Phys · renal
Glomerulonephritis (Nephritic Spectrum)
Also known as glomerulonephritis · GN · nephritic syndrome · rapidly progressive glomerulonephritis · RPGN · crescentic glomerulonephritis · IgA nephropathy · Berger disease · post-streptococcal glomerulonephritis · PSGN · lupus nephritis · ANCA-associated vasculitis · AAV · anti-GBM disease · Goodpasture syndrome · pauci-immune glomerulonephritis · granulomatosis with polyangiitis · GPA · microscopic polyangiitis · MPA · C3 glomerulopathy
Consultant-physician-depth guide to glomerulonephritis — nephritic syndrome, RPGN classification (anti-GBM, immune complex, pauci-immune/ANCA), IgA nephropathy (Oxford MEST-C), post-streptococcal GN, lupus nephritis (ISN/RPS), pulmonary-renal syndromes, complement interpretation, biopsy immunofluorescence, and evidence-based immunosuppression. Structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Glomerulonephritis (Nephritic Spectrum)
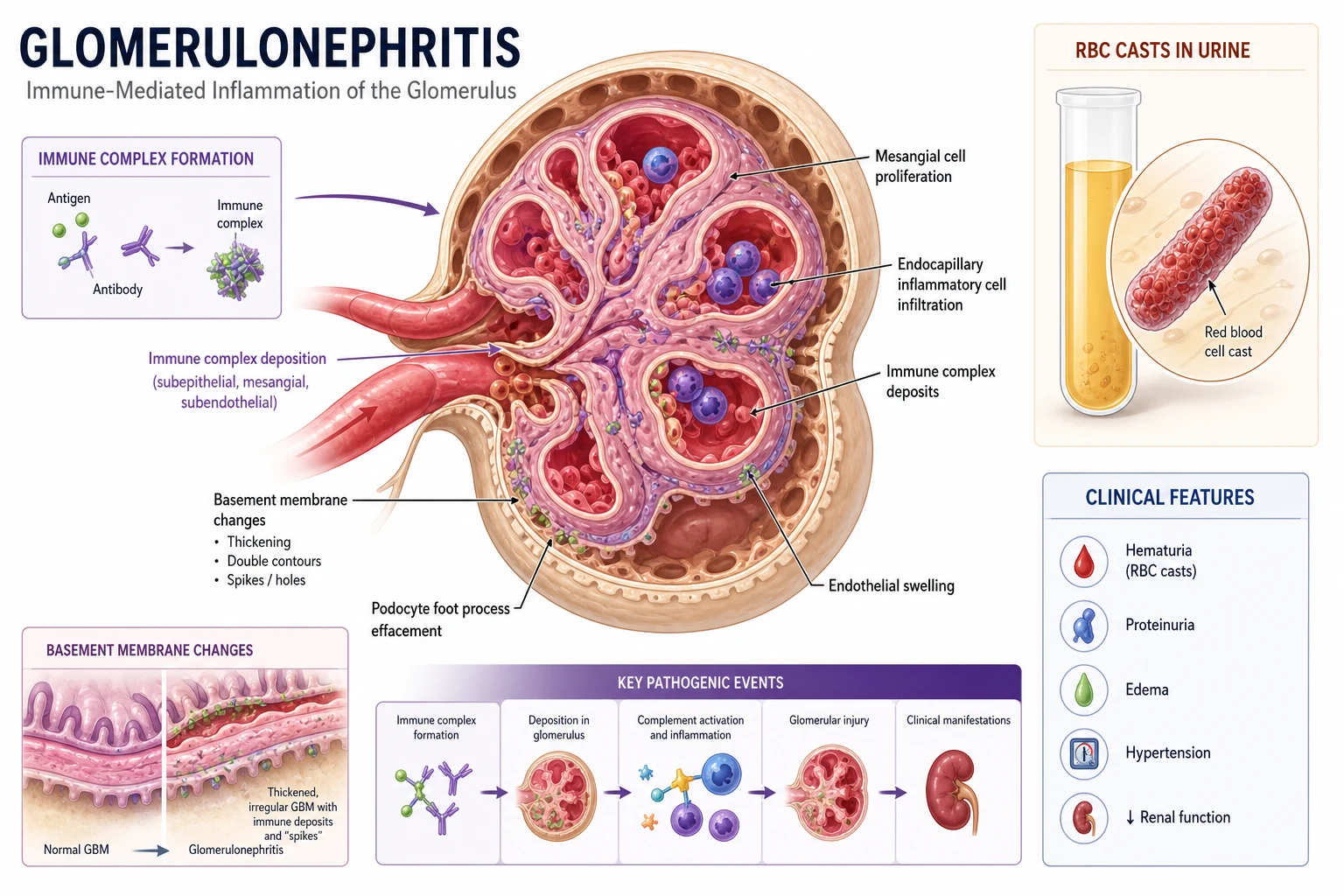
The answer first
Glomerulonephritis is immune-mediated inflammation of the glomerulus that presents across a clinical spectrum from asymptomatic microscopic haematuria to rapidly progressive glomerulonephritis (RPGN) causing acute kidney injury over days to weeks. The nephritic syndrome is the constellation of haematuria (dysmorphic red cells and red cell casts), proteinuria (usually sub-nephrotic), hypertension, oedema, and a rise in creatinine. The nephrotic syndrome sits at the other end of the same spectrum — heavy proteinuria above 3.5 g/day, hypoalbuminaemia, and oedema [1].
The single most important clinical task is to recognise RPGN as a renal emergency. A patient with a rapidly rising creatinine and an active urinary sediment has crescentic glomerulonephritis until proven otherwise. Irreversible glomerular destruction occurs over days to weeks. The mandate is: [1]
- Send the urgent serological panel immediately — ANA, anti-dsDNA, ANCA (MPO and PR3), anti-GBM antibody, C3 and C4, serum IgA, hepatitis B and C, HIV, and ASO/anti-DNase B if post-infectious is suspected.
- Biopsy within 24 to 48 hours — light microscopy, immunofluorescence, and electron microscopy.
- Start immunosuppression as soon as the diagnosis is secured — high-dose corticosteroids plus cyclophosphamide or rituximab, with plasma exchange for anti-GBM disease and selected cases of severe ANCA vasculitis.
- Treat pulmonary-renal syndromes as the highest priority — pulmonary haemorrhage from anti-GBM or ANCA vasculitis is immediately life-threatening. [1]
DWE high-yield: The complement profile is the single most discriminating serological test. Low C3 with normal C4 suggests alternative pathway activation (post-infectious GN, C3 glomerulopathy). Low C3 and low C4 suggests classical pathway activation (lupus, endocarditis, cryoglobulinaemia). Normal complement is found in IgA nephropathy, ANCA-associated vasculitis, and anti-GBM disease — do not be reassured by a normal complement. [1]
Nephritic versus nephrotic: the clinical spectrum
Glomerulonephritis is not one disease. It is a pattern of glomerular injury caused by a heterogeneous group of immune-mediated processes. Clinically, presentations fall on a spectrum: [1]
| Feature | Nephritic syndrome | Nephrotic syndrome |
|---|---|---|
| Haematuria | Prominent (macro or micro), dysmorphic RBC, RBC casts | Mild or absent |
| Proteinuria | Sub-nephrotic (less than 3.5 g/day) | Nephrotic range (above 3.5 g/day) |
| Creatinine | Reduced GFR, AKI pattern | Variable, may be normal |
| Oedema | Periorbital, peripheral, from sodium retention | Severe, from hypoalbuminaemia |
| Typical diseases | IgA nephropathy, post-strep GN, lupus, RPGN, AAV | Minimal change, FSGS, membranous |
Many diseases overlap — lupus nephritis and IgA nephropathy can produce mixed nephritic-nephrotic features. The clinical syndrome tells you the pattern of injury; serology and biopsy tell you the disease. [1]
Pathophysiology: how the immune system attacks the glomerulus

The four immunopathological mechanisms
Every glomerulonephritis is driven by one or more of these mechanisms. Understanding them explains the biopsy pattern and the treatment: [1]
1. Anti-GBM antibody disease (Goodpasture syndrome): Autoantibodies bind directly to the non-collagenous domain of the alpha-3 chain of type IV collagen in the glomerular basement membrane (GBM). Immunofluorescence shows a smooth, linear IgG deposition along the GBM — this is the pathognomonic finding. The antibody also attacks the alveolar basement membrane, producing pulmonary haemorrhage in 40 to 60% of cases. [1]
2. Immune complex deposition: Circulating immune complexes (antigen-antibody) lodge in the mesangium, subendothelial, or subepithelial space. Immunofluorescence shows granular deposition. Examples include IgA nephropathy (mesangial IgA), lupus nephritis ("full house" IgG, IgA, IgM, C3, C1q), and post-infectious GN ("humps" of subepithelial deposits). [1]
3. Pauci-immune / ANCA-associated vasculitis: Necrotising inflammation of small vessels (capillaries, arterioles, venules) with little or no immunoglobulin deposition on immunofluorescence ("pauci-immune"). ANCA antibodies (anti-MPO or anti-PR3) activate neutrophils, which degranulate and release proteases and reactive oxygen species. The alternative complement pathway is critically involved — C5a drives neutrophil priming, which is why complement-directed therapy has been investigated. [1]
4. Complement-mediated disease (C3 glomerulopathy): Dysregulation of the alternative complement pathway causes C3 deposition without immunoglobulin. C3 is low; C4 is normal (alternative pathway activation only). This includes dense deposit disease (DDD) and C3 glomerulonephritis. [1]
Crescent formation: the common endpoint of severe injury
A crescent is the histological hallmark of severe, rapidly progressive glomerular injury. It forms when the glomerular capillary wall ruptures, allowing fibrin, macrophages, and parietal epithelial cells to spill into Bowman's space. These cells proliferate and form a crescent-shaped mass that compresses and destroys the glomerular tuft. [1]
The key clinical point: crescents are reversible if treated early, but fibrotic (irreversible) within weeks. This is why RPGN is a renal emergency. A biopsy showing more than 50% of glomeruli with crescents carries a poor prognosis and demands immediate immunosuppression. [1]
The IgA nephropathy multi-hit hypothesis
IgA nephropathy follows a "multi-hit" model:
- Hit 1: Production of galactose-deficient IgA1 (Gd-IgA1), a genetically determined abnormality of O-glycosylation.
- Hit 2: Formation of autoantibodies against Gd-IgA1 (IgG or IgA).
- Hit 3: Formation of circulating immune complexes.
- Hit 4: Mesangial deposition, activation, and release of inflammatory mediators causing proliferation, scarring, and proteinuria. [1]
This model underpins modern therapies — the goal is to reduce Gd-IgA1 production (targeted-release budesonide), block immune complex effects (endothelin antagonists, complement inhibitors), and reduce glomerular pressure (RAAS blockade, SGLT2 inhibitors). [1]
RPGN classification: the framework you must know
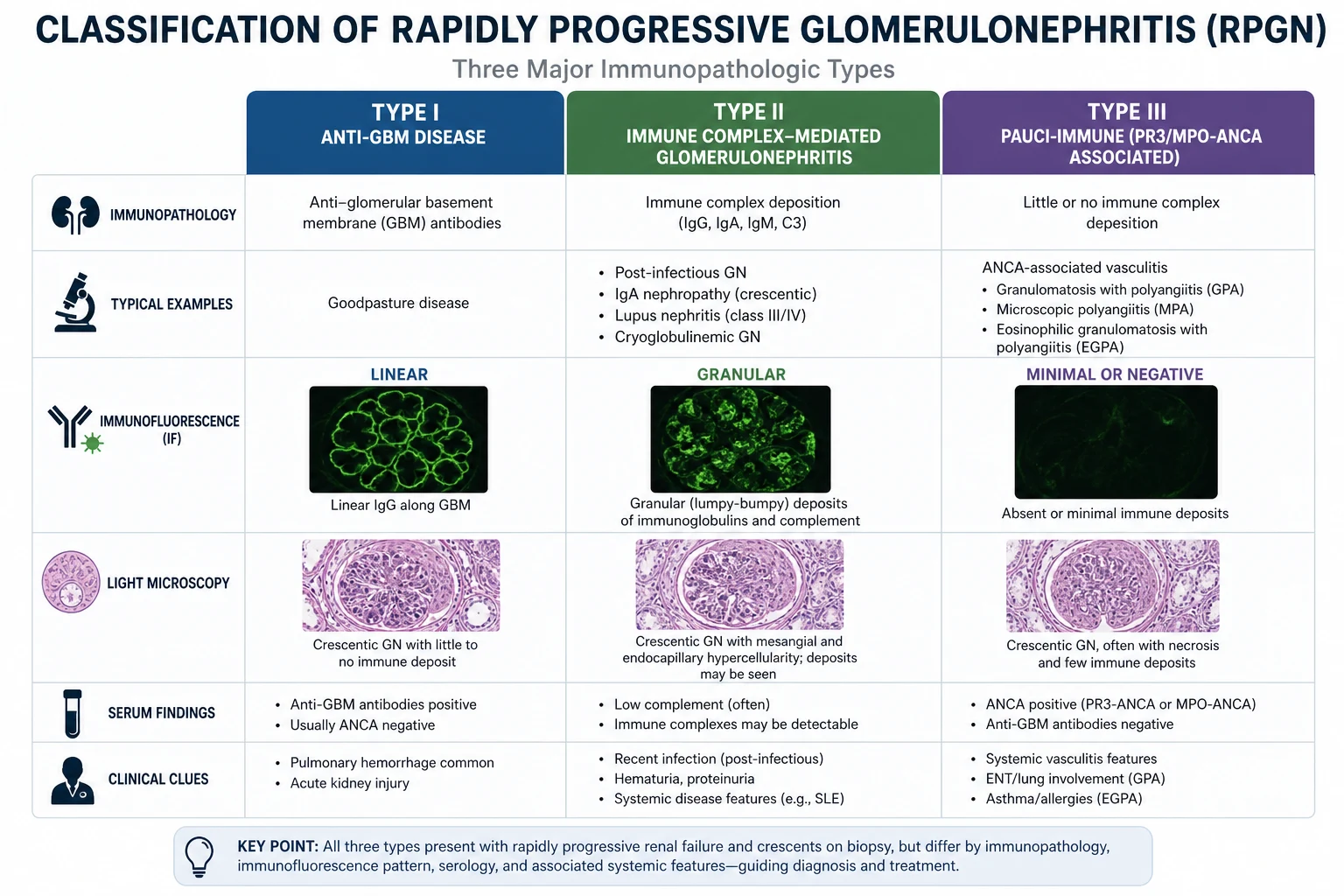
Rapidly progressive glomerulonephritis is defined clinically as a rapid decline in GFR (over days to weeks) with an active urinary sediment (dysmorphic RBC, RBC casts). The classification by immunofluorescence pattern is one of the highest-yield exam frameworks in nephrology: [1]
| Type | Mechanism | IF pattern | Complement | Key diseases |
|---|---|---|---|---|
| Type I | Anti-GBM antibody | Linear IgG | Normal | Goodpasture syndrome, renal-limited anti-GBM |
| Type II | Immune complex | Granular | Low (variable) | Lupus nephritis, post-infectious GN, IgA nephropathy, cryoglobulinaemia, endocarditis-associated |
| Type III | Pauci-immune / ANCA | Little/no deposition | Normal | GPA (Wegener), MPA, EGPA (Churg-Strauss) |
DCE trap — double-positive disease: Approximately 30 to 50% of patients with anti-GBM disease are also ANCA-positive (usually MPO). These "double-positive" patients behave clinically and prognostically like anti-GBM disease — they require plasma exchange regardless of the ANCA result. This is a frequently missed diagnosis that changes management fundamentally. [1]
IgA nephropathy — the most common primary glomerulonephritis
IgA nephropathy (Berger disease) is the most common primary glomerulonephritis worldwide. It is an under-recognised cause of ESKD — 20 to 40% of patients progress to end-stage over 20 years. [1]
Clinical presentation
- Macroscopic haematuria, synpharyngitic: Visible blood in the urine simultaneously with or within 1 to 2 days of an upper respiratory or gastrointestinal infection. This is the classic presentation in children and young adults. The key distinction from post-streptococcal GN is the timing — IgA is synpharyngitic (same time as infection); PSGN has a latency of 1 to 4 weeks.
- Asymptomatic microscopic haematuria and proteinuria: Detected on routine screening. This is the most common presentation in adults.
- CKD progression: Hypertension, progressive proteinuria, and rising creatinine over years.
- Crescentic IgA nephropathy (rapidly progressive): AKI with macroscopic haematuria, typically during an episode of gross haematuria. Biopsy shows crescents superimposed on mesangial IgA deposition. [1]
The Oxford MEST-C classification
The Oxford classification (2009, updated 2016) standardises IgA nephropathy biopsy reporting and provides prognostic information independent of clinical data [6]:
| Lesion | Score | Definition | Prognostic meaning |
|---|---|---|---|
| Mesangial hypercellularity | M0 / M1 | More than 50% of glomeruli with mesangial hypercellularity (M1) | Predicts progression; may predict response to immunosuppression |
| Endocapillary hypercellularity | E0 / E1 | Endocapillary hypercellularity present (E1) | Predicts progression; associated with more active inflammation |
| Segmental glomerulosclerosis | S0 / S1 | Any segmental scarring (S1) | Predicts progression and worse renal outcome |
| Tubulointerstitial damage | T0 / T1 / T2 | Interstitial fibrosis/tubular atrophy: 0 to 25% (T0), 26 to 50% (T1), above 50% (T2) | Strongest predictor of outcome — T2 carries the worst prognosis |
| Crescents (added 2016) | C0 / C1 / C2 | Cellular/fibrocellular crescents: none (C0), up to 25% of glomeruli (C1), above 25% (C2) | C2 predicts faster progression; influences immunosuppression decisions |
The MEST-C score is reported alongside clinical data (eGFR, proteinuria, blood pressure) to stratify risk and guide treatment. The S and T lesions are the strongest independent predictors of progression; T is the most powerful single predictor of outcome. [1]
Management of IgA nephropathy
KDIGO 2021 and the subsequent chapter updates define a tiered approach [1]:
Tier 1 — Optimised supportive care (all patients):
- RAAS blockade with an ACE inhibitor or ARB — titrated to maximum tolerated dose, regardless of blood pressure, if proteinuria is above 0.5 g/day. [1]- Blood pressure target below 130/80 mmHg (lower if tolerated, particularly with proteinuria).
- SGLT2 inhibitors — now recommended for IgA nephropathy with proteinuria based on DAPA-CKD and EMPA-KIDNEY subgroup data showing slowing of CKD progression. This is a Class 1 recommendation in the updated KDIGO IgA chapter.
- Lifestyle: sodium restriction, smoking cessation, weight management, fish oil (omega-3) — evidence for fish oil is mixed but it is low-risk and widely used. [1]
Tier 2 — Patients at high risk of progression despite 90 days of optimised supportive care (proteinuria above 0.75 to 1 g/day):
- Corticosteroids (TESTING protocol): The TESTING trial established that oral methylprednisolone reduces the risk of kidney failure, but the original high-dose protocol (0.8 mg/kg/day) caused excess serious infections including two deaths [7]. The trial was redesigned with a lower dose (0.4 mg/kg/day tapering over months) and PJP prophylaxis, and the final results (2022) confirmed benefit with an improved safety profile — hazard ratio 0.53 for the composite of 40% eGFR decline, kidney failure, or death [8].
- STOP-IgAN caveat: The STOP-IgAN trial showed that adding immunosuppression to optimised supportive care did not improve renal outcomes in a broadly lower-risk cohort [9]. This means steroids should be reserved for patients at genuine risk of progression — not given reflexively to everyone.
- Targeted-release budesonide (Nefecon): A gut-targeted formulation that modulates mucosal IgA production, approved for IgA nephropathy based on the NefIgArd trial showing proteinuria reduction and eGFR stabilisation.
Tier 3 — Emerging and refractory disease:
- Sparsentan (dual endothelin A and angiotensin receptor antagonist) — approved for IgA nephropathy based on PROTECT.
- Complement inhibitors (avacopan-like C5a inhibitors, iptacopan factor B inhibitor) — under investigation.
- Rapidly progressive / crescentic IgA nephropathy — treat as RPGN with corticosteroids and cyclophosphamide, analogous to other crescentic diseases. [1]
Exam trap: Do not prescribe high-dose pulsed methylprednisolone (the original TESTING protocol) for IgA nephropathy — it caused excess serious infections and two deaths. Use the modified low-dose protocol (0.4 mg/kg/day) with PJP prophylaxis, and only in patients at genuine risk of progression after optimised supportive care. [1]
Post-streptococcal glomerulonephritis
Post-streptococcal GN (PSGN) is the archetype of infection-related immune complex glomerulonephritis. It follows infection with nephritogenic strains of group A streptococcus — typically M types 1, 2, 4, 12, 49 (skin) or 1, 2, 12, 49, 57 (pharyngitis). [1]
Clinical presentation
- Latency: 1 to 3 weeks after pharyngitis, or 3 to 6 weeks after skin infection (impetigo). This latency is the key discriminator from IgA nephropathy, which is synpharyngitic.
- Nephritic syndrome: Macroscopic or microscopic haematuria, oedema, hypertension, oliguria, and a rise in creatinine. Children present more acutely; adults may have more subtle disease.
- Complement: C3 is low; C4 is typically normal (alternative pathway activation). Complement normalises within 6 to 8 weeks. If it does not, reconsider the diagnosis — consider C3 glomerulopathy.
- Serology: Elevated ASO (anti-streptolysin O) after pharyngitis; anti-DNase B after skin infection (ASO is often normal in skin infection). Both rise and fall over weeks. [1]
Biopsy findings (if performed, usually not needed in classic paediatric presentation)
- Light microscopy: Endocapillary proliferation and neutrophil infiltration ("exudative" GN) — diffuse and proliferative.
- Immunofluorescence: Coarse granular IgG and C3 deposition along the capillary loops.
- Electron microscopy: Subepithelial "humps" — the pathognomonic finding of PSGN. [1]
Management and prognosis
Management is supportive — control blood pressure, manage oedema with loop diuretics, treat any residual infection. Immunosuppression is not indicated. The prognosis is excellent in children — more than 95% recover renal function completely. Adults have a worse prognosis, particularly elderly patients and those with underlying CKD or diabetes, where persistent proteinuria and CKD progression occur in 20 to 50%. [1]
Lupus nephritis
Lupus nephritis develops in up to 60% of patients with systemic lupus erythematosus and is a major determinant of morbidity and mortality. Any patient with lupus should have urinalysis and proteinuria assessment at every visit — renal involvement may be silent. [1]
ISN/RPS classification (2003, updated 2018)
| Class | Pathology | Clinical pattern | Treatment approach |
|---|---|---|---|
| I | Minimal mesangial | Normal urine | Treat the lupus; no specific renal therapy |
| II | Mesangial proliferative | Microscopic haematuria, mild proteinuria | RAAS blockade; immunosuppression rarely needed |
| III | Focal proliferative (less than 50% of glomeruli) | Nephritic, proteinuria | Induction immunosuppression (see below) |
| IV | Diffuse proliferative (50% or more of glomeruli) | Nephritic-nephrotic, AKI | Induction immunosuppression — most aggressive and common severe class |
| V | Membranous | Nephrotic-range proteinuria | Induction for pure class V; combine with III/IV if mixed |
| VI | Advanced sclerosing (more than 90% glomeruli sclerosed) | CKD, low activity | No immunosuppression; supportive care / dialysis planning |
Class IV is the most clinically important — it is the most common severe class, carries the worst prognosis, and demands urgent induction. [1]
Induction therapy for class III/IV lupus nephritis
KDIGO 2021 and EULAR/ERA recommend two first-line induction regimens, both with corticosteroids [1][10][11]:
- Mycophenolate mofetil (MMF): Target dose 2 to 3 g/day orally, for 6 months. This is the most widely used first-line agent globally.
- Low-dose (Euro-Lupus) cyclophosphamide: 500 mg IV every 2 weeks for 6 doses (total 3 g). This is preferred in Caucasian populations (validated by the Euro-Lupus Nephritis Trial) and is as effective as the high-dose NIH regimen with markedly less toxicity [10].
Both are given with corticosteroids — methylprednisolone pulses (500 to 1000 mg IV daily for 3 days) followed by oral prednisone 0.5 to 1 mg/kg/day, tapering rapidly. The PEXIVAS trial demonstrated that a reduced-dose glucocorticoid regimen is noninferior to standard dose with fewer serious infections [3] — this has changed practice toward lower cumulative steroid exposure.
ALMS (2009) established that MMF is not superior to IV cyclophosphamide for induction but is an acceptable alternative with a different toxicity profile [11]. The ALMS maintenance phase showed MMF is superior to azathioprine for maintaining remission.
Rituximab (LUNAR trial): The LUNAR trial did not meet its primary endpoint of superior renal response when rituximab was added to MMF and steroids for induction [12]. Despite this, rituximab is widely used off-label for refractory or relapsing lupus nephritis, and for patients who cannot tolerate MMF or cyclophosphamide.
Maintenance therapy
After 6 months of induction, transition to maintenance with either MMF (1 to 2 g/day) or azathioprine (1 to 2 mg/kg/day), continued for at least 2 to 3 years. Rituximab (1 g x2, repeated as needed) is an option for maintenance in relapsing disease. Prednisone is tapered to the lowest effective dose — ideally below 5 to 7.5 mg/day to minimise toxicity. [1]
ANCA-associated vasculitis (AAV)
ANCA-associated vasculitis is a group of necrotising small-vessel vasculitides that cause pauci-immune glomerulonephritis. The three diseases are: [1]
| Disease | Typical ANCA | Classic features |
|---|---|---|
| GPA (granulomatosis with polyangiitis, formerly Wegener) | PR3 (c-ANCA) | ENT disease (nasal crusting, epistaxis, saddle nose deformity, otitis media), pulmonary nodules/cavities, RPGN |
| MPA (microscopic polyangiitis) | MPO (p-ANCA) | RPGN, pulmonary haemorrhage, neuropathy, purpura. No granulomatous ENT inflammation — this distinguishes it from GPA |
| EGPA (eosinophilic granulomatosis with polyangiitis, formerly Churg-Strauss) | MPO (p-ANCA) | Asthma, eosinophilia, sinusitis, neuropathy. Renal involvement is less common than in GPA/MPA |
Clinical presentation
AAV typically presents in middle-aged to older adults (peak 60 to 70 years). Presentations range from indolent (weight loss, fatigue, arthralgia) to fulminant (pulmonary haemorrhage, RPGN). Pulmonary haemorrhage is the most immediately life-threatening complication — it presents with dyspnoea, haemoptysis, diffuse alveolar infiltrates, and a rising transfer factor (DLCO) from alveolar blood. [1]
Investigations
- ANCA testing: Use both immunofluorescence (c-ANCA, p-ANCA) and ELISA for anti-PR3 and anti-MPO antibodies. PR3 is highly specific for GPA; MPO for MPA. Approximately 10 to 20% of patients with biopsy-proven AAV are ANCA-negative.
- Biopsy: Renal biopsy shows necrotising and crescentic GN with little or no immunoglobulin deposition ("pauci-immune"). Sinus or lung biopsy may show granulomatous inflammation in GPA.
- BVAS (Birmingham Vasculitis Activity Score): Used to quantify disease activity and guide treatment intensity. [1]
Induction therapy
Current induction (KDIGO 2021, ACR/VF 2021 guidelines) for organ-threatening or life-threatening AAV [1][4][5]:
- Rituximab: 375 mg/m2 IV weekly for 4 weeks, OR 1 g IV x2 two weeks apart. Established by RAVE (rituximab noninferior to cyclophosphamide, superior in relapsing disease) [4] and RITUXVAS (rituximab plus two pulses of cyclophosphamide, equivalent to standard cyclophosphamide) [5]. Rituximab is now preferred for relapsing disease and in younger patients where fertility preservation matters.
- Cyclophosphamide: IV pulse (15 mg/kg, adjusted for age and renal function) every 2 to 3 weeks for 3 to 6 months. Still used for severe or refractory disease and in centres where rituximab access is limited.
- Corticosteroids: Methylprednisolone pulses (500 to 1000 mg IV daily for 1 to 3 days) followed by oral prednisone 1 mg/kg/day (max 60 to 80 mg), tapering over months. PEXIVAS established that a reduced-dose steroid regimen is noninferior to standard dose with fewer serious infections [3] — this is now the standard.
Plasma exchange in AAV — the evidence
This is one of the most examined and clinically contested areas in nephrology: [1]
- MEPEX (2007): Demonstrated that plasma exchange reduced the risk of ESKD at 12 months compared to methylprednisolone pulses in patients with severe renal vasculitis (creatinine above 500 micromol/L) — 19% versus 43% dialysis-dependent at 12 months [2]. This established plasma exchange in clinical practice for severe renal AAV.
- PEXIVAS (2020): The largest trial ever in AAV (704 patients) found that plasma exchange did not reduce the composite of death or ESKD [3]. This has led to a significant reduction in the routine use of plasma exchange.
- Current practice (KDIGO 2024 chapter update): Plasma exchange is still indicated for: (a) anti-GBM disease or double-positive disease; (b) AAV with severe renal failure (creatinine above 500 micromol/L or dialysis-dependent) where rapid antibody removal may aid renal recovery; (c) AAV with life-threatening pulmonary haemorrhage, though the evidence here is weaker. The routine use for all severe AAV is no longer supported — the decision is individualised.
Maintenance therapy
After remission induction (3 to 6 months), transition to maintenance:
- Rituximab 1 g x2 (or 500 mg x2) every 4 to 6 months for at least 18 to 24 months — now the preferred maintenance, especially for PR3-positive disease (high relapse risk).
- Azathioprine 1 to 2 mg/kg/day — an alternative.
- Methotrexate or mycophenolate — alternatives if the first two are contraindicated. [1]
Relapse risk is highest in PR3-positive disease and in those who remain B-cell positive after rituximab. Monitor ANCA titres and clinical features (BVAS, renal function, urinalysis). [1]
Anti-GBM disease (Goodpasture syndrome)
Anti-GBM disease is the most aggressive of the pulmonary-renal syndromes. It is caused by autoantibodies against the alpha-3 chain of type IV collagen in the glomerular and alveolar basement membranes. [1]
Clinical presentation
- Peak incidence: 20 to 30 years (men) and 60 to 70 years (women). Smoking and respiratory infection increase the risk of pulmonary haemorrhage.
- Renal: Rapidly progressive GN — rising creatinine over days to weeks, with haematuria and proteinuria.
- Pulmonary: Haemoptysis (may be massive), dyspnoea, diffuse alveolar infiltrates on chest X-ray. Pulmonary involvement is present in 40 to 60% and is strongly associated with smoking and hydrocarbon exposure.
- Anti-GBM antibody: Positive in serum. The diagnosis is confirmed by biopsy showing linear IgG deposition along the GBM. [1]
Management
Anti-GBM disease is treated with plasma exchange (to remove circulating antibody), corticosteroids, and cyclophosphamide (to suppress antibody production): [1]
- Plasma exchange: Daily or alternate-day exchange of one plasma volume, replacing with 5% albumin (and fresh frozen plasma if bleeding risk), continued until anti-GBM antibody is undetectable (typically 2 to 3 weeks).
- Methylprednisolone pulses followed by oral prednisone 1 mg/kg/day.
- Cyclophosphamide 2 mg/kg/day orally for 3 to 6 months. [1]
Rituximab has a limited role — the goal is rapid antibody removal, which plasma exchange achieves directly. [1]
Prognosis and transplant considerations
If treated early (before oliguria or dialysis dependence), renal recovery is possible. Once a patient is dialysis-dependent at presentation with more than 85% crescents on biopsy, the chance of renal recovery is very low. Anti-GBM disease rarely recurs after transplantation if the antibody is undetectable for at least 6 to 12 months before transplant — this is a mandatory waiting period. [1]
Investigations: the integrated approach

Urine microscopy
The single most important bedside test. The nephritic sediment shows:
- Dysmorphic red blood cells — misshapen RBCs that have traversed the glomerular capillary wall (acanthocytes are highly specific for glomerular bleeding).
- Red blood cell casts — pathognomonic for glomerulonephritis. They dissolve rapidly, so the sample must be fresh and examined promptly by an experienced observer.
- Proteinuria — sub-nephrotic in pure nephritic, but can reach nephrotic range in mixed or advanced disease. [1]
Serological panel
Order the full panel at presentation — results take days, and treatment decisions cannot wait. [1]
| Test | What it tells you |
|---|---|
| C3 and C4 | Low C3 + normal C4: alternative pathway (PSGN, C3G). Low C3 + low C4: classical pathway (lupus, endocarditis, cryoglobulinaemia). Normal: IgA, AAV, anti-GBM |
| ANA and anti-dsDNA | Lupus nephritis (anti-dsDNA is highly specific) |
| ANCA (MPO and PR3) | ANCA-associated vasculitis (GPA is usually PR3; MPA and EGPA usually MPO) |
| Anti-GBM antibody | Goodpasture syndrome; check in all RPGN to identify double-positive disease |
| Serum IgA | Elevated in up to 50% of IgA nephropathy (not diagnostic alone) |
| Hepatitis B surface antigen, hepatitis C antibody, HIV | Screening for infection-associated GN (membranous, cryoglobulinaemia, MPGN-like) |
| ASO / anti-DNase B | Post-streptococcal GN (ASO for pharyngitis, anti-DNase B for skin infection) |
| Serum and urine electrophoresis, free light chains | Exclude paraprotein-associated disease in older patients |
| Cryoglobulins | Cryoglobulinaemic GN (hepatitis C-associated) |
Renal biopsy
The renal biopsy is the definitive diagnostic test. It requires:
- Light microscopy (haematoxylin-eosin, PAS, silver methenamine, trichrome, Jones) — to assess cellular proliferation, crescents, sclerosis, and tubulointerstitial damage.
- Immunofluorescence — to identify the pattern (linear, granular, pauci-immune) and the immunoglobulin and complement composition.
- Electron microscopy — to localise deposits (subepithelial, subendothelial, mesangial, intramembranous) and assess GBM architecture. [1]
Biopsy is indicated for: any unexplained RPGN; nephrotic syndrome in adults; persistent nephritic syndrome without a clear diagnosis; suspected lupus nephritis before committing to immunosuppression; and suspected vasculitis. It is generally not needed for classic paediatric PSGN with typical complement course. [1]
Long-term management and follow-up
Risk stratification and monitoring
Every patient with glomerulonephritis needs lifelong monitoring:
- Renal function (creatinine, eGFR) every 3 to 6 months, more frequently during active disease.
- Proteinuria (urine albumin-to-creatinine ratio or protein-to-creatinine ratio) — the most sensitive marker of disease activity and progression.
- Blood pressure — target below 130/80, or below 125/75 if proteinuric.
- ANCA / anti-dsDNA / anti-GBM titres — for the relevant disease, to detect relapse.
- Urine dipstick — at every visit, to detect recurrent haematuria or proteinuria. [1]
Progression to ESKD
The major glomerulonephritides carry different risks of ESKD:
- IgA nephropathy: 20 to 40% over 20 years (higher with proteinuria, hypertension, reduced GFR at diagnosis, and MEST-C T2/S1 lesions).
- Lupus nephritis class IV: 10 to 30% over 10 years despite treatment.
- ANCA vasculitis: 20 to 40% develop ESKD, highest with severe presentation (creatinine above 500 at diagnosis).
- Anti-GBM disease: 50 to 80% if dialysis-dependent at presentation. [1]
Transplant recurrence
Recurrence in the transplant graft is an important consideration: [1]
| Disease | Recurrence risk |
|---|---|
| IgA nephropathy | 20 to 50% histological recurrence, 10 to 15% clinical; recurrence is usually slow and graft-threatening over decades |
| Anti-GBM disease | Less than 5% if anti-GBM antibody undetectable for 6 to 12 months pre-transplant — mandatory waiting period |
| Lupus nephritis | Less than 5% — recurrence is uncommon |
| ANCA vasculitis | 15 to 20% clinical recurrence post-transplant; disease should be in remission |
| FSGS (for comparison) | 30 to 50% in primary FSGS — the highest recurrence rate |
Guideline controversies and regional differences
- Plasma exchange in AAV: MEPEX supported it; PEXIVAS challenged it. KDIGO 2024 retains plasma exchange for selected severe cases (creatinine above 500, dialysis-dependent, or pulmonary haemorrhage) but no longer recommends it routinely for all severe AAV.
- Corticosteroid dosing in AAV and lupus nephritis: PEXIVAS demonstrated that reduced-dose steroids are noninferior with fewer infections — practice has shifted to lower cumulative steroid exposure [3].
- Steroids in IgA nephropathy: TESTING supports low-dose steroids; STOP-IgAN challenges routine use. The consensus is targeted use in patients at genuine risk of progression, not reflexive prescribing [7][8][9].
- SGLT2 inhibitors in glomerular disease: DAPA-CKD and EMPA-KIDNEY included patients with IgA nephropathy and other chronic proteinuric glomerulonephritides, showing slowing of CKD progression independent of diabetes. SGLT2 inhibitors are now recommended in the KDIGO IgA chapter for proteinuric disease.
- Avacopan (ADVOCATE trial): The ADVOCATE trial of avacopan as a steroid-sparing agent in AAV was retracted in June 2026 after an FDA investigation found data integrity breaches (re-adjudication of primary endpoint assessments after database lock by unblinded sponsor personnel). Avacopan is no longer considered reliable evidence and should not be cited to justify steroid-sparing in AAV.
- Regional considerations: In ANZ, ANZSN and Kidney Health Australia endorse KDIGO with local protocols; the Euro-Lupus regimen is widely adopted; SGLT2 inhibitors are funded for proteinuric CKD. In the UK, NICE and the BSR/BHPR guidelines align with EULAR/ERA. In the US, the ACR/VF 2021 guidelines align with KDIGO.
DWE high-yield discriminators
- Synpharyngitic haematuria = IgA nephropathy. Latent haematuria (1 to 4 weeks after infection) = post-streptococcal GN. This timing distinction is one of the most tested facts.
- Low C3 + normal C4 = alternative pathway (PSGN, C3G). Low C3 + low C4 = classical pathway (lupus, endocarditis, cryoglobulinaemia). Normal complement = IgA, ANCA, anti-GBM.
- Linear IgG on IF = anti-GBM (Goodpasture). Granular = immune complex (lupus, IgA, PSGN). Pauci-immune = ANCA vasculitis.
- "Full house" immunofluorescence (IgG, IgA, IgM, C3, C1q all positive) = lupus nephritis.
- Subepithelial humps on EM = post-streptococcal GN.
- Double-positive ANCA + anti-GBM behaves as anti-GBM disease — plasma exchange is mandatory.
- PR3-ANCA = GPA (ENT disease, cavitating lung lesions). MPO-ANCA = MPA (renal-predominant, neuropathy, no granulomatous ENT disease).
- Crescents on biopsy = severe injury; demands urgent immunosuppression; fibrotic within weeks.
- Euro-Lupus regimen (500 mg cyclophosphamide IV q2weekly x6) is as effective as high-dose NIH cyclophosphamide with less toxicity — preferred for lupus nephritis induction in Caucasian populations.
- TESTING low-dose steroids (0.4 mg/kg/day) — the safe dose for IgA nephropathy; the high-dose protocol caused excess infections and deaths. [1]
DCE long-case integration
A complex glomerulonephritis long case will typically involve a patient with multisystem disease (lupus nephritis with hypertension, CKD, and immunosuppression complications; or ANCA vasculitis with pulmonary and renal involvement and treatment-related infections). The structured approach: [1]
- Opening statement (SASPOP): Symptoms, Age, Sex, Presentation, Occupation, Problems.
- Problem list — prioritised: the active disease (e.g., lupus nephritis class IV), the treatment complications (e.g., steroid-induced diabetes, PJP risk), the comorbidities (e.g., hypertension, CKD), the psychosocial (fertility, employment, adherence).
- Investigation interpretation — serology, biopsy, imaging.
- Integrated management plan — induction, supportive care, prophylaxis, monitoring, long-term follow-up.
- Communication — counselling on prognosis, fertility, immunosuppression risks, sick day rules. [1]
The key is to demonstrate that you can integrate nephrology, rheumatology, and general medicine — and that you understand the evidence behind every treatment decision. [1]
DCE short-case examination
In a nephrology short case, you may be asked to examine a patient with suspected glomerulonephritis or vasculitis: [1]
- General inspection: Cushingoid features (from steroids), oedema, pallor (anaemia of CKD), dialysis fistula.
- Hands: Nailfold infarcts (vasculitis), splinter haemorrhages (endocarditis or vasculitis), palmar erythema (lupus), joint deformities (lupus, rheumatoid).
- Face: Malar rash (lupus), oral ulcers (lupus, Behcet), saddle nose deformity (GPA), periorbital oedema.
- Neck: JVP (volume status), lymphadenopathy.
- Chest: Pulmonary crackles (pulmonary oedema or haemorrhage), pleural effusion (lupus, nephrotic).
- Abdomen: Ballotable kidneys (bilateral in polycystic disease, unilateral in tumour), renal bruits (renovascular), ascites (nephrotic), dialysis catheter or fistula.
- Neurological: Mononeuritis multiplex (vasculitis), sensory neuropathy.
- Legs: Palpable purpura (vasculitis), peripheral oedema. [1]
The presentation should lead with the findings, then offer a differential diagnosis, then the investigations you would order to confirm. See the viva and case artifacts for full model presentations. [1]
Key references and evidence summary
| Trial / Guideline | PMID | Key finding |
|---|---|---|
| KDIGO 2021 Glomerular Diseases | 34556256 | Comprehensive guidance for all glomerulonephritides [1] |
| MEPEX | 17582159 | Plasma exchange reduced ESKD in severe AAV (creatinine above 500) [2] |
| PEXIVAS | 32053298 | Plasma exchange did not reduce death/ESKD in AAV; reduced-dose steroids noninferior [3] |
| RAVE | 20647199 | Rituximab noninferior to cyclophosphamide for AAV induction; superior in relapse [4] |
| RITUXVAS | 20647198 | Rituximab plus cyclophosphamide pulses equivalent to standard cyclophosphamide in renal AAV [5] |
| Oxford classification | 19571790 | MEST-C scoring for IgA nephropathy prognosis [6] |
| TESTING (initial) | 28763548 | High-dose steroids reduced kidney failure but caused excess infections; trial halted [7] |
| TESTING (final) | 35579642 | Low-dose steroids (0.4 mg/kg/day) reduced kidney failure with improved safety [8] |
| STOP-IgAN | 26630142 | Immunosuppression added to supportive care did not improve renal outcomes [9] |
| Euro-Lupus Nephritis Trial | 19155235 | Low-dose cyclophosphamide as effective as high-dose for lupus nephritis [10] |
| ALMS (induction) | 19369404 | MMF equivalent to cyclophosphamide for lupus nephritis induction [11] |
| LUNAR | 22231479 | Rituximab did not meet primary endpoint in lupus nephritis induction [12] |
KDIGO 2021 Glomerular Diseases Guideline; EULAR/ERA Lupus Nephritis and AAV Recommendations; Kidney Health Australia Glomerulonephritis Guidance; ANZSN Clinical Practice Statements. [1]
References
- [1]Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases Kidney Int, 2021.PMID 34556256
- [2]Jayne DR, Gaskin G, Rasmussen N, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis J Am Soc Nephrol, 2007.PMID 17582159
- [3]Walsh M, Merkel PA, Peh CA, et al. Plasma Exchange and Glucocorticoids in Severe ANCA-Associated Vasculitis N Engl J Med, 2020.PMID 32053298
- [4]Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis N Engl J Med, 2010.PMID 20647199
- [5]Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis N Engl J Med, 2010.PMID 20647198
- [6]Roberts IS, Cook HT, Troyanov S, et al. The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility Kidney Int, 2009.PMID 19571790
- [7]Lv J, Zhang H, Wong MG, et al. Effect of Oral Methylprednisolone on Clinical Outcomes in Patients With IgA Nephropathy: The TESTING Randomized Clinical Trial JAMA, 2017.PMID 28763548
- [8]Lv J, Wong MG, Hladunewich MA, et al. Effect of Oral Methylprednisolone on Decline in Kidney Function or Kidney Failure in Patients With IgA Nephropathy: The TESTING Randomized Clinical Trial JAMA, 2022.PMID 35579642
- [9]Rauen T, Eitner F, Fitzner C, et al. Intensive Supportive Care plus Immunosuppression in IgA Nephropathy N Engl J Med, 2015.PMID 26630142
- [10]Houssiau FA, Vasconcelos C, D'Cruz D, et al. The 10-year follow-up data of the Euro-Lupus Nephritis Trial comparing low-dose and high-dose intravenous cyclophosphamide Ann Rheum Dis, 2010.PMID 19155235
- [11]Appel GB, Contreras G, Dooley MA, et al. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis J Am Soc Nephrol, 2009.PMID 19369404
- [12]Rovin BH, Furie R, Latinis K, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study Arthritis Rheum, 2012.PMID 22231479