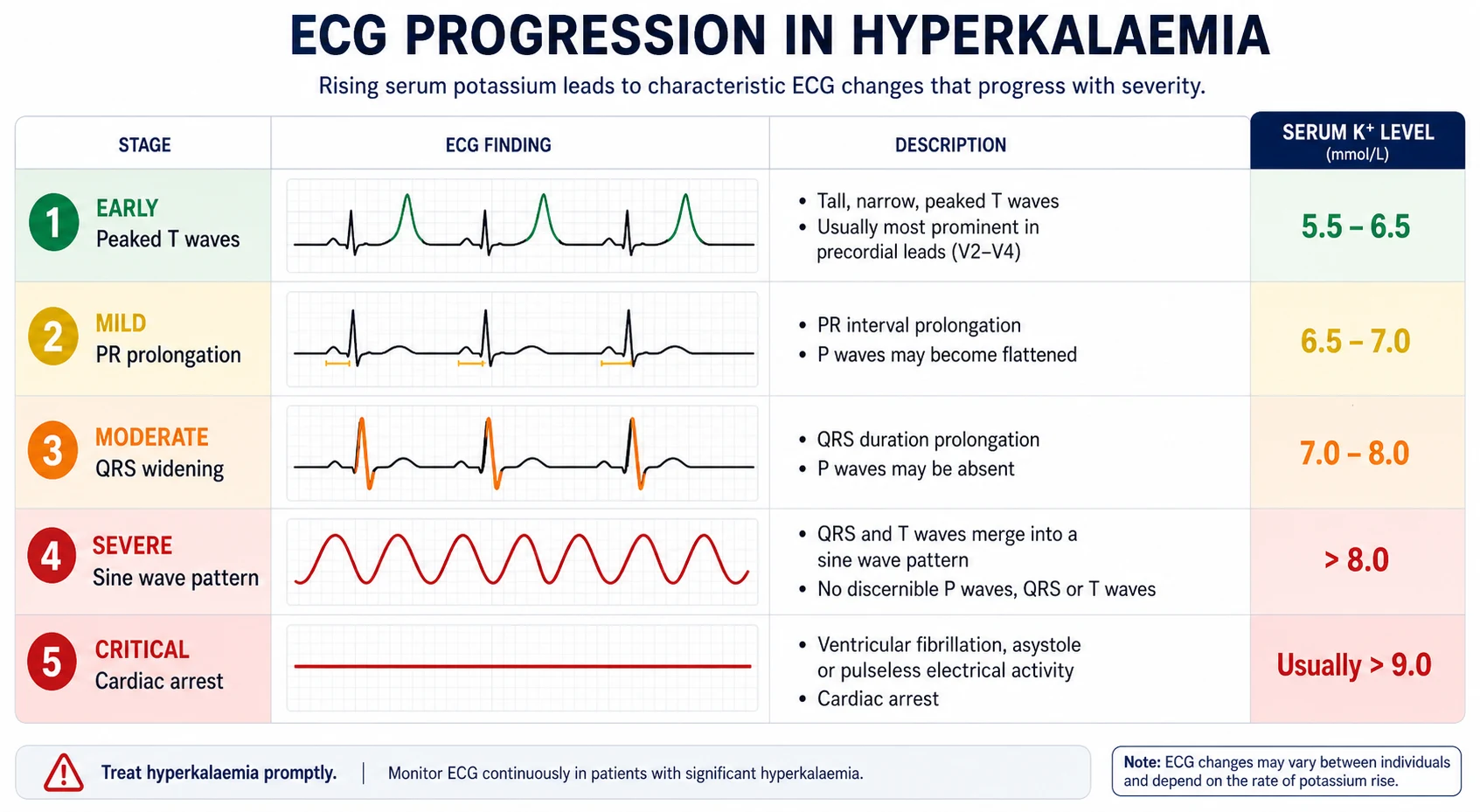
| Stage | Serum K+ (approximate) | ECG change | Mechanism |
|---|---|---|---|
| Early | 5.5 to 6.5 | Peaked T waves — tall, narrow-based, symmetric "tenting", best in precordial leads; shortened QT | Accelerated terminal repolarisation |
| Intermediate | 6.5 to 7.5 | PR prolongation, flattening then loss of P waves; lengthening QRS | Slowed atrial and AV conduction |
| Advanced | 7.5 to 8.5 | QRS widening into the T wave; conduction blocks; ventricular ectopy; bradycardia; escape rhythms | Generalised conduction slowing |
| Pre-terminal | Above 8.5 | Sine wave — the QRS and T wave merge into a continuous sine pattern; this precedes arrest | Myocardial standstill |
| Arrest | Any | Ventricular fibrillation, pulseless electrical activity, asystole | Loss of organised electrical activity |
The peaked T wave is the classic early sign. The definition varies, but a practical rule is a T wave taller than 6 mm in the limb leads or 10 mm in the precordial leads, with a narrow base and symmetrical upstroke and downstroke. The sine wave is the pre-terminal pattern and indicates that calcium and definitive therapy cannot wait. [1]
Hypocalcaemia prolongs the QT interval and amplifies the cardiac risk of hyperkalaemia — relevant in tumour lysis syndrome (where hypocalcaemia coexists) and in CKD (where both are common). Conversely, hypercalcaemia can partly protect against hyperkalaemic toxicity, which is one rationale for giving calcium in treatment. [1]
The DWE high-yield associations
- A diabetic patient with CKD on an ACE inhibitor and spironolactone who presents with weakness — the classic polypharmacy hyperkalaemia. Stop the K-sparing agent, consider the ACE inhibitor dose, treat per the algorithm.
- Addison's disease: hyperkalaemia with hyponatraemia, hypoglycaemia, hypotension, hyperpigmentation — check a morning cortisol and give parenteral hydrocortisone; do not wait for the result if the patient is unwell.
- Rhabdomyolysis: hyperkalaemia with a markedly raised creatine kinase, elevated AST and ALT (both leak from muscle), pigmented urine, and a dipstick positive for blood but with no red cells on microscopy (myoglobin). Aggressive IV fluids are the first intervention; treat the hyperkalaemia per the algorithm.
- Tumour lysis syndrome: hyperkalaemia with hyperphosphataemia, hypocalcaemia, and AKI within 12 to 72 hours of starting cytotoxic therapy for a high-grade lymphoma or leukaemia. Pre-empt with rasburicase and IV fluids in high-risk patients.
- Digoxin toxicity: hyperkalaemia (the digoxin inhibits the Na+/K+-ATPase) with gastrointestinal symptoms, visual disturbance (yellow vision, xanthopsia), and arrhythmia (atrial tachycardia with block is classic). Avoid calcium gluconate in digoxin toxicity — historically thought to cause "stone heart" via intracellular calcium overload, though this is now debated; the safer move is digoxin Fab antibody fragments. [1]
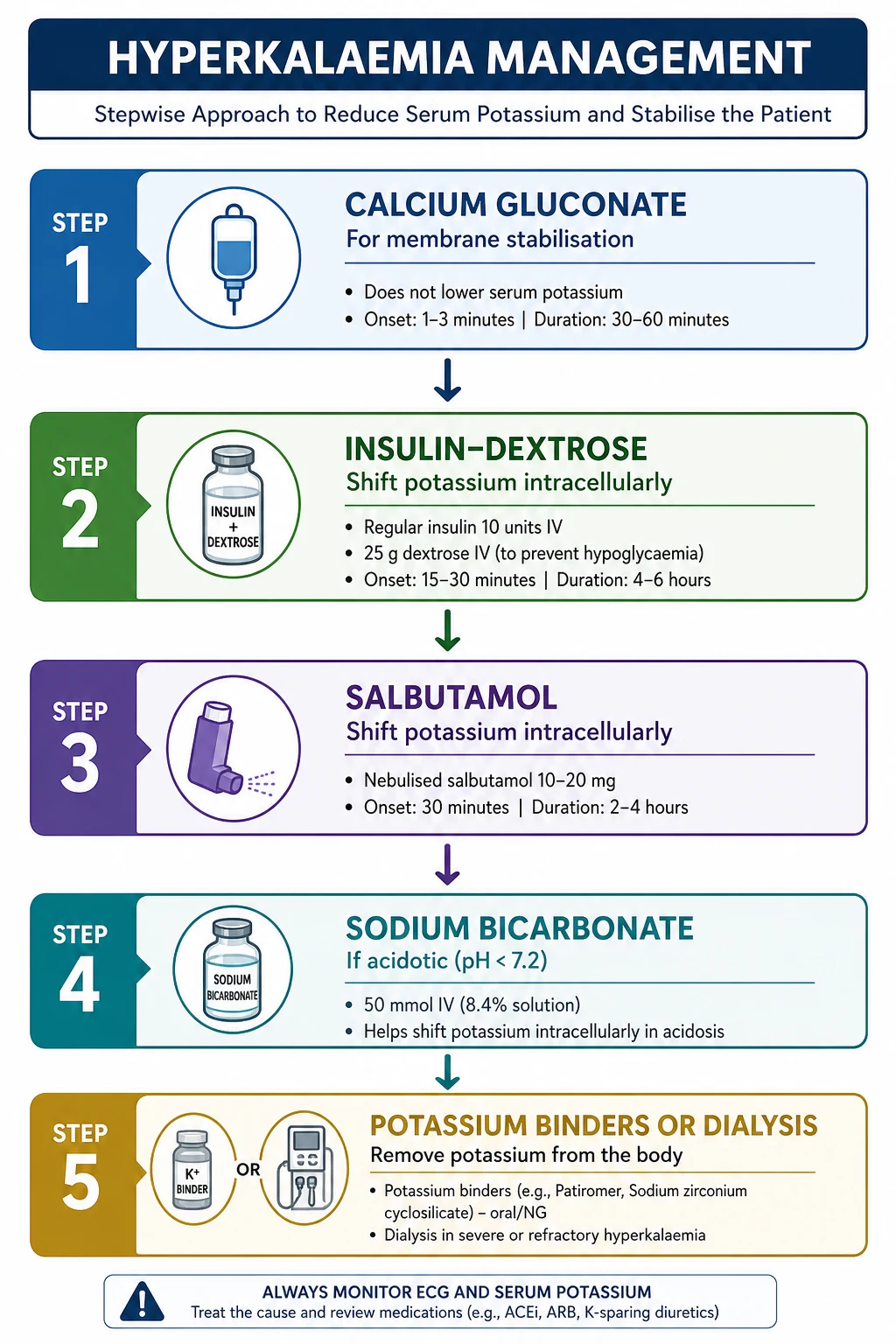
Emergency management of hyperkalaemia — the four pillars
The treatment of severe or ECG-changed hyperkalaemia has four simultaneous goals. They have different onsets, durations, and mechanisms, and you deploy them together, not in sequence. [1]
Pillar 1 — Stabilise the myocardium (onset within minutes). Intravenous calcium raises the threshold for excitation and reverses the membrane effects of hyperkalaemia on conduction. It does not lower the potassium; it buys time by preventing arrhythmia while the shift and removal therapies work. [1]
- Calcium gluconate 10%, 10 mL (1 g or 2.25 mmol of calcium) intravenously over 2 to 5 minutes, with ECG monitoring. Onset within 1 to 3 minutes, duration 30 to 60 minutes. Repeat once after 5 minutes if ECG changes persist [9].
- Calcium chloride contains three times more calcium per millilitre but is intensely vesicant and must be given via a central line — reserve for cardiac arrest. In the arrest context, give 10 mL of 10% calcium chloride IV.
- Re-dose calcium every 30 to 60 minutes if ECG changes recur or persist while waiting for definitive therapy.
- The historical caution against calcium in digoxin toxicity is contested, but the safer approach in suspected digoxin toxicity is to use digoxin Fab antibody fragments rather than rely on calcium.
Pillar 2 — Shift potassium into cells (onset 15 to 30 minutes, duration 4 to 6 hours). This temporarily lowers the serum potassium; it does not remove potassium from the body. The two first-line agents are insulin-dextrose and salbutamol; use both. [1]
- Insulin with dextrose. Give 10 units of rapid-acting (soluble) insulin IV with 25 g of glucose — typically 25 mL of 50% dextrose or 50 mL of 50% dextrose, given as a bolus or over 15 to 30 minutes. The insulin stimulates the Na+/K+-ATPase and shifts potassium into cells. Onset 15 minutes, peak 30 to 60 minutes, lowers K+ by approximately 0.5 to 1.2 mmol/L, duration 4 to 6 hours [5][10].
- Hypoglycaemia is the commonest serious adverse event — occurring in up to 20 percent of patients with the traditional high-dose insulin regimen, and it can occur up to 6 hours after the dose. Check a blood glucose at baseline, at 30 minutes, at 1 hour, hourly for 4 to 6 hours, and treat hypoglycaemia promptly. Lower-dose regimens (5 units) lower potassium as effectively with less hypoglycaemia, and are increasingly recommended [5].
- In the hyperglycaemic patient (DKA, HHS) who is already insulin-deficient, give the insulin alone and titrate against the glucose — the dextrose is unnecessary and harmful when the glucose is high.
- Salbutamol (albuterol). Give 10 to 20 mg nebulised (5 to 10 times the bronchodilator dose) or 0.5 mg IV. The beta-2 agonist stimulates the Na+/K+-ATPase and shifts potassium into cells, lowering K+ by 0.5 to 1.0 mmol/L over 30 minutes, with effect for 2 to 4 hours [10].
- Additive to insulin-dextrose — give both for a combined effect of 1.0 to 1.5 mmol/L.
- Cautions: tachycardia, tremor, and (rarely) myocardial ischaemia in patients with coronary disease; use cautiously in those with known cardiac disease. Less effective in beta-blocked patients.
- Sodium bicarbonate. The role is limited to the acidotic patient. Bicarbonate shifts potassium into cells as it corrects acidosis, and is most useful when the serum bicarbonate is low (less than 18 mmol/L). The traditional dose is 1 to 2 ampoules (50 to 100 mmol) of 8.4% sodium bicarbonate IV over 15 to 30 minutes, or as an isotonic infusion in 5% dextrose. Bicarbonate is not effective as monotherapy in non-acidotic hyperkalaemia, and should not replace insulin-dextrose; the modern consensus is that its role is confined to the acidotic or acidaemic patient [7]. It carries a sodium and volume load and can cause hypernatraemia and fluid overload in CKD/heart failure.
Pillar 3 — Remove potassium from the body (onset hours, sustained). Shift therapy redistributes potassium but does not eliminate it; the patient will rebound unless removal is addressed. [1]
- Loop diuretics. Frusemide 40 to 80 mg IV promotes potassium excretion in the patient with residual renal function; useful in volume-overloaded CKD and heart failure. Ineffective in oligoanuria.
- Gastrointestinal cation exchangers (potassium binders). These bind potassium in the gut lumen and increase faecal elimination. The three agents in current use:
- Patiromer — a non-absorbed polymer that binds potassium in the colon. Dose 8.4 g orally once daily (range 8.4 to 25.2 g daily, divided). Onset over hours; the OPAL-HK trial demonstrated a sustained 0.8 mmol/L reduction [3]. Allows continuation of RAAS inhibitors in CKD. Hypomagnesaemia and gastrointestinal effects (constipation, diarrhoea) are the main adverse effects.
- Sodium zirconium cyclosilicate (SZC) — an inorganic crystal that exchanges sodium or hydrogen for potassium throughout the GI tract. Dose 10 g three times daily for up to 48 hours for acute correction, then 5 g once daily for maintenance. The HARMONIZE trial showed a rapid reduction within 4 hours and sustained control at 28 days [4]. Carries a sodium load — caution in heart failure and hypertension. Has a faster onset than patiromer and is approved for acute use in some regions.
- Sodium polystyrene sulfonate (resonium / SPS) — an older resin that exchanges sodium for potassium. Dose 15 to 30 g orally (or 30 g retention enema). Slower (24 to 48 hours) and less predictable than the newer agents. Has been associated with intestinal necrosis, especially when given as a retention enema in the post-operative or ileus patient — the FDA issued a warning. Largely superseded by patiromer and SZC in modern practice but still used where newer agents are unavailable or unaffordable.
- Binders are not appropriate as sole therapy for severe or ECG-changed hyperkalaemia — they are too slow. Their role is in the subacute and chronic management, particularly to enable RAAS inhibitor continuation in CKD and heart failure [1][8].
- Dialysis. The definitive removal modality when the patient is refractory to medical therapy, has severe life-threatening hyperkalaemia (especially with ECG changes and oligoanuria), or has severe CKD/AKI in which medical therapy is failing. Haemodialysis lowers potassium faster than peritoneal dialysis (a 1 to 1.5 mmol/L fall in the first hour). Plan early — vascular access, anticoagulation, and the logistics take time. Continuous renal replacement therapy (CVVHDF) is preferred in the haemodynamically unstable ICU patient, with a slower but sustained potassium removal. Be aware of rebound after dialysis, particularly in the patient with ongoing potassium release (rhabdomyolysis, tumour lysis, GI bleed, acidosis) — recheck the potassium after dialysis.
Pillar 4 — Identify and address the cause; prevent recurrence. Review the medication chart (the commonest reversible contributor). Treat Addison's, digoxin toxicity, rhabdomyolysis, tumour lysis, and acidosis specifically. In the CKD patient on RAAS inhibitors, weigh the cardiorenal benefit of continuing the drug against the hyperkalaemia — the newer binders (patiromer, SZC) may allow continuation, but the threshold to hold or reduce depends on context, and the decision is made with the nephrology and cardiology teams [1][8].
A worked acute management sequence
For a patient with severe hyperkalaemia (K+ above 6.5 or ECG changes), in the first 30 minutes: [1]
- Assess ABC; place on cardiac monitor; secure IV access; repeat the potassium urgently (VBG) to confirm and exclude pseudohyperkalaemia.
- Calcium gluconate 10 mL of 10% IV over 2 to 5 minutes if any ECG change (peaked T, PR prolongation, QRS widening, sine wave) — repeat once at 5 minutes if no ECG improvement. [1]3. Insulin 10 units with 25 g IV dextrose, plus salbutamol 10 to 20 mg nebulised (give both — additive effect).
- If acidotic (bicarbonate below 18): sodium bicarbonate 50 mmol of 8.4% IV over 15 to 30 minutes.
- Begin potassium removal: frusemide 40 to 80 mg IV if the patient makes urine; a binder (SZC 10 g TDS or patiromer 8.4 g) for subacute lowering; call nephrology for dialysis if refractory, oligoanuric, or severe with ECG changes.
- Address the cause: stop K-sparing drugs; review RAAS inhibitors; treat Addison's, digoxin toxicity, tumour lysis, rhabdomyolysis as indicated.
- Recheck K+ at 1, 2, 4 hours; monitor the ECG and glucose continuously (hypoglycaemia surveillance). Have a clear plan for who will dialyse and when. [1]
Hypokalaemia
Definition and severity
Hypokalaemia is a serum potassium below 3.5 mmol/L. A practical severity framework: [1]
| Severity | Serum K+ (mmol/L) | Action |
|---|---|---|
| Mild | 3.0 to 3.5 | Often asymptomatic; identify cause; oral replacement if symptomatic or at risk |
| Moderate | 2.5 to 3.0 | Symptoms likely (weakness, cramps, palpitations); oral replacement, ± IV if symptomatic; investigate the cause |
| Severe | Below 2.5 | Cardiac and neuromuscular risk; admit; IV replacement under cardiac monitoring; aggressive investigation of the cause [2] |
A serum potassium below 3.0 mmol/L typically represents a total body deficit of 200 to 400 mmol. The serum is a poor index of the deficit because of transcellular shifts: a patient with hypokalaemic periodic paralysis may have a serum of 1.5 with a near-normal total body potassium. [1]
Causes — the high-yield classification
Hypokalaemia arises from one of three mechanisms: inadequate intake, transcellular shift into cells, or excessive loss (renal or gastrointestinal). In the ward patient, renal and GI losses dominate; in the outpatient, surreptitious diuretic use, vomiting, and the inherited tubulopathies come into play. [1]
Decreased intake is rarely the sole cause in a patient with normal kidneys — the kidney can conserve potassium to less than 20 mmol per day. It becomes relevant in the anorexic patient, the alcoholic, the parenteral nutrition patient without potassium supplementation, and the "tea and toast" or poor-intake elderly patient — and it is the cause of the hypokalaemia of re-feeding syndrome (insulin surge shifts potassium into cells). [1]
Transcellular shift into cells: [1]
| Category | Specific causes |
|---|---|
| Alkalosis | Metabolic alkalosis (vomiting, diuretics) shifts potassium into cells in exchange for hydrogen; for every 0.1 unit rise in pH, the potassium falls by approximately 0.3 to 0.6 mmol/L |
| Beta-2 agonists | Salbutamol, terbutaline, clenbuterol; also tocolytics; the mechanism of "salbutamol-induced hypokalaemia" in severe asthma |
| Hypokalaemic periodic paralysis | Familial (calcium or sodium channel mutation) or thyrotoxic periodic paralysis (more common in Asian males with Graves' disease) — sudden profound hypokalaemia from a transcellular shift; treat cautiously because potassium rebounds |
| Vitamin B12 therapy | Rapid haematopoietic response with cellular uptake of potassium |
| Hypothermia | Re-warming shifts potassium back out |
Excessive loss — gastrointestinal: [1]
- Vomiting — the gastric fluid potassium is only 5 to 10 mmol/L, so vomiting causes hypokalaemia mainly through volume depletion, hypochloraemia, metabolic alkalosis, and secondary hyperaldosteronism that drives renal potassium loss (the kidney, not the gut, is the major route of potassium loss in vomiting). The hypokalaemia of vomiting is a renal phenomenon mediated by aldosterone.
- Diarrhoea — colonic fluid is rich in potassium (30 to 50 mmol/L), so diarrhoea causes true gastrointestinal potassium loss; the metabolic acidosis (rather than alkalosis) and the low bicarbonate distinguish it from vomiting.
- Laxative abuse, villous adenoma, VIPoma (WDHA syndrome), ileostomy, prolonged nasogastric suction, fistulae — all are GI potassium loss states. [1]
Excessive loss — renal (the largest and most diagnostically rich category): [1]
| Category | Specific causes | Distinguishing feature |
|---|---|---|
| Diuretics | Loop (frusemide, bumetanide), thiazide (hydrochlorothiazide, indapamide, chlorthalidone); the commonest drug cause | Increased distal sodium delivery and flow; metabolic alkalosis; high urine potassium |
| Mineralocorticoid excess | Primary aldosteronism (Conn's) — adenoma or hyperplasia; Cushing's syndrome (cortisol acts on the mineralocorticoid receptor when present in excess); secondary hyperaldosteronism (renal artery stenosis, malignant hypertension, renin-secreting tumour); liquorice (inhibits 11-beta-hydroxysteroid dehydrogenase, allowing cortisol to act as a mineralocorticoid); exogenous mineralocorticoid | Hypertension (Conn's and Cushing's), metabolic alkalosis, low renin (primary) or high renin (secondary) |
| Bartter syndrome | Inherited defect of the thick ascending limb (Na-K-2Cl cotransporter, the loop-of-Henle target of frusemide) — "pharmacological frusemide" | Hypokalaemia, metabolic alkalosis, normotensive, high renin and aldosterone, hypercalciuria; presents in childhood |
| Gitelman syndrome | Inherited defect of the distal convoluted tubule (Na-Cl cotransporter, the thiazide target) — "pharmacological thiazide" | Hypokalaemia, metabolic alkalosis, normotensive, high renin and aldosterone, hypomagnesaemia and hypocalciuria; presents later (adolescence or adulthood); the more benign of the two [11] |
| Renal tubular acidosis | Proximal (type 2) and distal (type 1) RTA both cause renal potassium wasting with metabolic acidosis; type 2 with bicarbonaturia; type 1 with impaired acid secretion and high urine pH | Metabolic acidosis (distinguishes from Bartter/Gitelman) |
| Magnesium deficiency | The key to refractory hypokalaemia — hypomagnesaemia causes renal potassium wasting by disinhibiting ROMK; the hypokalaemia will not correct until the magnesium is repleted | Hypomagnesaemia; hypokalaemia resistant to potassium replacement |
| DKA recovery | After insulin and fluid, the total-body potassium deficit is revealed; treatment unmasks hypokalaemia | Diabetic, insulin-treated |
| Amphotericin B, cisplatin, aminoglycosides, ifosfamide | Drug-induced tubular injury with renal potassium (and magnesium) wasting | Drug exposure; coexisting hypomagnesaemia |
| Cetuximab, panitumumab (anti-EGFR) | Block magnesium and potassium reabsorption | Colorectal cancer therapy; refractory hypokalaemia and hypomagnesaemia |
The clinical presentation
The clinical features of hypokalaemia reflect the hyperpolarisation of excitable tissues: [1]
- Cardiac: palpitations, ectopic beats, atrial arrhythmias (atrial fibrillation, atrial tachycardia), ventricular arrhythmias, torsades de pointes (hypokalaemia is a leading precipitant, especially with concurrent QT-prolonging drugs), and cardiac arrest. The risk is amplified by digoxin (hypokalaemia potentiates digoxin toxicity and reduces the threshold for digoxin-related arrhythmia).
- Neuromuscular: skeletal muscle weakness (the classic feature, often proximal and ascending), cramps, myalgia, fasciculations, paralysis (in severe cases — periodic paralysis or profound hypokalaemia), impaired concentrating ability (nephrogenic DI from chronic hypokalaemia), rhabdomyolysis (severe hypokalaemia impairs muscle blood flow and predisposes to rhabdomyolysis, which then worsens the picture if hyperkalaemia ensues), and ileus (smooth muscle involvement — hypokalaemia contributes to post-operative ileus).
- Renal: chronic hypokalaemia causes a vacuolar tubulopathy, nephrogenic diabetes insipidus (impaired concentrating ability with polyuria and polydipsia), and is associated with cyst formation (medullary sponge kidney, and rarely a hypokalaemic nephropathy progressing to CKD). [1]
ECG changes
The ECG hallmark of hypokalaemia is repolarisation abnormality: [1]
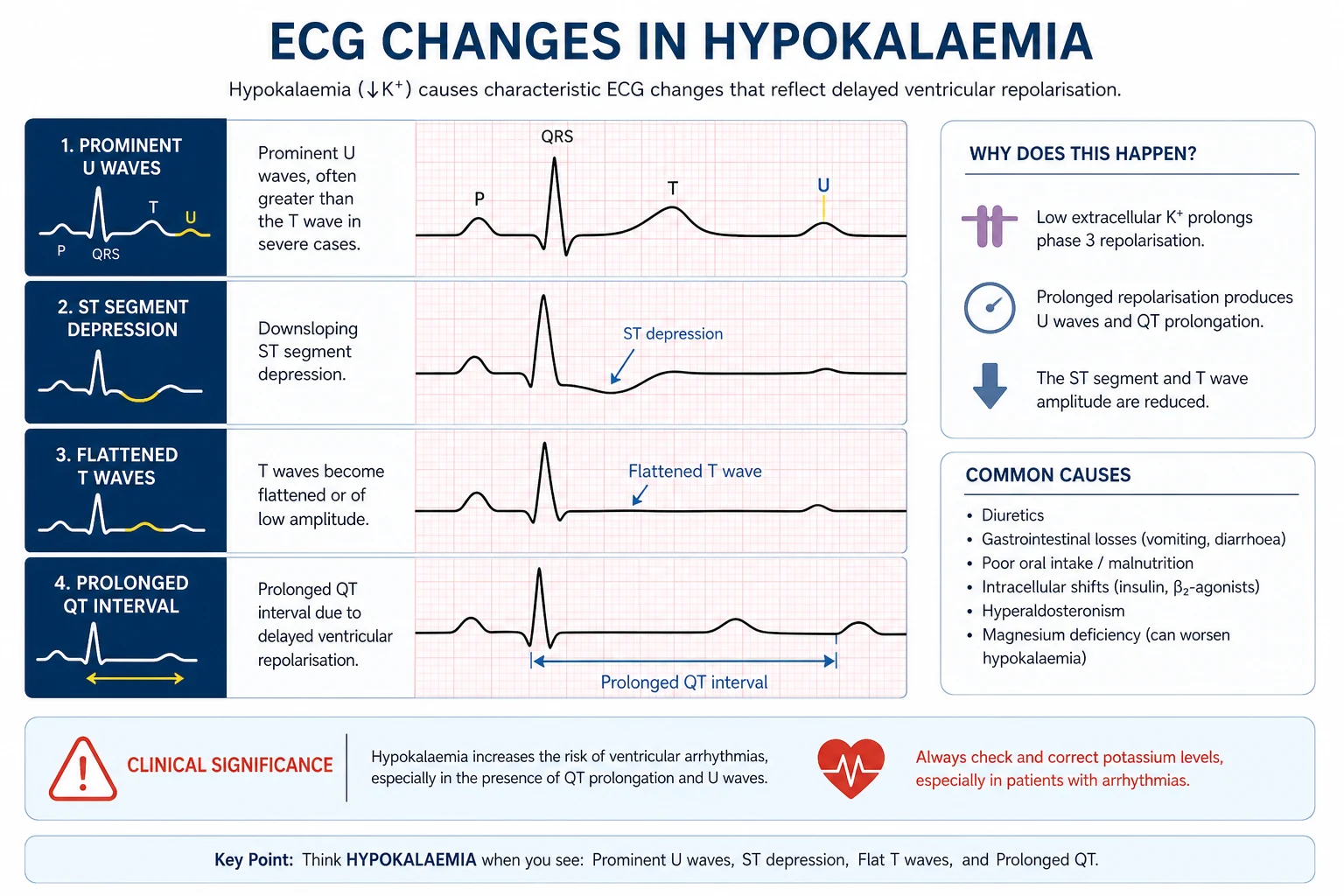
| Severity | ECG change |
|---|---|
| Mild (K+ 3.0 to 3.5) | T wave flattening or inversion; minor ST depression |
| Moderate (K+ 2.5 to 3.0) | Prominent U waves — the hallmark; best seen in V2 to V4; the U wave may merge with the T wave giving an apparent long QT (it is a long QU) |
| Severe (K+ below 2.5) | QT (QU) prolongation, premature atrial and ventricular complexes, torsades de pointes, ventricular tachycardia, ventricular fibrillation |
The U wave is the diagnostic feature — a small positive deflection after the T wave, best seen in the mid-precordial leads. A U wave taller than the preceding T wave is highly suggestive of significant hypokalaemia. The apparent QT prolongation is often a QU fusion; the true corrected QT is sometimes normal, but the practical point is that hypokalaemia prolongs ventricular repolarisation and predisposes to torsades, especially with concurrent QT-prolonging drugs (macrolides, fluoroquinolones, antipsychotics, methadone, ondansetron) [2].
The DWE high-yield associations
- A hypertensive patient with hypokalaemia: think Conn's syndrome (primary aldosteronism) — the most common cause of secondary hypertension. Screen with a morning aldosterone-to-renin ratio (ARR) while correcting for confounders (sit quietly, no diuretics or beta-blockers; spironolactone and ACE inhibitors interfere). A high aldosterone with a suppressed renin is the diagnostic pattern.
- A normotensive patient with hypokalaemia, metabolic alkalosis, and a high urine potassium: think Bartter or Gitelman syndrome. Distinguish by calcium: Bartter has hypercalciuria, Gitelman has hypocalciuria and hypomagnesaemia [11].
- A hypokalaemic patient resistant to potassium replacement: check the magnesium and replete it first. Hypomagnesaemia disinhibits ROMK and causes ongoing renal potassium wasting; the potassium will not rise until the magnesium is normal.
- An Asian male presenting with acute proximal weakness and a low potassium after a carbohydrate load: think thyrotoxic periodic paralysis. Treat cautiously with small doses of potassium and a non-selective beta-blocker (propranolol) — the potassium rebounds and hyperkalaemia is a recognised complication of over-treatment.
- A patient with severe asthma on salbutamol and steroids: salbutamol-induced potassium shift plus steroid-related mineralocorticoid effect. Monitor the potassium.
- A patient on a thiazide or loop diuretic: the single commonest cause of iatrogenic hypokalaemia. Replace potassium; consider a potassium-sparing diuretic (amiloride) if the diuretic must continue; always co-prescribe a potassium-sparing agent when combining diuretics or in heart failure.
Management of hypokalaemia
Replace potassium; correct the magnesium; treat the cause. The route, rate, and dose depend on severity, symptoms, and the ability to take oral therapy. [1]
Oral replacement is preferred whenever possible — it is safer (no risk of fatal hyperkalaemia from a rapid infusion) and effective. Potassium chloride is the standard preparation. Slow-release potassium chloride tablets (e.g., Chlorvescent, Span-K, K-Continus) — 8 to 24 mmol per dose, up to 40 to 80 mmol per day in divided doses with food. Oral potassium is irritant to the gastric mucosa and to the small bowel — give with food, warn about nausea, and (for slow-release preparations) acknowledge the small risk of small-bowel ulceration and strictures. Liquid potassium is faster-acting but unpalatable. In the asymptomatic patient with mild to moderate hypokalaemia, oral replacement over days is appropriate. [1]
Intravenous replacement is reserved for severe hypokalaemia (K+ below 2.5 to 3.0), symptomatic hypokalaemia (arrhythmia, marked weakness), or the patient who cannot tolerate oral therapy. [1]
| Parameter | Recommendation |
|---|---|
| Concentration | Maximum 40 mmol per litre of compatible fluid (usually normal saline; avoid dextrose-containing fluids because the insulin response to dextrose can lower the potassium further) |
| Monitoring | Continuous ECG monitoring for rates above 10 mmol/h; check serum K+ every 2 to 4 hours |
| Vein care | Potassium is intensely irritant — a large peripheral vein or central line; assess the cannula site for extravasation |
- Never give potassium as an IV bolus — it is fatal. Even in cardiac arrest from hypokalaemia, give potassium via infusion, not a push.
- Correct the magnesium first. In refractory hypokalaemia, give magnesium sulphate 2 to 4 g IV (8 to 16 mmol) over 1 to 2 hours and continue oral or IV magnesium supplementation; the potassium will only rise once the magnesium is replete.
- Recheck the potassium frequently during IV replacement — every 2 to 4 hours depending on severity and rate.
- In DKA and HHS, the patient is total-body potassium-depleted despite a normal or high serum potassium (acidosis and insulin deficiency shift potassium out of cells). Begin potassium replacement when the serum potassium falls below 5.2 to 5.3 mmol/L; aim to maintain 4.0 to 5.0 mmol/L during insulin therapy. This is the rationale for the "5.0 mmol/L trigger" in DKA protocols. [1]
The workup of unexplained potassium disorder
For the patient with recurrent or unexplained hypo- or hyperkalaemia, a structured workup identifies the mechanism and the cause. [1]
For hypokalaemia — the "where is the potassium going?" workup
| Test | What it tells you |
|---|---|
| 24-hour urine potassium (or spot urine K/creatinine) | If urine K is less than 20 to 25 mmol/day, the kidney is conserving potassium — the loss is extrarenal (GI) or a transcellular shift. If urine K is greater than 25 to 30 mmol/day in a hypokalaemic patient, the kidney is inappropriately wasting potassium — renal loss [2] |
| Blood pressure | Hypertension with hypokalaemia points to mineralocorticoid excess (Conn's, Cushing's, secondary hyperaldosteronism, liquorice, exogenous mineralocorticoid); normotensive hypokalaemia with alkalosis points to Bartter, Gitelman, diuretic abuse, or vomiting |
| Acid-base status (venous blood gas and serum bicarbonate) | Metabolic alkalosis (vomiting, diuretics, Bartter, Gitelman, mineralocorticoid excess) versus metabolic acidosis (diarrhoea, renal tubular acidosis, DKA) |
| Serum magnesium | The cause of refractory hypokalaemia — replete first |
| Renin and aldosterone (ideally a morning ARR) | Distinguishes primary aldosteronism (low renin, high aldosterone) from secondary hyperaldosteronism (high renin, high aldosterone) from the inherited tubulopathies (high renin, high aldosterone, normotensive) [11] |
| Urine chloride | Distinguishes vomiting (low urine chloride, less than 20) from Bartter/Gitelman and diuretic abuse (high urine chloride, greater than 40) |
| Urine calcium | Distinguishes Bartter (hypercalciuria) from Gitelman (hypocalciuria) |
| Toxicology screen | Diuretic screen for surreptitious use; consider laxative screen |
| TSH | Thyrotoxic periodic paralysis — check thyroid function in any young male presenting with acute hypokalaemic weakness |
For hyperkalaemia — the "why is it not being excreted?" workup
| Test | What it tells you |
|---|---|
| Repeat the potassium (VBG) immediately | Exclude pseudohyperkalaemia before anything else |
| Medication review | The commonest contributor — ACE inhibitors, ARBs, spironolactone, eplerenone, amiloride, triamterene, trimethoprim (a potassium-sparing effect), heparin, calcineurin inhibitors, NSAIDs, beta-blockers, digoxin |
| Renal function (urea, creatinine, eGFR) | CKD and AKI are the structural basis of impaired potassium excretion |
| Acid-base and bicarbonate | Metabolic acidosis (especially hyperchloraemic) shifts potassium out of cells and is a feature of CKD and type 4 RTA |
| Glucose and blood gas | DKA and HHS; insulin deficiency drives hyperkalaemia |
| Creatine kinase | Rhabdomyolysis — the CK is in the thousands; check if there is any suggestion of muscle injury |
| Phosphate, calcium, urate, lactate dehydrogenase | Tumour lysis syndrome — hyperphosphataemia, hypocalcaemia, high LDH, with AKI |
| Morning cortisol (± short Synacthen test) | Addison's disease — hyperkalaemia with hyponatraemia, hypotension, hypoglycaemia, hyperpigmentation; the cortisol is low and fails to respond to Synacthen |
| Digoxin level | If the picture suggests digoxin toxicity (visual disturbance, arrhythmia, gastrointestinal symptoms, hyperkalaemia in a patient on digoxin) |
| ECG | Determines urgency — see the hyperkalaemia section |
DWE high-yield discriminators and exam traps
- Treat the ECG and the trend, not the single value. A patient with K+ of 6.8 and a normal ECG is still an emergency, but a patient with K+ of 6.2 and a sine wave is in extremis.
- Pseudohyperkalaemia is the first thing to exclude. Haemolysis, fist clenching, severe leucocytosis or thrombocytosis, and delay in processing. A VBG run immediately resolves it.
- Hypoglycaemia after insulin-dextrose is the commonest serious complication of hyperkalaemia treatment — monitor glucose for 4 to 6 hours. Lower-dose insulin (5 units) is as effective with less hypoglycaemia [5].
- Calcium does not lower potassium. It stabilises the membrane. The potassium is unchanged — you must still shift and remove.
- Bicarbonate is not effective as monotherapy in non-acidotic hyperkalaemia. Reserve it for the acidotic patient.
- Always correct magnesium first in refractory hypokalaemia. This is one of the most frequently tested points.
- The potassium-sparing diuretic plus ACE inhibitor plus CKD combination is the classic DWE drug-induced hyperkalaemia stem.
- Conn's syndrome is the classic DWE cause of hypertension with hypokalaemia — screen with an ARR.
- Gitelman syndrome has hypocalciuria and hypomagnesaemia; Bartter has hypercalciuria. This calcium distinction is a high-yield discriminator [11].
- The maximum IV potassium rate is 10 mmol/h peripheral, 20 mmol/h central. Never bolus. Continuous ECG monitoring at the higher rates.
- Thyrotoxic periodic paralysis is treated with caution — small doses of potassium and propranolol; the potassium rebounds and over-correction causes hyperkalaemia.
- Patiromer and SZC are the modern binders; sodium polystyrene sulfonate (resonium) is older and associated with intestinal necrosis, especially via the rectal route.
- Recheck the potassium after treatment — rebound hyperkalaemia (in treated hypokalaemia, or after potassium-lowering therapy in patients with ongoing release) and rebound hypokalaemia (after insulin-dextrose wears off at 4 to 6 hours) are both recognised.
Multimorbidity and the complex medical patient
- CKD and heart failure. The RAAS inhibitor (ACE inhibitor, ARB, or ARNI) and the mineralocorticoid receptor antagonist (spironolactone, eplerenone, finerenone) are the cornerstone of heart failure and CKD therapy — and each raises the potassium. The modern approach is to titrate to the highest tolerated dose, accept mild hyperkalaemia (K+ 5.5 to 6.0), and add a binder (patiromer, SZC) to enable continuation, rather than to abandon these life-saving drugs at the first rise [1][8].
- Diabetes. Diabetic nephropathy is the leading cause of type 4 RTA (hyporeninaemic hypoaldosteronism) and contributes to hyperkalaemia; the diabetic is also prone to hypokalaemia during DKA treatment and to the cardiovascular consequences of both disturbances.
- The elderly patient with polypharmacy. The combination of ACE inhibitor, K-sparing diuretic, NSAID, and CKD is a near-guarantee of hyperkalaemia. Review the chart, stop the offending agent, treat per the algorithm.
- Perioperative. Suxamethonium-induced hyperkalaemia in the denervated patient (burns, prolonged immobility, neuromuscular disease, crush) is an anaesthetic emergency. Hypokalaemia perioperatively increases arrhythmia and ileus risk — replace before elective surgery if K+ is below 3.5.
- End-of-life and palliative. Hyperkalaemia in advanced CKD may be a terminal event; the decision to dialyse or not is made in the context of goals of care.
Communication and shared decision-making
- In the acute setting, the emergency interventions (calcium, insulin-dextrose, dialysis) are explained briefly and consented under the doctrine of necessity. The patient and family are kept informed as the picture evolves.
- In the CKD patient with recurrent hyperkalaemia, the decision about whether to continue, dose-reduce, or cease a RAAS inhibitor is a shared one — balancing the cardiorenal benefit against the hyperkalaemia risk and the option of a binder. Explain the trade-off and the monitoring plan.
- For the patient with Gitelman or Bartter syndrome, the explanation is of a lifelong inherited tubular condition managed with potassium and magnesium supplementation and (in Bartter) often a prostaglandin synthesis inhibitor; the prognosis in Gitelman is good with treatment.
- For Addison's disease presenting as hyperkalaemia, the diagnosis is explained as adrenal insufficiency requiring lifelong glucocorticoid and mineralocorticoid replacement, with a sick-day escalation plan and a medical alert.
- For the patient with periodic paralysis, the explanation includes avoiding triggers (carbohydrate load, exertion, cold), the role of prophylaxis, and the importance of cautious treatment of acute attacks. [1]
Long-term outcomes, follow-up, and surveillance
- Hyperkalaemia outcome depends on cause and severity — acute mortality is from arrhythmia; long-term prognosis depends on the underlying CKD, heart failure, or Addison's. Patients with recurrent severe hyperkalaemia on RAAS inhibitors should be on a binder if continuation is essential.
- Hypokalaemia outcome depends on cause and correction — most diuretic-related cases resolve with replacement and adjustment; the inherited tubulopathies (Bartter, Gitelman) are chronic; chronic untreated hypokalaemia causes nephropathy and cyst formation; severe acute hypokalaemia can cause rhabdomyolysis, ileus, and arrhythmia.
- Surveillance. In CKD and heart failure on RAAS inhibitors, check the potassium and renal function 1 to 2 weeks after initiation or dose change, then regularly. In the patient on long-term potassium supplementation, check the potassium and magnesium periodically. In Gitelman and Bartter, monitor potassium, magnesium, calcium excretion, and renal function lifelong. [1]
Cross-links
This page integrates with acute kidney injury, chronic kidney disease, and diabetic kidney disease (the structural renal conditions that predispose to hyperkalaemia); electrolyte-sodium-disorders (the parallel electrolyte disturbance that frequently coexists — Addison's presents with both hypo-Na and hyper-K); acid-base-disorders (acidosis drives hyperkalaemia and alkalosis drives hypokalaemia); and adrenal-insufficiency (the Addison's crisis is the high-yield endocrine emergency with hyperkalaemia). [1]
Key references
Kovesdy, Management of hyperkalaemia in chronic kidney disease (Nat Rev Nephrol 2014) [1]; Gennari, Hypokalemia (NEJM 1998) [2]; Weir et al, Patiromer OPAL-HK (NEJM 2015) [3]; Kosiborod et al, SZC HARMONIZE (JAMA 2014) [4]; Moussavi et al, Insulin dosing meta-analysis (Pharmacotherapy 2021) [5]; Lemoine et al, ED management of acute hyperkalaemia (J Emerg Med 2021) [6]; Long et al, Controversies in hyperkalaemia management (J Emerg Med 2018) [7]; Rafique et al, Expert panel recommendations hyperkalaemia (JMCP 2017) [8]; Maxwell et al, Management of hyperkalaemia (J R Coll Physicians Edinb 2013) [9]; Mahoney et al, Cochrane emergency interventions for hyperkalaemia (2005) [10]; Fulchiero and Seo-Mayer, Bartter and Gitelman syndromes (Pediatr Clin North Am 2019) [11]; Viera and Wouk, Potassium disorders (Am Fam Physician 2015) [12]; Lindner et al, KDIGO consensus on acute hyperkalaemia (Eur J Emerg Med 2020) [13]; KDIGO; UK Renal Association; CARI Guidelines; Kidney Health Australia; NICE CKS.
References
- [1]Kovesdy CP Management of hyperkalaemia in chronic kidney disease Nat Rev Nephrol, 2014.PMID 25223988
- [2]Gennari FJ Hypokalemia N Engl J Med, 1998.PMID 9700180
- [3]Weir MR, Bakris GL, Bushinsky DA, et al. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors N Engl J Med, 2015.PMID 25415805
- [4]Kosiborod M, Rasmussen HS, Lavin P, et al. Effect of sodium zirconium cyclosilicate on potassium lowering for 28 days among outpatients with hyperkalemia: the HARMONIZE randomized clinical trial JAMA, 2014.PMID 25402495
- [5]Moussavi K, Niksuikan A,dirge E, et al. Reduced alternative insulin dosing in hyperkalemia: A meta-analysis of effects on hypoglycemia and potassium reduction Pharmacotherapy, 2021.PMID 33993515
- [6]Lemoine L, Thompson D, Grosse J, et al. An Evidence-Based Narrative Review of the Emergency Department Management of Acute Hyperkalemia J Emerg Med, 2021.PMID 33423833
- [7]Long B, Warix JR, Koyfman A Controversies in Management of Hyperkalemia J Emerg Med, 2018.PMID 29731287
- [8]Rafique Z, Liu F, Clark AL, et al. Expert Panel Recommendations for the Identification and Management of Hyperkalemia and Role of Patiromer in Patients with Chronic Kidney Disease and Heart Failure J Manag Care Spec Pharm, 2017.PMID 28485203
- [9]Maxwell AP, Linden K, O'Donnell S, Fogarty DG Management of hyperkalaemia J R Coll Physicians Edinb, 2013.PMID 24087806
- [10]Mahoney BA, Smith WAD, Lo DS, Tsoi K, Tonelli M, Clase CM Emergency interventions for hyperkalaemia Cochrane Database Syst Rev, 2005.PMID 15846652
- [11]Fulchiero R, Seo-Mayer P Bartter Syndrome and Gitelman Syndrome Pediatr Clin North Am, 2019.PMID 30454738
- [12]Viera AJ, Wouk N Potassium Disorders: Hypokalemia and Hyperkalemia Am Fam Physician, 2015.PMID 26371733
- [13]Lindner G, Burdmann EA, Clase CM, et al. Acute hyperkalemia in the emergency department: a summary from a Kidney Disease: Improving Global Outcomes conference Eur J Emerg Med, 2020.PMID 32852924