Phys · renal
Sodium Disorders — Hyponatraemia and Hypernatraemia
Also known as hyponatraemia · hyponatremia · hypernatraemia · hypernatremia · SIADH · syndrome of inappropriate antidiuresis · osmotic demyelination syndrome · central pontine myelinolysis · diabetes insipidus · arginine vasopressin deficiency · arginine vasopressin resistance · cerebral salt wasting · psychogenic polydipsia · tea and toast hyponatraemia · beer potomania · reset osmostat
Consultant-physician-depth guide to sodium and water homeostasis. Covers hyponatraemia (the commonest inpatient electrolyte disorder): symptom-stratified urgency, volume-status classification, serum and urine osmolality with urine sodium interpretation, severe symptomatic management with 3% hypertonic saline boluses, the 8-10 mmol/L in 24 hours correction ceiling and osmotic demyelination syndrome, SIADH, and the euvolaemic/hypervolaemic differentials. Covers hypernatraemia as a water-deficit state: central versus nephrogenic diabetes insipidus, the water deprivation test and copeptin, free water deficit calculation, and slow oral or D5W correction over 48-72 hours. Structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Sodium Disorders — Hyponatraemia and Hypernatraemia
The answer first
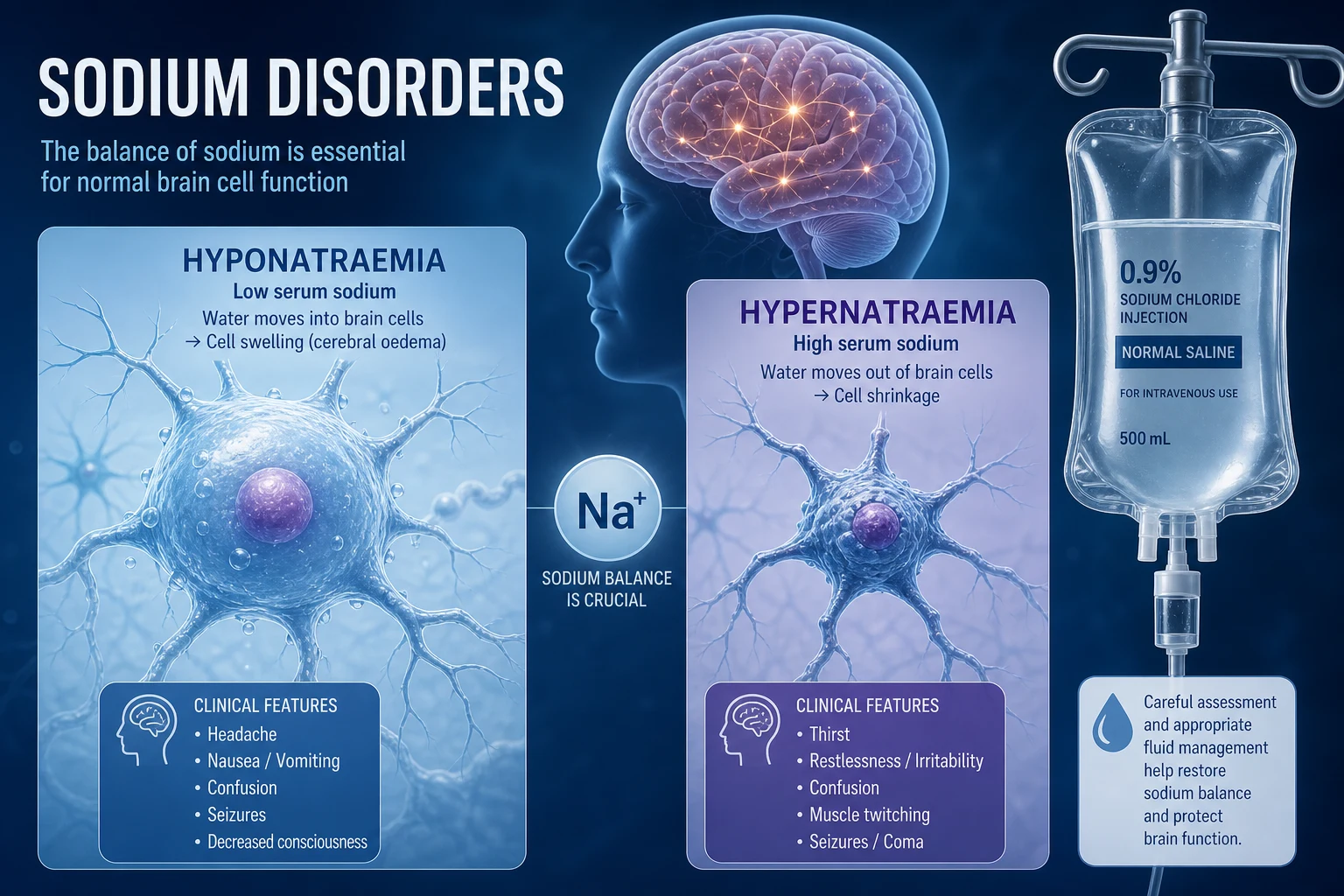
Sodium disorders are water disorders. Serum sodium reflects the ratio of total body exchangeable sodium to total body water — it tells you about water balance, not sodium balance. A normal sodium means water and sodium are matched; an abnormal sodium means they are mismatched. The clinician's job is to work out which side of the equation is wrong. [1]
Hyponatraemia (Na less than 135 mmol/L) is the commonest electrolyte disorder in hospitalised patients, present in 15 to 30 percent of inpatients [4]. It is almost always a problem of relative water excess — the kidney cannot excrete free water because arginine vasopressin (AVP, also called antidiuretic hormone or ADH) is inappropriately present. Even hypovolaemic hyponatraemia is driven by non-osmotic AVP release: the body defends circulating volume at the cost of tonicity.
Hypernatraemia (Na greater than 145 mmol/L) is almost always a problem of relative water deficit, and it almost always implies impaired access to water — the very old, the intubated, the neurologically impaired, the post-operative. A patient with an intact thirst mechanism and free access to water does not develop sustained hypernatraemia. Hypernatraemia is iatrogenic more often than it is admitted [5].
The two clinical questions that dominate management: [1]
- How fast did it develop, and is the brain threatened? Acute hyponatraemia (under 48 hours) risks cerebral oedema and herniation — correct promptly. Chronic hyponatraemia (over 48 hours) risks osmotic demyelination if corrected too fast — correct slowly.
- What is the volume status, and what is driving the abnormal vasopressin? That single bedside assessment — hypovolaemic, euvolaemic, or hypervolaemic — divides the differential into three families and determines treatment. [1]
The management mandate, in priority order: [1]
- Severely symptomatic hyponatraemia (seizure, coma) is an emergency — give 3% hypertonic saline, 100 mL bolus over 10 minutes, repeat up to three times, aiming for a 4 to 5 mmol/L rise in the first 4 hours. Do not wait for bloods.
- Respect the correction ceiling — no more than 8 to 10 mmol/L in 24 hours, and 18 mmol/L in 48 hours, to avoid osmotic demyelination syndrome (ODS) [1][2].
- Treat the cause — stop the thiazide, the SSRI, the carbamazepine; give glucocorticoids for adrenal insufficiency; treat heart failure; remove the water load.
The single most important exam principle: the risk of treating hyponatraemia is ODS, and the risk of not treating it is cerebral herniation. Both kill. The skill is judging which risk dominates for the patient in front of you, based on symptoms and chronicity. [1]
Sodium and water physiology — the short version
The hypothalamic osmostat holds serum osmolality near 285 mOsm/kg by controlling thirst and AVP release. A 1 to 2 percent rise in osmolality triggers thirst and AVP; a similar fall suppresses AVP. AVP binds V2 receptors in the collecting duct, inserting aquaporin-2 channels and reabsorbing free water. With AVP fully suppressed, the kidney can dilute urine to 50 mOsm/kg and excrete up to 20 litres of free water per day. [1]
This means hyponatraemia is, with rare exceptions, a state where AVP is non-suppressible — the kidney cannot excrete the ingested water load. The exceptions (true AVP-independent hyponatraemia) are primary polydipsia, where the water intake overwhelms even maximal dilution, and advanced renal failure, where the diluting capacity itself is lost. [1]
Hypernatraemia, conversely, requires either absent AVP (central diabetes insipidus) or renal resistance to AVP (nephrogenic diabetes insipidus), OR impaired access to water. The kidney is concentrating urine maximally but cannot defend tonicity alone — the thirst mechanism and access to water are the final defence. [1]
Hyponatraemia — pathophysiology of brain adaptation
The brain sits in a fixed skull. As extracellular tonicity falls, water moves into brain cells, raising intracranial pressure. Within hours, the brain sheds osmoles — first electrolytes (rapid adaptation), then organic osmolytes such as glutamate, taurine, and myo-inositol (slow adaptation, over 24 to 48 hours). This adaptive loss of intracellular osmoles is what makes chronic hyponatraemia tolerable at very low sodium levels, and what makes rapid correction catastrophic. [1]

Two competing risks flow directly from this adaptation: [1]
- Cerebral oedema occurs when hyponatraemia develops faster than the brain can adapt — typically within 48 hours. Post-operative hyponatraemia, MDMA-induced hyponatraemia, primary polydipsia, and colonoscopy prep hyponatraemia are the classic acute causes. The brain is swollen; herniation can occur.
- Osmotic demyelination syndrome (ODS) occurs when a chronically adapted brain is corrected too quickly. The brain has shed its osmoles; a rapid rise in extracellular tonicity drags water out of brain cells faster than they can re-accumulate osmoles. Astrocytes die, then oligodendrocytes, producing demyelination — classically in the pons (central pontine myelinolysis) but also extra-pontine [3].
DWE exam trap: The symptoms of hyponatraemia are non-specific (headache, nausea, malaise, confusion) and overlap with the symptoms of the underlying cause. Do not attribute altered cognition to hyponatraemia without considering an alternative. Severe symptoms — seizure, coma, vomiting, respiratory arrest — are the trigger for urgent hypertonic saline, regardless of the absolute sodium value. [1]
Step 1 — assess symptoms first (severity stratifies everything)
The first question is not "what is the sodium" but "is this patient's brain under threat right now." Symptom severity, not the absolute number, determines the urgency and the rate of correction [1][7].
| Severity | Symptoms | Action |
|---|---|---|
| Severe (cerebral oedema) | Seizure, coma, Cheyne-Stokes respiration, vomiting, decerebrate posturing | 3% saline 100 mL bolus over 10 minutes, repeat to a maximum of three boluses; aim for 4 to 5 mmol/L rise in 4 hours |
| Moderately severe | Confusion, disorientation, lethargy, nausea, headache, falls | Urgent assessment; correct slowly; admit; investigate the cause |
| Mild / asymptomatic | Detected on routine bloods; patient well | Investigate the cause; correct slowly; rarely needs urgent intervention |
The presence of severe symptoms mandates hypertonic saline immediately — do not wait for urine osmolality or a computed volume status. The 100 mL bolus approach has replaced the older weight-based continuous infusion in most guidelines because it is safer (lower risk of overcorrection) and achieves a predictable 1 to 2 mmol/L rise per bolus [2].
Chronicity matters and is often inferred rather than known. If the sodium was normal within 48 hours (post-operative, recent admission, MDMA use), treat as acute — the brain has not adapted, cerebral oedema is the threat, and faster correction is tolerated and required. If chronic or unknown, assume chronic — ODS is the threat, and the correction ceiling is strict. [1]
Step 2 — volume status (the single most useful bedside assessment)
Once the brain is safe, classify the patient by volume status. This is done at the bedside, not in the laboratory, and it divides the differential into three families. [1]

| Volume status | Bedside signs | Key physiology |
|---|---|---|
| Hypovolaemic | Dry mucous membranes, reduced skin turgor, postural drop, tachycardia, low JVP | True sodium and water loss; non-osmotic AVP release retains water in excess of sodium |
| Euvolaemic | No signs of overload or depletion; normal JVP, normal perfusion | Water balance disordered with near-normal total body sodium; SIADH is the prototype |
| Hypervolaemic | Raised JVP, peripheral or sacral oedema, ascites, crackles, S3 | Total body sodium and water both increased, but water increased more; oedema-forming states |
The bedside examination is imperfect — particularly in the elderly, where dry mucous membranes are common and skin turgor is unreliable. The biochemical confirmatory tests (urine sodium, urine osmolality, paired serum osmolality) resolve the ambiguity. [1]
Step 3 — serum and urine biochemistry
Serum osmolality — the first laboratory fork
Hyponatraemia is only meaningful as a hypo-osmolar state. Measure serum osmolality first. [1]
| Serum osmolality | Interpretation |
|---|---|
| Low (less than 275 mOsm/kg) — true hypotonic hyponatraemia | The common and clinically important form; proceed to volume status assessment |
| Normal (275 to 295) — pseudohyponatraemia or isotonic hyponatraemia | Pseudohyponatraemia from hyperlipidaemia, hyperproteinaemia (old assays, now rare); or isotonic water shift from hyperglycaemia (correct Na up by 2 mmol/L for every 5.6 mmol/L glucose above 5.6) or mannitol |
| High (greater than 295) — hypertonic hyponatraemia | Hyperglycaemia (the classic DKA/HHS scenario), mannitol, glycine irrigation (TURP syndrome) |
Correcting for hyperglycaemia is a high-yield DWE point: measured sodium falls by approximately 2 mmol/L for every 5.6 mmol/L (100 mg/dL) that glucose rises above normal. Modern sodium-specific ion-selective electrodes have largely eliminated pseudohyponatraemia, but the concept still appears in questions. [1]
Urine osmolality — is AVP present?
| Urine osmolality | Interpretation |
|---|---|
| Less than 100 mOsm/kg (maximally dilute) | AVP is fully suppressed — primary polydipsia, low solute intake (beer potomania, tea and toast), or resetting after correction |
| Greater than 100 mOsm/kg | AVP is present — the kidney is retaining water; this is the vast majority of hyponatraemia |
A urine osmolality below 100 in a hyponatraemic patient is the signature of AVP-independent hyponatraemia and narrows the differential to psychogenic polydipsia, beer potomania, or reset osmostat with dilute intake. [1]
Urine sodium — distinguishes the cause and the volume status
| Urine sodium | In a hypovolaemic patient | In a euvolaemic patient |
|---|---|---|
| Less than 20 mmol/L | Extra-renal loss (vomiting, diarrhoea, burns, third spacing) — kidney avidly retains sodium | Rare in true euvolaemia; suggests low solute intake |
| Greater than 20 (often greater than 40) mmol/L | Renal loss (thiazide, loop diuretic, mineralocorticoid deficiency, cerebral salt wasting, bicarbonaturia) | SIADH, glucocorticoid deficiency, hypothyroidism, reset osmostat |
The combined picture — urine osmolality high (AVP present) plus urine sodium over 20 with normal renal function — points to SIADH, provided hypothyroidism and glucocorticoid deficiency have been excluded. A urine sodium over 40 in a patient who appears hypovolaemic should prompt consideration of adrenal insufficiency (cortisol is needed to suppress AVP) and cerebral salt wasting. [1]
The supporting panel
- Serum glucose — corrects for hypertonic hyponatraemia.
- Serum urate — typically low (less than 0.24 mmol/L) in SIADH; helps distinguish from hypovolaemia where urate is normal or high. [1]- TSH and morning cortisol — exclude hypothyroidism and adrenal insufficiency before diagnosing SIADH. Adrenal insufficiency causes hyponatraemia through cortisol lack and aldosterone lack (hyperkalaemia is the clue).
- Urea and creatinine — exclude renal failure; help calculate the free water deficit in hypernatraemia.
- Lipids and protein — if pseudohyponatraemia is suspected. [1]
Hyponatraemia by category
Hypovolaemic hyponatraemia
The patient has lost both sodium and water, but the sodium loss is relatively greater, and non-osmotic AVP release retains free water in compensation. Clinical features of volume depletion dominate. [1]
Extra-renal losses (urine sodium less than 20): vomiting, diarrhoea, burns, pancreatitis, bowel obstruction, third-space losses. The kidney is sodium-avid because the patient is volume-depleted. [1]
Renal losses (urine sodium greater than 20): thiazide diuretic (the single commonest drug cause — see below), loop diuretic (less common, because loop diuretics impair the concentrating mechanism itself), mineralocorticoid deficiency (Addison's disease — hyperkalaemia, hypoglycaemia, hyperpigmentation), cerebral salt wasting (rare, seen in subarachnoid haemorrhage and traumatic brain injury — see below), bicarbonaturia (proximal renal tubular acidosis, post-hypocapnia), ketonuria. [1]
Management: stop the loss, replace with isotonic saline. The sodium rises as AVP is suppressed by volume repletion. The exam trap is overcorrection — a volume-depleted patient on a thiazide can correct rapidly once the drug is stopped and the kidney resumes free water excretion; monitor every 2 to 4 hours and have DDAVP ready to relower if correction exceeds the ceiling. [1]
DWE high-yield — thiazide-induced hyponatraemia: The thiazide is the classic DWE culprit in an older woman with a low sodium, normal or low potassium, and a slightly high or normal urea. The mechanism is impaired dilution (the distal tubule is the diluting segment) plus potassium depletion (which drives sodium into cells and stimulates thirst). Stop the thiazide permanently — rechallenge is dangerous. Check the cortisol, because Addison's presents identically and is lethal if missed. [1]
Euvolaemic hyponatraemia — SIADH and its mimics
Total body water is increased but total body sodium is near normal; the patient looks euvolaemic. SIADH (syndrome of inappropriate antidiuresis — the modern preferred term is syndrome of inappropriate antidiuresis, SIA) is the prototype [4][7].
Diagnostic criteria for SIADH:
- Hypotonic hyponatraemia (serum osmolality less than 275 mOsm/kg).
- Inappropriately concentrated urine (urine osmolality greater than 100 mOsm/kg).
- Urine sodium greater than 20 to 30 mmol/L with normal intake. [1]4. Clinically euvolaemic.
- Normal thyroid, adrenal, and renal function.
- No diuretic use. [1]
Causes of SIADH — organise by system: [1]
| System | Causes |
|---|---|
| Malignancy (ectopic AVP) | Small cell lung cancer (the classic), head and neck cancer, oesophageal, pancreatic, prostate, lymphoma, thymoma |
| CNS disorders | Meningitis, encephalitis, subarachnoid haemorrhage, traumatic brain injury, stroke, brain tumour, Guillain-Barre syndrome, acute intermittent porphyria |
| Pulmonary | Pneumonia (the commonest inpatient cause), mechanical ventilation, asthma exacerbation, tuberculosis, abscess |
| Drugs | SSRIs (the commonest drug cause in the modern era), carbamazepine, oxcarbazepine, vincristine, cyclophosphamide, MDMA, antipsychotics (especially phenothiazines), MDMA, desmopressin (and other AVP analogues) |
| Other | Pain, nausea, post-operative state, endurance exercise, HIV infection |
Management of SIADH (chronic / moderate): the European guideline prioritises fluid restriction as first-line; the American expert panel is more willing to use vaptans early [1][7].
- Fluid restriction — 500 mL below the previous day's urine and insensible losses, typically 800 to 1000 mL per day. Effective in about one-third; fails when the urine-to-serum electrolyte ratio exceeds 1 (a marker of high urine tonicity).
- High solute intake — salt tablets (2 to 3 g per day) with a loop diuretic to enable excretion of the water load; or oral urea (15 to 30 g per day, unpalatable but effective and cheap).
- Vaptans — tolvaptan is the V2-receptor antagonist used in ANZ. SALT-1 and SALT-2 showed a mean sodium rise of 4 to 5 mmol/L over 30 days [6]. Cautions: expensive, must NOT be used with hypertonic saline (combined overcorrection risk), requires inpatient initiation with sodium monitoring every 6 hours, and is contraindicated in hypovolaemic hyponatraemia and in anuria. Avoid in liver disease (hepatotoxicity at high dose, as seen in the polycystic kidney trials) and do not fluid-restrict on the day of initiation.
- Treat the cause — resect the tumour, treat the pneumonia, stop the drug.
DWE exam trap: Do NOT give normal saline to a patient with SIADH — it worsens the hyponatraemia. The urine osmolality in SIADH is fixed by AVP, so the kidney retains the water and excretes the sodium (urine sodium rises further). The sodium drops. This is the "desalination" phenomenon and is a classic DWE distractor. [1]
Euvolaemic mimics that are NOT SIADH — exclude before labelling: [1]
- Glucocorticoid deficiency (secondary adrenal insufficiency). Cortisol normally exerts tonic inhibition of AVP release; its absence produces a syndrome indistinguishable from SIADH biochemically. A morning cortisol must be checked. Primary adrenal insufficiency (Addison's) adds aldosterone deficiency and presents with hyperkalaemia and hypotension; secondary adrenal insufficiency (pituitary) spares aldosterone and presents as isolated hyponatraemia.
- Hypothyroidism. Low cardiac output and reduced GFR stimulate non-osmotic AVP release; free water excretion is impaired. Check TSH in every hyponatraemic patient.
- Primary polydipsia (psychogenic polydipsia). Seen in schizophrenia; intake of 5 to 10 litres per day overwhelms even maximal dilution. Urine osmolality is low (less than 100), distinguishing it from SIADH.
- Low solute intake (beer potomania, tea and toast). The diluting mechanism requires solute delivery to the distal nephron. With minimal solute intake, even modest water intake causes hyponatraemia. Urine osmolality is low.
- Reset osmostat. A stable, chronic, modest hyponatraemia (typically 125 to 130) where the osmostat has reset downward. The kidney can dilute and concentrate appropriately around the new set point. No treatment is needed; the diagnosis is one of stability over months to years with normal response to water loading. [1]
Hypervolaemic hyponatraemia
Total body sodium and water are both increased, but water has increased more, producing dilution. The patient is oedematous. Effective arterial blood volume is low (despite total volume overload), so AVP is non-osmotically stimulated, and free water is retained. [1]
The three causes are heart failure, cirrhosis, and advanced chronic kidney disease. The clue is the oedema and the underlying disease. [1]
Management is of the underlying state — fluid restriction, loop diuretics, and disease-modifying therapy (RAAS blockade, SGLT2 inhibitors, beta-blockers for heart failure; see the heart failure topic). Vaptans have a limited role (tolvaptan is approved for heart failure hyponatraemia but does not improve survival and is not first-line). Avoid hypertonic saline except for severe symptoms. [1]
Cerebral salt wasting
A rare cause of hypovolaemic hyponatraemia seen after subarachnoid haemorrhage, traumatic brain injury, and neurosurgery. The mechanism is release of natriuretic peptides causing renal sodium loss and volume depletion, with secondary AVP release. It mimics SIADH biochemically (high urine sodium, concentrated urine) but the patient is volume-depleted — postural drop, low JVP, high urea. Management is salt and volume replacement, sometimes with fludrocortisone. Confusing it with SIADH and fluid-restricting is dangerous. [1]
Severe symptomatic hyponatraemia — the emergency pathway
A patient with a seizure, coma, or signs of cerebral herniation and a low sodium needs 3% hypertonic saline now. The European guideline and the American expert panel converge on a bolus-based approach that is safer than the older continuous infusion [1][2][8].
The bolus regimen: [1]
- 100 mL of 3% saline (514 mmol/L sodium) over 10 minutes, through a central or large peripheral line.
- Recheck sodium at the end of the bolus (or after the first hour).
- Repeat the bolus up to a maximum of three boluses, until symptoms resolve or the sodium has risen by 4 to 5 mmol/L.
- Target: a 4 to 5 mmol/L rise in the first 4 hours is sufficient to relieve cerebral oedema. Do not exceed 8 to 10 mmol/L in 24 hours. [1]
A 100 mL bolus of 3% saline raises the serum sodium by approximately 1 to 2 mmol/L acutely. Three boluses typically achieve the 4 to 5 mmol/L target. The bolus approach is preferred because it is titratable, avoids the steady overshoot of continuous infusion, and can be given in any setting [2].
What to do once symptoms resolve: stop the hypertonic saline, transition to a holding strategy (fluid restriction, or normal saline if volume-depleted), investigate the cause, and monitor the sodium every 2 to 4 hours for the first 24 hours. The risk of overcorrection is highest in the first day, particularly in thiazide-induced and post-operative hyponatraemia. [1]
DCE long-case viva line: "In severe symptomatic hyponatraemia, I give 100 mL of 3% saline over 10 minutes and repeat up to three times to achieve a 4 to 5 mmol/L rise in the first four hours. I do not use a continuous infusion as the first-line, because the bolus approach is more controllable. I monitor the sodium every two to four hours and have DDAVP available to relower if correction exceeds the ceiling." [1]
The correction ceiling and osmotic demyelination syndrome
ODS is the catastrophic complication of overcorrecting chronic hyponatraemia. It presents days to a week after a rapid correction with a biphasic course: initial improvement as the sodium rises, followed by deterioration with dysarthria, dysphagia, quadriparesis, locked-in syndrome, seizures, coma, and often death [3]. MRI shows characteristic pontine and extra-pontine demyelination, though imaging may lag the clinical picture by days.
The correction limits — memorise these: [1]
| Timeframe | Maximum rise |
|---|---|
| First 4 hours (for severe symptoms) | 4 to 5 mmol/L — the symptomatic target |
| 24 hours | 8 to 10 mmol/L |
| Any 48-hour period | 18 mmol/L |
These limits are tighter for patients at highest risk of ODS: sodium under 105 mmol/L, hypokalaemia, alcohol use disorder, malnutrition, anorexia, advanced liver disease, burns, and the elderly. In these patients aim for the lower end (8 mmol/L in 24 hours). [1]
What to do if correction exceeds the ceiling: relower the sodium with DDAVP (desmopressin) 1 to 2 micrograms subcutaneously or intravenously every 8 hours, plus free water (oral or 5% dextrose IV). This is a salvage manoeuvre — the evidence suggests that prompt relowering reduces the risk of ODS [2][8]. The strategy is to recreate a controlled hyponatraemic state and then correct within the ceiling.
Prevention is the priority. Identify high-risk patients, use the bolus approach, monitor frequently, anticipate the autonomous water diuresis that follows stopping a thiazide or SSRI or treating SIADH, and have DDAVP ready. [1]
Hypernatraemia — pathophysiology
Hypernatraemia (Na greater than 145 mmol/L) is a water-deficit state [5]. The body has lost water in excess of sodium, or, less commonly, gained sodium in excess of water (salt poisoning, hypertonic saline, hyperaldosteronism). The crucial clinical point: hypernatraemia implies a defective water intake or access mechanism, because a patient with an intact thirst centre and free access to water will drink to normality.
The brain adapts to hypernatraemia by accumulating osmoles (the reverse of hyponatraemia) — electrolytes rapidly, then idiogenic osmoles over 24 to 48 hours. This is why rapid correction of chronic hypernatraemia risks cerebral oedema (the mirror image of ODS): the brain has built up osmoles, a rapid fall in tonicity drives water in, and the brain swells. [1]
| Severity | Sodium | Clinical context |
|---|---|---|
| Mild | 146 to 149 mmol/L | Often incidental; correct the cause |
| Moderate | 150 to 159 mmol/L | Symptomatic (thirst, lethargy, confusion); investigate and treat |
| Severe | 160 mmol/L or above | High mortality; ICU; correct slowly over 48 to 72 hours |
Hypernatraemia is iatrogenic in a high proportion of hospitalised cases — inadequate free water provision to the intubated patient, hypertonic sodium bicarbonate, tube-feeding without water supplementation, and the post-operative patient on normal saline alone. [1]
Causes of hypernatraemia — by mechanism
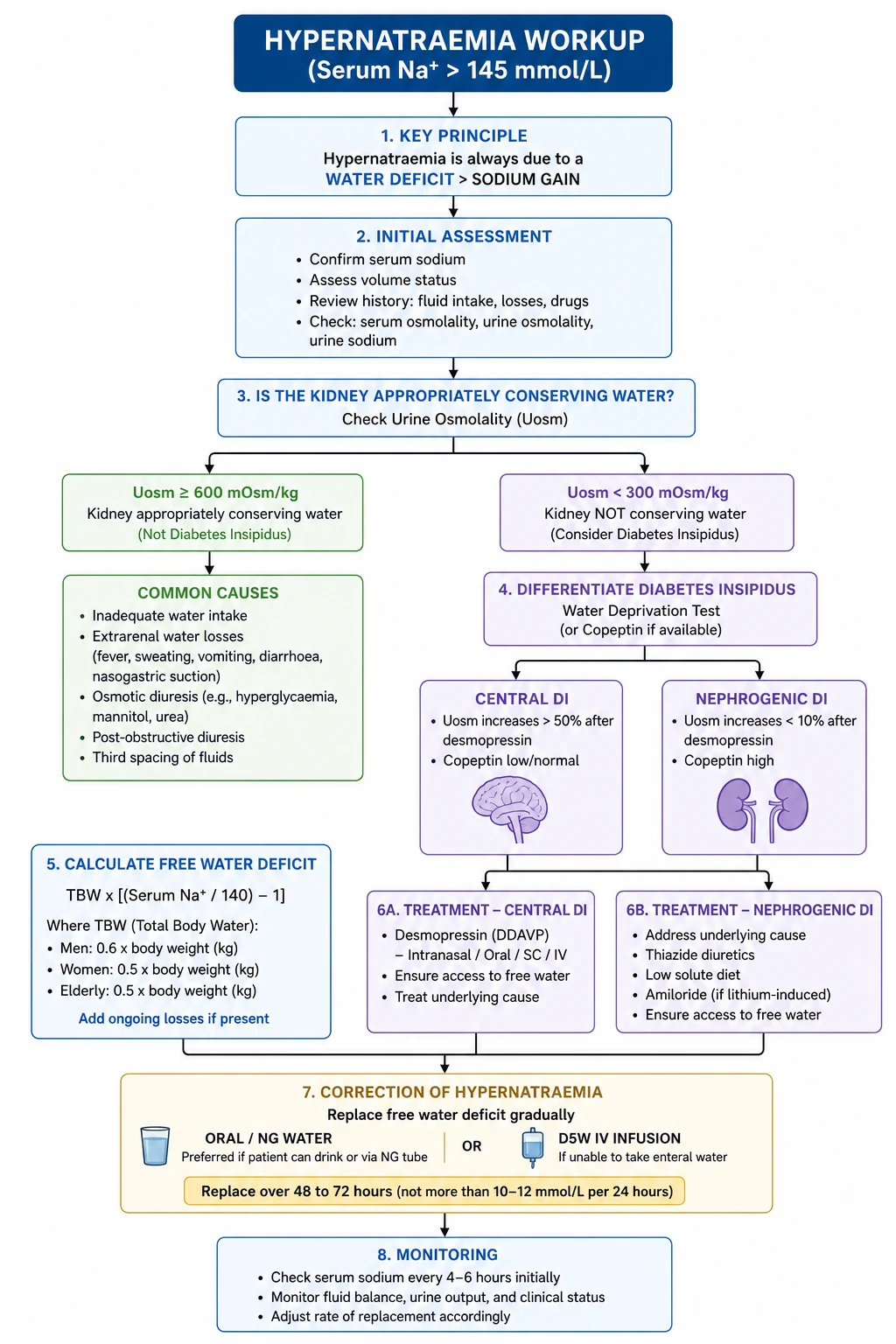
Water loss (the vast majority)
Extra-renal water loss (urine osmolality very high, over 700 to 800 mOsm/kg — the kidney is concentrating maximally): [1]
- Insensible loss — fever, heat exposure, tachypnoea, severe burns, open wounds.
- Gastrointestinal loss — diarrhoea (osmotic, particularly lactulose in hepatic encephalopathy), vomiting (less, because it is isotonic).
- Dermal loss — burns, severe exfoliative rashes. [1]
Renal water loss (urine osmolality inappropriately low or normal despite hypernatraemia): [1]
- Central diabetes insipidus (AVP deficiency). Hypothalamic-pituitary disease — post-neurosurgery (transsphenoidal), head trauma, tumours (craniopharyngioma, metastases), infiltrative (histiocytosis, sarcoidosis, lymphocytic hypophysitis), Sheehan syndrome, idiopathic, autoimmune. Recent consensus proposes renaming central DI arginine vasopressin deficiency (AVP-D).
- Nephrogenic diabetes insipidus (AVP resistance). The kidney cannot respond to AVP. Causes: lithium (the commonest acquired cause — affects about 20 percent of long-term users, may be partially reversible), hypercalcaemia, hypokalaemia, congenital (AVPR2 mutation X-linked, aquaporin-2 mutation autosomal recessive), demeclocycline (now rarely used), ifosfamide, foscarnet, amyloidosis, Sjogren syndrome, sickle cell disease. Recent consensus proposes renaming this arginine vasopressin resistance (AVP-R).
- Osmotic diuresis — uncontrolled diabetes (glucose), mannitol, high-protein tube feeds (urea diuresis), post-obstructive diuresis.
- Reset osmostat (rare in hypernatraemia). [1]
Sodium gain (uncommon)
- Hypertonic saline or sodium bicarbonate — iatrogenic, particularly during resuscitation.
- Salt poisoning — accidental (infant formula error), deliberate (rare).
- Mineralocorticoid excess — primary hyperaldosteronism, Cushing syndrome; usually mild hypernatraemia, because thirst compensates. [1]
Diabetes insipidus — the discriminating assessment
The clinical question in hypernatraemia from renal water loss is: is the AVP axis intact, and is the kidney responsive to AVP? [1]
Classic clinical features of DI: polyuria (more than 3 litres per day, often much more, with dilute urine), polydipsia, nocturia, preference for cold water. In conscious DI the thirst mechanism protects against hypernatraemia; hypernatraemia develops only when access to water is impaired (the unconscious patient, the intubated patient, the elderly, the infant). [1]
The water deprivation test (the traditional approach)
Withhold fluids and measure serial weights, urine volumes, urine and serum osmolality, and sodium. Stop when: body weight falls by 3 to 5 percent, serum sodium exceeds 145 mmol/L, or serum osmolality exceeds 300 mOsm/kg. Then give desmopressin (DDAVP) 2 micrograms intramuscularly and measure urine osmolality for 2 hours. [1]
| Response to DDAVP | Diagnosis |
|---|---|
| Urine osmolality rises to over 750 mOsm/kg | Central DI (the kidney can concentrate when given AVP) |
| Little or no rise (stays under 300) | Nephrogenic DI (the kidney cannot respond to AVP) |
| Intermediate rise (300 to 750) | Partial central DI, or partial nephrogenic DI — distinguish clinically and with direct AVP measurement |
| Already concentrated (over 750) before DDAVP | Primary polydipsia (the patient was water-loading; once deprived, they concentrate normally) |
Copeptin (the modern approach): Copeptin is the C-terminal fragment of the AVP prohormone, released stoichiometrically with AVP and far more stable in the lab. A stimulated copeptin (with hypertonic saline infusion) above 21.4 pmol/L excludes central DI; below 3.8 pmol/L confirms it; in between requires the water deprivation test. The direct copeptin test is increasingly used in specialist centres and is a high-yield DWE discriminator. [1]
The post-pituitary-surgery triphasic response
After transsphenoidal surgery, DI can follow a triphasic pattern over 1 to 2 weeks: [1]
- Phase 1 (days 0 to 3): central DI from axonal shock and failure of AVP release — polyuria, hypernatraemia. Treat with DDAVP.
- Phase 2 (days 4 to 10): SIADH from unregulated release of stored AVP from degenerating posterior pituitary neurones — hyponatraemia. STOP DDAVP; fluid restrict.
- Phase 3 (after day 10): permanent central DI — AVP production is gone. Resume lifelong DDAVP. [1]
Failing to recognise phase 2 produces dangerous hyponatraemia in a patient just treated for hypernatraemia. This is a classic viva trap. [1]
Free water deficit calculation
The free water deficit is the volume of pure water needed to return serum sodium to 140 mmol/L [5].
Formula: [1]
Water deficit = TBW x (serum Na / 140 − 1) [1]
where TBW (total body water) = weight (kg) x fraction (0.6 for adult men and non-elderly women, 0.5 for elderly men, 0.45 for elderly women). [1]
Worked example: A 70 kg elderly man, serum Na 168.
- TBW = 70 x 0.5 = 35 litres.
- Deficit = 35 x (168/140 − 1) = 35 x 0.20 = 7 litres of free water. [1]
Critical caveats: [1]
- The formula calculates the deficit to 140, but you correct to a target, not all at once — typically lower sodium by no more than 10 mmol/L in 24 hours, over 48 to 72 hours, to avoid cerebral oedema [5].
- The formula does not include ongoing losses — insensible, urine, GI. These must be added and replaced as they occur. The commonest error is calculating the deficit, replacing it, and watching the sodium rebound because ongoing polyuria was ignored.
- Use the formula as a guide, not a prescription — re-measure the sodium every 4 to 6 hours and adjust.
In practice, the Adrogue-Madias change in sodium per litre of fluid formula is also useful for selecting the replacement fluid: [1]
Change in Na per litre infused = (infusate Na + infusate K − serum Na) / (TBW + 1) [1]
For D5W (sodium 0), the change is roughly −(serum Na)/(TBW + 1), which is why D5W is the most sodium-lowering fluid. For 0.9% saline (sodium 154), the change is small and may be positive if serum sodium is below 154 — normal saline is NOT the right fluid for most hypernatraemia [5].
Correction of hypernatraemia
Principles: [1]
- Correct the underlying cause — stop the offending drug (lithium, demeclocycline), treat the hypercalcaemia or hypokalaemia, resect the tumour, give DDAVP for central DI.
- Replace the free water deficit slowly — over 48 to 72 hours, aiming for a sodium fall of no more than 10 mmol/L in 24 hours (0.5 mmol/L per hour) in chronic hypernatraemia. Faster correction risks cerebral oedema, seizure, and death. [1]3. Replace ongoing losses as they occur.
- Oral or enteral water is the preferred route — the safest and most physiological. Give via NG or orally if the patient can drink.
- If IV is needed, use 5% dextrose (D5W) — not normal saline. D5W provides free water once the glucose is metabolised. Monitor glucose closely (risk of hyperglycaemia).
- In central DI, give DDAVP — desmopressin 1 to 2 micrograms IV/SC every 8 to 12 hours, or 10 to 20 micrograms intranasally. Titrate to urine output. Use the lowest dose that controls polyuria; overshoot causes hyponatraemia.
- In nephrogenic DI, treat the cause and use adjuncts — stop lithium if possible, correct calcium and potassium; thiazide diuretics paradoxically reduce polyuria (the mechanism: mild volume depletion activates proximal tubular reabsorption of sodium and water, reducing delivery to the defective collecting duct). Add a low-sodium diet and a thiazide (e.g., hydrochlorothiazide 25 mg, or amiloride for lithium-induced DI — amiloride blocks lithium entry through the epithelial sodium channel). NSAIDs (indometacin) can reduce polyuria by inhibiting renal prostaglandins, but are rarely used because of renal and gastrointestinal toxicity. [1]
DWE exam trap — the thiazide paradox: A patient with nephrogenic DI from lithium is polyuric. You give a thiazide diuretic — and the urine output falls. This seems paradoxical (a diuretic reducing urine output) but the mechanism is sound: thiazide-induced mild volume contraction increases proximal tubular reabsorption, so less filtrate reaches the defective collecting duct. Amiloride is preferred in lithium-induced nephrogenic DI because it blocks lithium entry into the principal cell via the epithelial sodium channel, partially protecting the kidney. [1]
Drugs and sodium disorders — the high-yield list
Drugs causing hyponatraemia: [1]
- Thiazide diuretics — the single commonest drug cause; older women; presents within weeks of starting; may coexist with hypokalaemia. Stop permanently.
- SSRIs — the commonest psychiatric drug cause; typically within the first month; SIADH mechanism. Particular caution in the elderly.
- Carbamazepine and oxcarbazepine — SIADH mechanism; check sodium before and after starting.
- ** vincristine, cyclophosphamide** — oncology patients.
- MDMA — acute, severe hyponatraemia from SIADH plus excessive water intake; high mortality.
- Desmopressin — prescribed for nocturia, enuresis, von Willebrand; iatrogenic SIADH; avoid in the elderly for nocturia.
- Antipsychotics (phenothiazines, haloperidol) — less common.
- PPIs — weak association.
- MDMA, opiates — acute. [1]
Drugs causing (or treating) hypernatraemia: [1]
- Lithium — nephrogenic DI in about 20 percent of long-term users; may be partially reversible with early cessation.
- Demeclocycline — historically used to TREAT SIADH by inducing nephrogenic DI; now largely abandoned because of hepatotoxicity and nephrotoxicity.
- Foscarnet, cidofovir, amphotericin B, ifosfamide — renal concentrating defect.
- ** Mannitol** — osmotic diuresis. [1]
DWE integration — high-yield patterns
The DWE tests reasoning across symptom severity, volume status, urine biochemistry, and correction rate. Common stem patterns: [1]
- "An elderly woman on a thiazide, Na 118, confused." Stop the thiazide, correct slowly, monitor for overcorrection, check cortisol and TSH.
- "A young man post-MDMA, seizing, Na 116." Acute — give 3% saline boluses now; the brain has not adapted; cerebral oedema is the threat.
- "A lung cancer patient, Na 122, euvolaemic, urine Na 45, urine osm 350." SIADH from small cell lung cancer; fluid restrict, salt tablets, consider tolvaptan; treat the cancer; check the futility discussion.
- "A cirrhotic patient, Na 124, ascites." Hypervolaemic hyponatraemia; fluid restrict, loop diuretic, treat the liver disease; avoid hypertonic saline except for severe symptoms; transplant assessment.
- "A post-pituitary-surgery patient, day 5, Na 128." Phase 2 SIADH of the triphasic response; fluid restrict; stop DDAVP.
- "An intubated ICU patient, Na 162, urine osm 600." Extra-renal water loss (insensible, inadequate free water); give D5W or enteral water slowly over 48 hours; audit the fluid prescription.
- "A bipolar patient on lithium, Na 156, polyuria 6 L/day, urine osm 180." Nephrogenic DI; stop lithium if possible; thiazide plus amiloride, low-sodium diet, ensure water access.
- "Hyponatraemia corrected from 108 to 132 in 24 hours; patient now dysarthric." ODS from overcorrection; relower with DDAVP and free water; supportive care; MRI to confirm. [1]
DCE integration — long and short case
Long case — hyponatraemia in a complex patient: a 78-year-old woman with hypertension (on a thiazide), osteoporosis, mild cognitive impairment, and a recent fall, presenting with confusion and Na 119. She is mildly volume-depleted, potassium 3.2, urea 9. The problem list: thiazide-induced hyponatraemia with hypokalaemia (a risk factor for ODS), volume depletion, cognitive impairment with a fall (assess for injury, delirium, dementia), osteoporosis with a fall risk, polypharmacy. The integrated plan: stop the thiazide permanently, replace potassium and volume cautiously with normal saline, monitor sodium every 4 hours with DDAVP available to relower, reassess antihypertensives (an alternative for her hypertension), falls workup, cognitive assessment, medication reconciliation. The model opening: "I will manage this as acute-on-chronic thiazide-induced hyponatraemia with two competing risks — cerebral oedema from the acute component and osmotic demyelination from overcorrection, compounded by hypokalaemia which raises the ODS risk." [1]
Short case — volume status assessment: "Assess this hyponatraemic patient's volume status and look for signs suggesting the cause." The systematic routine: hands (pigmentation of Addison's, pulse, signs of dehydration), neck (JVP — low in hypovolaemia, high in hypervolaemia), cardiovascular (postural blood pressures, signs of heart failure), respiratory (consolidation suggesting pneumonia as an SIADH cause, crackles suggesting heart failure), abdomen (ascites and liver stigmata suggesting cirrhosis, ballotable masses suggesting polycystic kidneys), legs (oedema), and general (cachexia of malignancy or Addison's, signs of hypothyroidism). The findings — low JVP, postural drop, dry mucous membranes — point to hypovolaemia; the prescription chart reveals the thiazide. [1]
Common exam traps and high-yield discriminators
- Symptom severity, not the absolute sodium, drives urgency. A seizing patient with Na 124 needs 3% saline; an asymptomatic patient with Na 120 does not.
- Acute versus chronic changes everything. Acute hyponatraemia (under 48 hours) tolerates and requires faster correction; chronic hyponatraemia must be corrected slowly. When chronicity is unknown, assume chronic.
- Normal saline worsens SIADH. The "desalination" phenomenon — the kidney retains the water, excretes the sodium, and the sodium falls further.
- Always check cortisol and TSH before diagnosing SIADH. Adrenal insufficiency and hypothyroidism are treatable mimics; missing Addison's is fatal.
- The correction ceiling is 8 to 10 mmol/L in 24 hours. Overcorrection from any cause — boluses, water diuresis after stopping a thiazide — requires prompt relowering with DDAVP and free water. [1]6. Hypokalaemia, alcoholism, malnutrition, liver disease, and Na under 105 raise the ODS risk — aim for the lower end of the correction range.
- Hypernatraemia is iatrogenic more often than admitted. Audit the fluid chart in every inpatient case.
- The thiazide paradox and the DDAVP relowering manoeuvre are classic viva discriminators — know the mechanisms.
- Urine osmolality is the key fork in both dysnatraemias — low (under 100) in primary polydipsia and beer potomania; high in SIADH and most other causes of hyponatraemia; inappropriately low in diabetes insipidus despite hypernatraemia.
- Correct hypernatraemia slowly — no more than 10 mmol/L in 24 hours — to avoid cerebral oedema, the mirror image of ODS. Oral or enteral water is preferred; D5W if IV is needed. [1]
Sources
European Clinical Practice Guideline on Hyponatraemia (Spasovski) [1]; Sterns, Disorders of plasma sodium (NEJM 2015) [2]; Sterns et al, Osmotic demyelination syndrome (NEJM 1986) [3]; Adrogue and Madias, Hyponatremia (NEJM 2000) [4]; Adrogue and Madias, Hypernatremia (NEJM 2000) [5]; Schrier et al, SALT tolvaptan trials (NEJM 2006) [6]; Verbalis et al, Expert panel recommendations (Am J Med 2013) [7]; Sterns et al, Treatment of hyponatremia (Semin Nephrol 2009) [8]; NICE CG174 (Intravenous fluid therapy); CARI Guidelines.
References
- [1]Spasovski G, Vanholder R, Allolio B, et al. Clinical practice guideline on diagnosis and treatment of hyponatraemia Nephrol Dial Transplant, 2014.PMID 24569496
- [2]Sterns RH Disorders of plasma sodium--causes, consequences, and correction N Engl J Med, 2015.PMID 25551526
- [3]Sterns RH, Riggs JE, Schochet SS Jr Osmotic demyelination syndrome following correction of hyponatremia N Engl J Med, 1986.PMID 3713747
- [4]Adrogué HJ, Madias NE Hyponatremia N Engl J Med, 2000.PMID 10824078
- [5]Adrogué HJ, Madias NE Hypernatremia N Engl J Med, 2000.PMID 10816188
- [6]Schrier RW, Gross P, Gheorghiade M, et al. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia N Engl J Med, 2006.PMID 17105757
- [7]Verbalis JG, Goldsmith SR, Greenberg A, et al. Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations Am J Med, 2013.PMID 24074529
- [8]Sterns RH, Nigwekar SU, Hix JK The treatment of hyponatremia Semin Nephrol, 2009.PMID 19523575