Phys · renal
Nephrotic Syndrome
Also known as nephrosis · nephrotic-range proteinuria · minimal change disease · MCD · nil disease · focal segmental glomerulosclerosis · FSGS · membranous nephropathy · membranous glomerulonephritis · primary membranous nephropathy · PLA2R-associated membranous nephropathy · membranoproliferative glomerulonephritis · MPGN · diabetic nephropathy · amyloidosis · AL amyloid · AA amyloid · lupus nephritis · renal vein thrombosis · HIV-associated nephropathy · HIVAN · collapsing glomerulopathy
Consultant-physician-depth guide to nephrotic syndrome — definition (proteinuria greater than 3.5 g/day, hypoalbuminaemia, oedema, hyperlipidaemia), podocyte and slit-diaphragm pathophysiology, cause-by-age differential (minimal change disease, FSGS, membranous nephropathy with PLA2R antibody, membranoproliferative GN; secondary diabetic nephropathy, amyloidosis, lupus nephritis, viral-associated, malignancy-associated), complications (renal vein thrombosis, encapsulated-organism infection, hyperlipidaemia, AKI), investigation pathway (24-hour protein/PCR, biopsy, PLA2R antibody, viral and amyloid screens) and management (ACEi/ARB, cause-specific immunosuppression including rituximab for membranous — MENTOR and GEMRITUX). Structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Nephrotic Syndrome
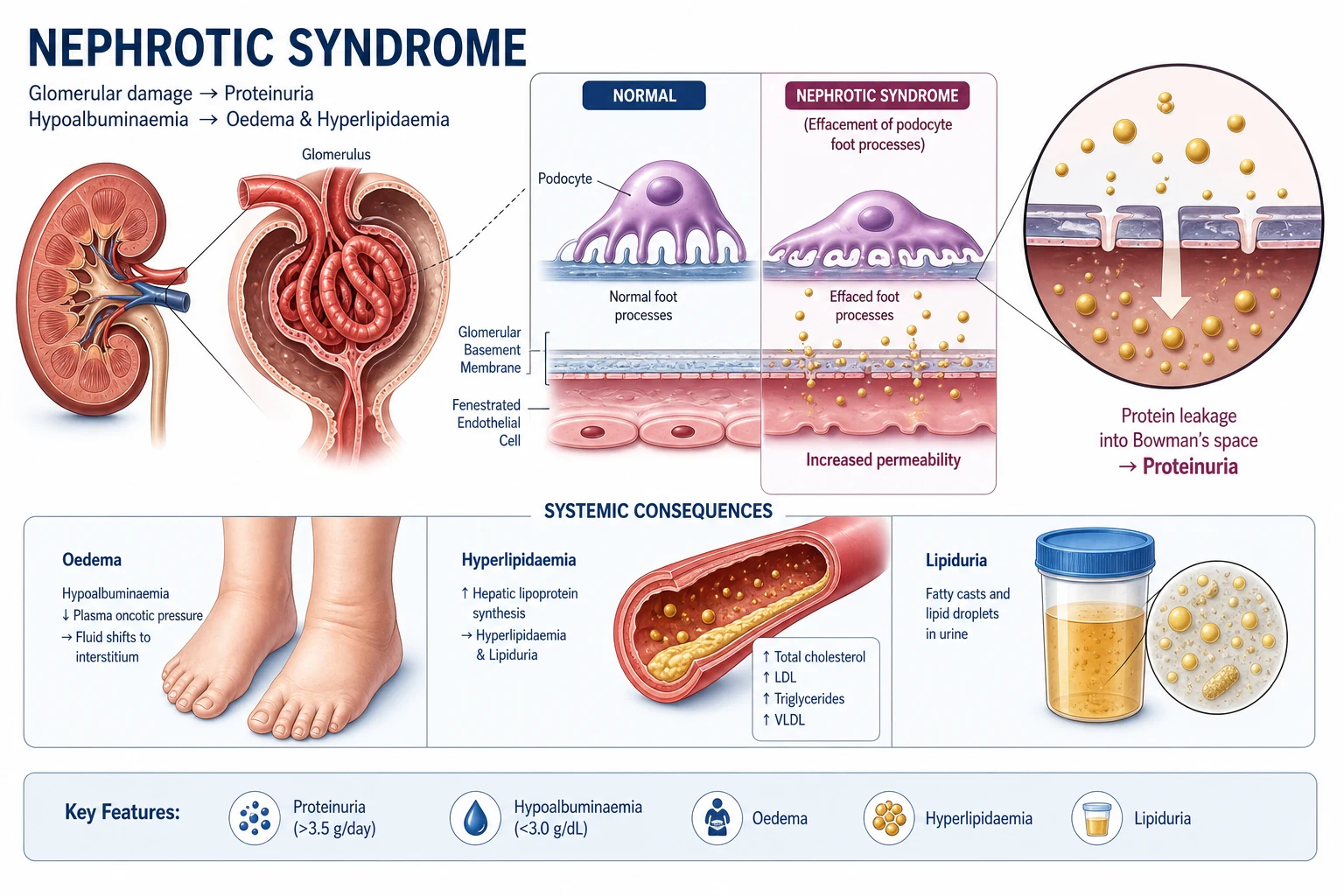
The answer first
Nephrotic syndrome is the clinical consequence of a disrupted glomerular filtration barrier producing nephrotic-range proteinuria — greater than 3.5 g per 24 hours (or a protein-to-creatinine ratio above approximately 350 mg/mmol) — accompanied by hypoalbuminaemia (albumin less than 30 g/L), peripheral oedema, and hyperlipidaemia [1].
Nephrotic syndrome is not a diagnosis. It is a glomerular phenotype, the way a pneumonia is a lung phenotype. Your job is to find the cause, because the cause determines the immunosuppression, the cancer screen, the prognosis, and the surveillance plan. [1]
The two clinical questions that dominate management: [1]
- What is causing the protein leak, and is it a primary glomerular disease or secondary to a systemic process? Primary causes (minimal change disease, FSGS, membranous nephropathy, membranoproliferative GN) have distinct immunosuppressive pathways. Secondary causes (diabetic nephropathy, amyloidosis, lupus, viral, malignancy) require treatment of the underlying disease.
- What complications of the nephrotic state are present or imminent, and how do I prevent them? Thromboembolism (especially renal vein thrombosis in membranous nephropathy), infection (encapsulated organisms from urinary immunoglobulin loss), hyperlipidaemia, AKI, and progressive CKD. [1]
The management mandate, in priority order: [1]
- Confirm the nephrotic state with quantified proteinuria (24-hour protein or PCR), serum albumin, renal function, and urine microscopy. Decide whether biopsy is indicated.
- Treat the cause — cause-specific immunosuppression for primary glomerular disease (steroids for minimal change disease, rituximab for membranous nephropathy per MENTOR [2]); treat the underlying systemic disease for secondary causes.
- Reduce proteinuria with an ACE inhibitor or ARB in every patient, titrated to maximum tolerated dose. This is a renal-protective intervention independent of blood pressure.
- Prevent and manage complications — anticoagulate when albumin is below 25 g/L (especially in membranous nephropathy), give pneumococcal and influenza vaccination, manage oedema with salt restriction and loop diuretics, treat hyperlipidaemia with a statin.
- Protect the long-term kidney — manage volume and blood pressure, avoid nephrotoxins (NSAIDs), and surveil for progression to CKD.
The single most important exam principle: the biopsy answer, the PLA2R antibody, and the cause-by-age distribution are the three highest-yield discriminators. A child with nephrotic syndrome is treated empirically with steroids; an adult with new nephrotic syndrome is biopsied and screened for secondary causes before any immunosuppression. [1]
Definition and the glomerular filtration barrier
The diagnostic tetrad
Nephrotic syndrome is defined by a reproducible set of findings that reflect a single defect — failure of the glomerular filtration barrier to retain albumin-sized proteins: [1]
| Feature | Threshold | Mechanism |
|---|---|---|
| Proteinuria | Greater than 3.5 g/24h or PCR greater than 350 mg/mmol (approximately ACR greater than 220 mg/mmol) | Loss of size-selective barrier |
| Oedema | Dependent, often periorbital in children; anasarca in severe cases | Hypoalbuminaemia lowers oncotic pressure; primary renal sodium retention |
| Hyperlipidaemia | Total cholesterol and LDL elevated; often markedly | Hepatic upregulation of lipoprotein synthesis in response to low oncotic pressure |
Two additional features clinch the diagnosis: lipiduria (oval fat bodies, "Maltese cross" appearance under polarised light) and a tendency to a thrombotic and infective diathesis. [1]
DWE exam trap: The 3.5 g/24h threshold is per 1.73 m² body surface area, but in practice the uncorrected value is used in adults. The threshold defines nephrotic-range proteinuria; full nephrotic syndrome also requires hypoalbuminaemia and oedema. A patient leaking 4 g of protein with a normal albumin (e.g., early diabetic nephropathy) has nephrotic-range proteinuria, not nephrotic syndrome. [1]
Nephrotic versus nephritic — the bedside discriminator
The glomerular pattern of injury falls on a spectrum. Nephrotic and nephritic syndromes are the two ends, and many diseases (membranoproliferative GN, lupus nephritis) sit in between. The distinction guides the differential and the biopsy urgency. [1]
| Feature | Nephrotic | Nephritic |
|---|---|---|
| Proteinuria | Nephrotic range (greater than 3.5 g/day) | Sub-nephrotic (less than 3.5 g/day) |
| Blood pressure | Often normal | Hypertension |
| GFR | Normal or low | Low (acute fall) |
| Oedema | Soft, dependent, periorbital | Present |
| Complement | Usually normal | Often low (low C3 in MPGN, post-infectious; low C3-C4 in lupus, cryoglobulinaemia) |
A patient with heavy proteinuria, brown urine, red cell casts, hypertension and a rising creatinine has a nephritic or mixed picture. That pushes you towards rapidly progressive glomerulonephritis, lupus, IgA nephropathy, or post-infectious GN — and demands an urgent biopsy, not empiric steroid treatment. [1]
Pathophysiology — the podocyte and the slit diaphragm
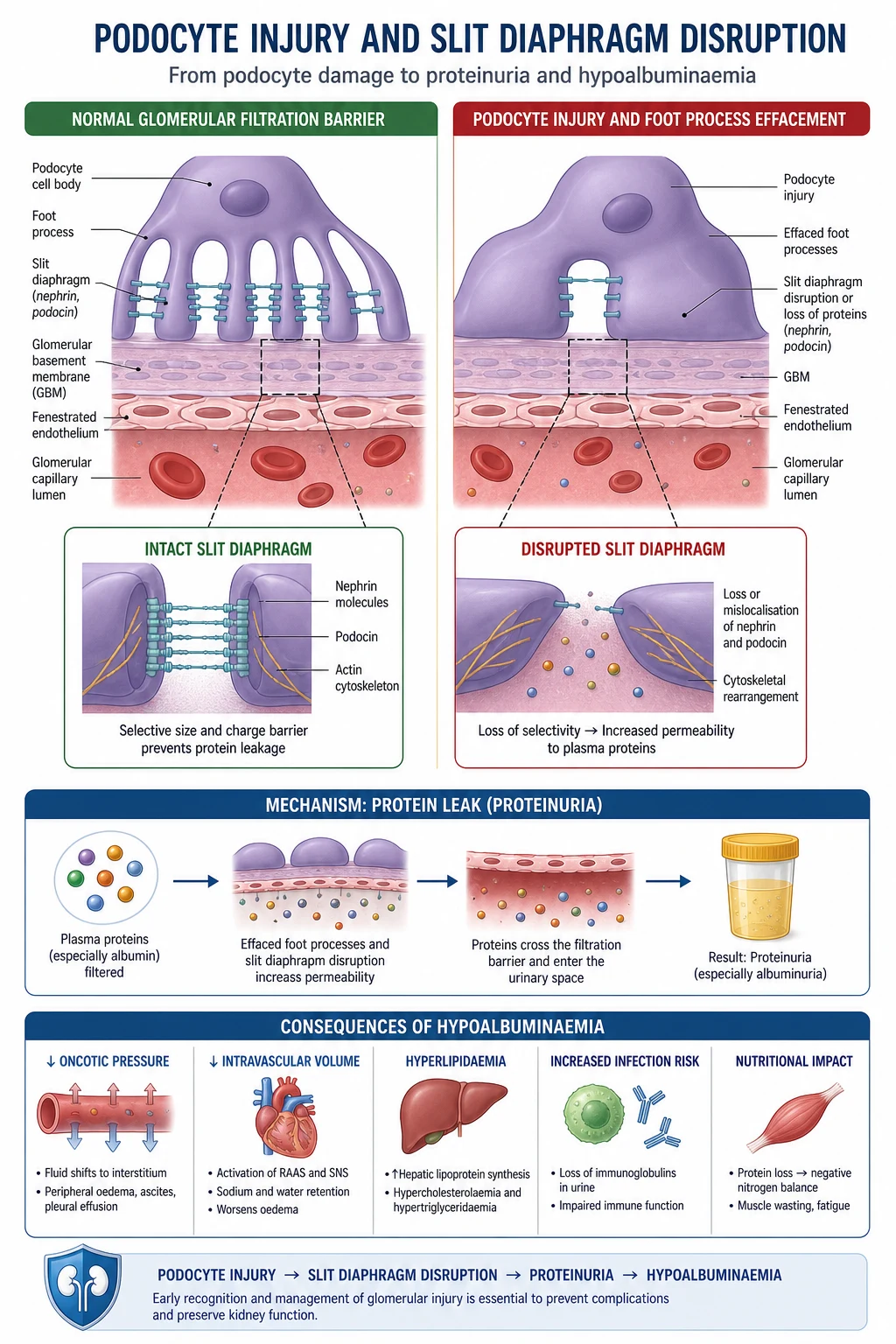
The glomerular filtration barrier
The barrier that holds back albumin has three layers. All three must be intact: [1]
- Fenestrated endothelium — the inner cell layer with pores that allow water and small solutes through but resist cells.
- Glomerular basement membrane (GBM) — a negatively charged collagen-IV and proteoglycan meshwork. Charge selectivity repels albumin (which is negatively charged at physiological pH).
- Podocytes — epithelial cells with interdigitating foot processes connected by the slit diaphragm, a specialised protein complex (nephrin, podocin, CD2AP, alpha-actinin-4). [1]
Nephrotic-range proteinuria arises when any layer is disrupted. In practice, the podocyte and its slit diaphragm are the common final pathway. The hallmark of a proteinuric glomerular disease on electron microscopy is foot process effacement — the delicate interdigitating foot processes flatten and fuse, the slit diaphragm is lost, and albumin pours through. [1]
Why podocyte injury is central
Podocytes are terminally differentiated epithelial cells with a very limited capacity to replicate. When they are injured — by immune attack (membranous, minimal change), by genetic defects (nephrin, podocin, alpha-actinin-4 mutations), by circulating permeability factors (FSGS), by viral infection (HIV), or by hyperfiltration and mechanical stress — they undergo one of three responses: [1]
- Foot process effacement — reversible, the signature of minimal change disease and the active phase of any proteinuric disease.
- Detachment from the GBM — the podocyte is lost into the urine. The denuded GBM adheres to the Bowman capsule, and a focal segmental scar (FSGS) forms.
- Apoptosis or maladaptive proliferation — collapsing glomerulopathy, where podocytes dedifferentiate and proliferate, collapsing the capillary tuft. [1]
This is why podocyte loss is irreversible. The clinical implication: the longer a patient leaks heavy protein, the more podocytes are lost, the more sclerosis accrues, and the harder it is to recover kidney function. Reducing proteinuria — with an ACE inhibitor, with immunosuppression, with an SGLT2 inhibitor — protects podocytes. [1]
The sodium-retention mechanism of oedema
Oedema in nephrotic syndrome was once taught as a simple "underfill" model: albumin leaks, plasma oncotic pressure falls, fluid shifts to the interstitium, effective circulating volume falls, the kidney retains sodium. This is part of the story, but modern evidence favours a primary renal sodium retention mechanism — the damaged glomerulus leaks proteases (plasmin) that activate epithelial sodium channels (ENaC) in the collecting duct, driving sodium retention directly. The two mechanisms coexist. The practical implication: a loop diuretic alone may not suffice; combination with an aldosterone antagonist or a thiazide may be needed for diuretic-resistant oedema. [1]
Causes by age and type
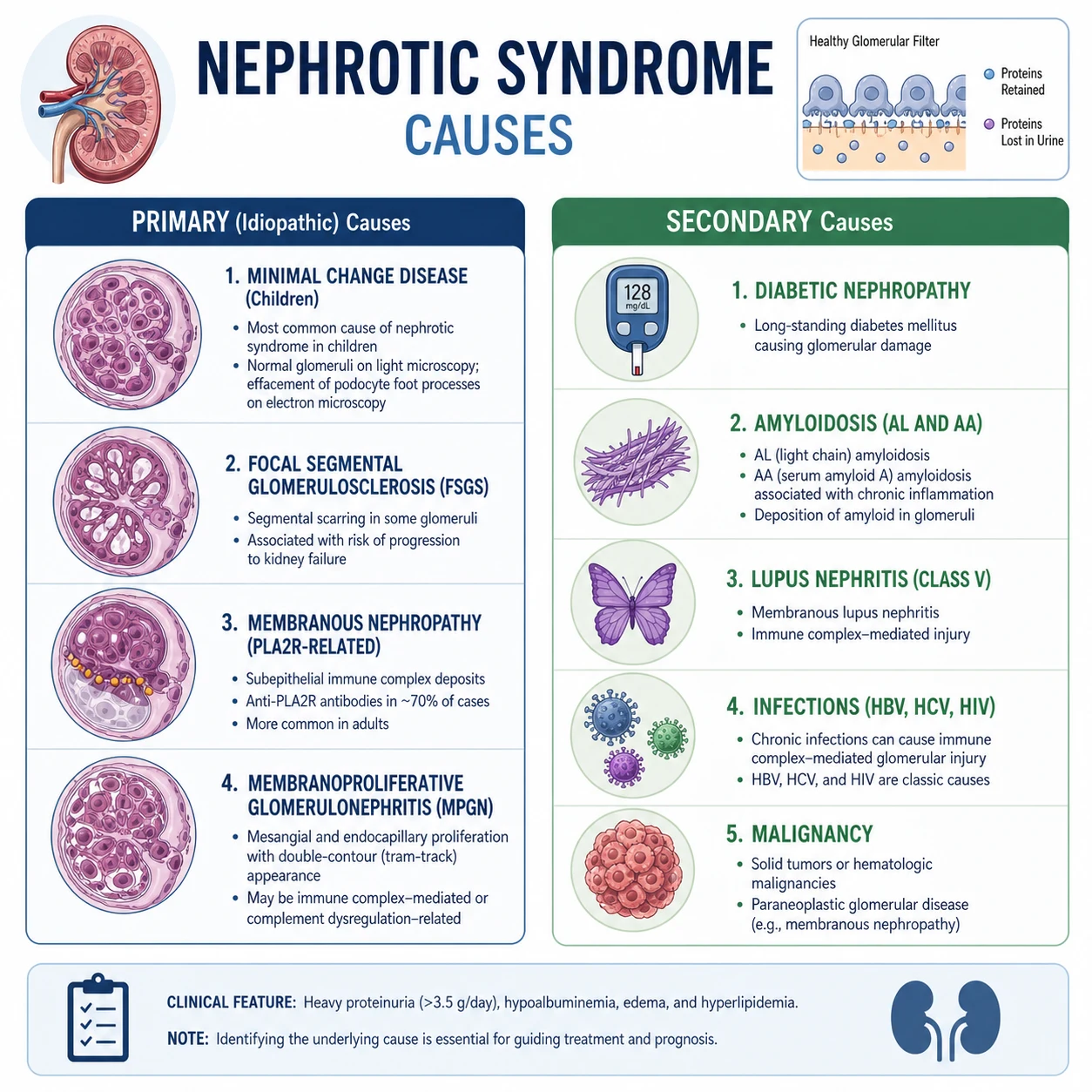
The single highest-yield discriminator in any nephrotic syndrome stem is the patient's age and the primary-versus-secondary split. Memorise the table below. [1]
Children (1 to 10 years)
In a child presenting with their first episode of nephrotic syndrome, the overwhelming likelihood is minimal change disease (MCD) — approximately 70 to 80 per cent of paediatric cases [1]. MCD is steroid-responsive in over 90 per cent, so the standard ANZ and international practice is to treat empirically with high-dose oral prednisolone and biopsy only if steroid-resistant or atypical (haematuria, low complement, hypertension, renal impairment, age over 10 or under 1). The other paediatric causes are FSGS (10 to 15 per cent, and the most common cause of steroid resistance) and membranoproliferative GN.
Adults
The adult distribution inverts. Membranous nephropathy is the most common cause of primary nephrotic syndrome in Caucasian adults; FSGS is the most common in African-American and Hispanic populations and is rising globally; minimal change disease accounts for 10 to 15 per cent in adults. Secondary causes become prominent — diabetic nephropathy is the most common cause of nephrotic-range proteinuria in adults overall, and amyloidosis, lupus nephritis, and viral-associated disease (hepatitis B and C, HIV) must always be sought. [1]
The complete differential
| Cause | Age peak | Hallmarks | Key test |
|---|---|---|---|
| Minimal change disease | Children 2 to 6; adults 30 to 40 | Selective proteinuria, normal BP, normal GFR, no haematuria; steroid-responsive | Trial of steroids; biopsy if atypical or resistant |
| FSGS (primary) | Young adults; African ancestry | Non-selective proteinuria, may have haematuria, hypertension, reduced GFR; steroid-resistant in many | Biopsy; APOL1 genotyping in African ancestry |
| Collapsing FSGS / HIVAN | African ancestry, HIV | Rapid progression to kidney failure; collapsing tufts on biopsy | HIV test; APOL1 |
| Membranous nephropathy (primary) | Adults 40 to 60 | Insidious oedema, highest thrombotic risk (renal vein thrombosis) | Anti-PLA2R antibody [4] |
| Membranoproliferative GN | Adolescents, young adults | Mixed nephritic-nephrotic; low complement | C3, C4; biopsy; cryoglobulins, HCV |
| Diabetic nephropathy | Adults with long-standing diabetes | Diabetic retinopathy, slowly progressive albuminuria, no need to biopsy if classic | Long-standing diabetes, retinopathy |
| Amyloidosis (AL) | Older adults (over 50) | Massive proteinuria, minimal haematuria, multi-organ (heart, nerve, gut) | Serum/urine free light chains; biopsy with Congo red |
| Amyloidosis (AA) | Any age with chronic inflammation | Chronic inflammatory disease (rheumatoid, IBD, familial Mediterranean fever) | Serum amyloid A; biopsy |
| Lupus nephritis (class V) | Young women | Multi-system, ANA, anti-dsDNA, low complement | ANA, anti-dsDNA, C3, C4; biopsy |
| Hepatitis B-associated | Adults from endemic regions | Membranous or MPGN pattern; chronic HBV | HBsAg, anti-HBc |
| Hepatitis C-associated | Adults; cryoglobulinaemia | MPGN with low C3-C4; cryoglobulins | Anti-HCV, HCV RNA, cryoglobulins |
| Malignancy-associated | Adults over 50 | Membranous in solid tumours (lung, colon, prostate, breast); MCD in Hodgkin lymphoma | Age-appropriate cancer screen |
DWE exam trap: The most common cause of nephrotic syndrome in children is minimal change disease. The most common cause of primary nephrotic syndrome in adults is membranous nephropathy. The most common cause of nephrotic-range proteinuria in adults overall is diabetic nephropathy. Three different answers — read the question carefully. [1]
Minimal change disease — the steroid-responsive archetype
MCD is defined histologically by normal light microscopy (hence "minimal change" and the older name "nil disease"), no immune deposits on immunofluorescence, and diffuse foot process effacement on electron microscopy. The pathophysiology is thought to be a T-cell-mediated release of a circulating permeability factor (the candidate is haemopoietic growth factor — but the precise factor remains unconfirmed). [1]
Clinically: a child (typically 2 to 6 years) presents with rapid onset of periorbital and dependent oedema, often after a viral upper respiratory tract infection. Blood pressure and renal function are usually normal. The proteinuria is selective (mostly albumin, little IgG). Treatment is oral prednisolone 60 mg/m²/day (maximum 80 mg/day) for 4 to 6 weeks, then alternate day taper — over 90 per cent achieve complete remission within 8 weeks. Adults are treated with the same agent but the response is slower (up to 16 weeks) and biopsy is performed first. [1]
Relapse is common (over 70 per cent of children relapse). Frequently relapsing or steroid-dependent disease moves to steroid-sparing agents: calcineurin inhibitors (cyclosporin, tacrolimus), mycophenolate mofetil, and rituximab — the Iijima trial established rituximab as effective in frequently-relapsing childhood nephrotic syndrome [6].
FSGS — the podocyte scar
FSGS is a histological pattern, not a single disease. A segment of a glomerulus is sclerosed (scarred), and the scarring is focal (some glomeruli) and segmental (part of each tuft). The shared lesion is podocyte loss. The Columbia classification recognises five variants: collapsing, tip, cellular, perihilar, and not-otherwise-specified. The variant matters — the collapsing variant is aggressive, often HIV-associated, APOL1-driven in African ancestry, and progresses rapidly to kidney failure without intervention. [1]
Primary (idiopathic) FSGS is thought to be caused by a circulating permeability factor, which is why it can recur within hours of kidney transplantation. Secondary FSGS is an adaptive response to reduced nephron number (obesity, reflux nephropathy, sickle cell, single kidney) or to adaptive hyperfiltration — these forms do not respond to immunosuppression and are managed with ACE inhibition and addressing the underlying driver. [1]
Treatment of primary FSGS: steroids first-line (prednisolone 1 mg/kg/day for up to 16 weeks), with calcineurin inhibitors or mycophenolate for steroid-resistant or steroid-dependent disease. Prognosis correlates with proteinuria — patients who achieve remission do well; those with persistent nephrotic-range proteinuria progress to kidney failure in 5 to 10 years. [1]
Membranous nephropathy — the PLA2R archetype
Primary membranous nephropathy (PMN) is an autoimmune disease. In 2009, Beck and colleagues identified the M-type phospholipase A2 receptor (PLA2R) on podocytes as the target antigen in approximately 70 to 80 per cent of primary cases [4]. Anti-PLA2R antibodies are now a diagnostic, activity-monitoring, and relapse-predicting biomarker — they appear before clinical relapse, fall with immunosuppression, and are absent in secondary membranous nephropathy. A second antigen, THSD7A, accounts for a further 2 to 5 per cent of primary cases.
Histologically: diffuse thickening of the GBM on light microscopy, granular IgG and C3 deposition along the capillary loops on immunofluorescence, and subepithelial immune deposits with spike formation on electron microscopy. [1]
Clinically: an adult (peak 40 to 60) presents with insidious onset of oedema, nephrotic-range proteinuria, and the highest thrombotic risk of any glomerular disease — renal vein thrombosis is sufficiently common that any acute flank pain in membranous nephropathy demands imaging. About one-third of patients undergo spontaneous remission; one-third have persistent proteinuria; one-third progress to kidney failure. [1]
The anti-PLA2R antibody test has transformed diagnosis: a positive anti-PLA2R in the right clinical context can secure the diagnosis without biopsy, though biopsy remains the gold standard and is required to grade and exclude superimposed lesions. Crucially, anti-PLA2R must be checked before diagnosing primary disease — its presence argues strongly for primary membranous nephropathy; its absence obliges you to search for a secondary cause (malignancy, infection, drugs, autoimmune). [1]
Membranoproliferative GN — the mixed pattern
MPGN is the classic nephritic-nephrotic overlap. Light microscopy shows mesangial hypercellularity and GBM doubling ("tram-track" appearance). Historically classified by electron microscopy into Type I (subendothelial immune deposits), Type II (dense deposit disease, driven by complement dysregulation), and Type III. The modern immunofluorescence-based classification divides MPGN into immune-complex mediated (usually infection- or autoimmune-driven, immunoglobulin-dominant) and complement-mediated (C3-dominant, driven by alternative pathway dysregulation). This matters because complement-mediated disease is treated with eculizumab or other complement inhibitors, not conventional immunosuppression. [1]
Low complement is the clue: low C3 with normal C4 suggests alternative pathway / classical bypass (C3 glomerulopathy, post-streptococcal in late recovery); low C3 and low C4 suggests classical pathway activation (lupus, cryoglobulinaemia, hepatitis C, endocarditis). [1]
Secondary causes — the search that must not be missed
Before any immunosuppression, every adult with new nephrotic syndrome must be screened for secondary causes. Treating the primary disease without addressing a secondary driver is a fundamental error. [1]
Diabetic nephropathy
The most common cause of nephrotic-range proteinuria in adults. The diagnosis is clinical in a patient with long-standing diabetes (typically over 10 years), diabetic retinopathy, slowly progressive albuminuria progressing to nephrotic-range, and no need to biopsy if the picture is classic. Biopsy is reserved for atypical features (rapid onset, active sediment, falling GFR out of proportion, absence of retinopathy, signs of another systemic disease). Management is glycaemic and blood pressure control, ACE inhibitor or ARB, and now an SGLT2 inhibitor per KDIGO 2024 [9]. See the dedicated diabetic nephropathy topic.
Amyloidosis
Amyloidosis produces massive, often nephrotic-range proteinuria with remarkably little haematuria — the kidney is stiffened by extracellular amyloid deposition. Two types: [1]
- AL amyloid (primary) — a plasma-cell dyscrasia producing monoclonal light chains that misfold into amyloid. Screen with serum and urine electrophoresis plus serum free light chains in any adult over 50 with nephrotic syndrome and other organ involvement (cardiomyopathy with restrictive physiology, macroglossia, peripheral or autonomic neuropathy, easy bruising).
- AA amyloid (secondary) — produced in chronic inflammatory states (rheumatoid arthritis, inflammatory bowel disease, chronic infection, familial Mediterranean fever, osteomyelitis). Screen with serum amyloid A; look for a chronic inflammatory history. [1]
Diagnosis is by biopsy with Congo red staining (apple-green birefringence under polarised light) and amyloid typing by mass spectrometry. Kidney biopsy shows amyloid in the mesangium and vessels; electron microscopy shows characteristic randomly arranged 8 to 12 nm fibrils. Treatment is of the underlying plasma-cell clone (AL — bortezomib-based, now daratumumab) or inflammatory drive (AA — anti-IL-1, anti-IL-6, anti-TNF). Prognosis has improved markedly with modern haematology regimens. [1]
Lupus nephritis
Systemic lupus erythematosus with glomerular involvement produces a spectrum from class I (mesangial) to class VI (advanced sclerosing). Class V lupus nephritis (membranous) produces a pure nephrotic syndrome and is clinically and histologically similar to primary membranous nephropathy — the discriminator is the ANA, anti-dsDNA, and low complement (low C3, low C4). Biopsy with immunofluorescence shows "full house" deposition (IgG, IgA, IgM, C3, C1q) — the C1q positivity distinguishes lupus from primary membranous. Treatment is immunosuppression (mycophenolate mofetil or cyclophosphamide with corticosteroids for induction, mycophenolate or azathioprine for maintenance). [1]
Viral-associated nephrotic syndrome
- Hepatitis B — classically causes membranous nephropathy in children from endemic regions, and MPGN in adults. Screen with HBsAg and anti-HBc.
- Hepatitis C — causes MPGN with cryoglobulinaemia, low C3 and C4, and a mixed nephritic-nephrotic picture. Screen with anti-HCV and HCV RNA. Direct-acting antiviral cure of HCV can resolve the glomerular disease.
- HIV — causes HIV-associated nephropathy (HIVAN), a collapsing variant of FSGS seen overwhelmingly in patients of African ancestry (APOL1 risk alleles). Presents with heavy proteinuria and rapid progression to kidney failure. The treatment is antiretroviral therapy plus an ACE inhibitor — the introduction of ART has dramatically reduced HIVAN incidence. [1]
Malignancy-associated nephrotic syndrome
A paraneoplastic membranous nephropathy occurs in solid tumours (lung, colon, prostate, breast, stomach), typically in adults over 50 — the nephrotic syndrome may precede the cancer diagnosis by months. Hodgkin lymphoma is classically associated with minimal change disease (the lymphoma produces a permeability factor). Any older patient with membranous nephropathy and a negative anti-PLA2R, or MCD with systemic symptoms, should have an age-appropriate malignancy screen — and re-screening at intervals, because occult cancers can declare later. [1]
Complications of the nephrotic state
The nephrotic state carries predictable complications independent of the underlying cause. Every nephrotic patient must be assessed for each. [1]
Thromboembolism and renal vein thrombosis
Nephrotic syndrome is an acquired hypercoagulable state. The risk of venous thromboembolism (DVT, pulmonary embolism, and renal vein thrombosis) is highest in membranous nephropathy, where the annual incidence of thromboembolic events approaches 10 per cent. [1]
The mechanism is multifactorial and classically taught as: [1]
- Urinary loss of antithrombin (molecular weight 58 kDa, small enough to leak) — reduced plasma antithrombin lowers anticoagulant capacity [7].
- Urinary loss of free protein S — a cofactor in the protein C anticoagulant pathway.
- Hepatic upregulation of procoagulants (fibrinogen, factor VIII, von Willebrand factor) in response to low oncotic pressure.
- Platelet activation and aggregation, impaired fibrinolysis, and volume contraction.
Modern evidence has nuanced the antithrombin story — multi-cohort work suggests antithrombin levels do not perfectly correlate with thrombotic risk, and the mechanism is more complex than the single-factor model [8]. For the exam, however, the antithrombin loss in urine remains the expected answer for "why is nephrotic syndrome hypercoagulable?".
Renal vein thrombosis (RVT) is the signature complication. It presents with acute or subacute flank pain, gross or microscopic haematuria, and a sudden fall in GFR — though it can be entirely asymptomatic and found on imaging. Membranous nephropathy is the highest-risk substrate. Diagnosis is by CT renal venography or magnetic resonance venography (ultrasound is insensitive). Treatment is anticoagulation with warfarin or a DOAC for at least the duration of the nephrotic state, often long-term. [1]
Prophylactic anticoagulation is recommended by KDIGO and major guidelines for patients at high risk — albumin less than 25 to 30 g/L, particularly in membranous nephropathy. Several risk models exist (the Lee / Sarasin model uses albumin, proteinuria, and history of thromboembolism to estimate risk) to guide the decision. [1]
Infection — encapsulated organisms
Nephrotic patients are immunocompromised through urinary loss of immunoglobulins (IgG), alternative complement pathway factors (factor B, factor D), and the immunosuppressive therapy they often receive. The classic infective threats are: [1]
- Encapsulated bacteria — Streptococcus pneumoniae, Haemophilus influenzae, Neisseria — from loss of IgG and alternative pathway factors. Spontaneous bacterial peritonitis (in nephrotic children with ascites), cellulitis, and pneumonia are the classic presentations.
- Peritonitis — a nephrotic child with abdominal pain and ascites has spontaneous bacterial peritonitis until proven otherwise; treat empirically.
- Viral — cellulitis from infection, and in immunosuppressed patients, opportunistic infection (PCP, CMV). [1]
Prevention is mandatory: pneumococcal vaccination (23-valent polysaccharide, with 13-valent conjugate in some schedules), annual influenza vaccination, hepatitis B vaccination, and varicella vaccination in children between relapses. Avoid live vaccines during active immunosuppression. [1]
Hyperlipidaemia and cardiovascular risk
The nephrotic state upregulates hepatic lipoprotein synthesis, producing marked hypercholesterolaemia and elevated LDL, with elevated triglycerides and reduced HDL. This is atherosclerotic and accelerates cardiovascular risk. Treat with a statin per standard cardiovascular risk guidelines; the threshold is lower in nephrotic patients because of the combined proteinuria and lipid risk. The lipid abnormality resolves with remission of the nephrotic state. [1]
Acute kidney injury
AKI complicates nephrotic syndrome through several mechanisms: intravascular volume depletion from over-diuresis, intrinsic tubular injury (the heavy protein load is tubulotoxic), bilateral renal vein thrombosis, interstitial nephritis (often from diuretics, antibiotics, or PPIs), and superimposed ATN from sepsis or hypoperfusion. The approach is to reassess volume, hold nephrotoxins, image for RVT if the GFR fall is unexplained, and support the kidney while treating the cause. Bilateral RVT is a reversible cause of AKI and must not be missed. [1]
Vitamin D deficiency, bone disease and other losses
Nephrotic syndrome loses vitamin D-binding protein (and hence 25-hydroxyvitamin D) and transferrin (contributing to a microcytic anaemia that is not iron-responsive in the usual way), thyroxine-binding globulin (altering thyroid function tests), and ceruloplasmin (altering caeruloplasmin-based copper indices). Vitamin D deficiency and secondary hyperparathyroidism are common; supplement cholecalciferol. [1]
Investigations — the nephrotic workup
Quantify the proteinuria
| Test | What it tells you | Practical point |
|---|---|---|
| 24-hour urine protein | The gold standard for nephrotic-range (greater than 3.5 g/24h) | Collect accurately; simultaneous creatinine clearance checks adequacy |
| Albumin-to-creatinine ratio (ACR) | Preferred for diabetic nephropathy and early detection | ACR greater than 220 mg/mmol is roughly nephrotic-range |
| Urine microscopy | Distinguishes nephrotic (bland) from nephritic (dysmorphic red cells, casts) | Oval fat bodies and Maltese crosses under polarised light are lipiduria |
Blood panel
- Full blood count — anaemia (may be from inflammation, iron loss, or amyloid bone marrow infiltration); a high eosinophil count suggests eosinophilic interstitial nephritis.
- Albumin, total protein, renal function, electrolytes — define severity and renal function.
- Complement C3 and C4 — low C3 suggests MPGN, post-infectious GN, C3 glomerulopathy; low C3 and C4 suggests lupus, cryoglobulinaemia, hepatitis C, endocarditis. Normal complement favours MCD, FSGS, membranous.
- ANA, anti-dsDNA, ANA pattern — lupus screen.
- Serum and urine electrophoresis, serum free light chains — plasma-cell dyscrasia, AL amyloid.
- Hepatitis B (HBsAg, anti-HBc), hepatitis C (anti-HCV), HIV — viral-associated nephropathy.
- Anti-PLA2R antibody — primary membranous nephropathy [4]; obviates biopsy if positive in a typical presentation.
- Fasting lipids, HbA1c — cardiovascular risk and diabetes screen.
- Serum amyloid A — if AA amyloid suspected.
- Cryoglobulins — if MPGN with low complement, hepatitis C, or purpura.
Renal biopsy
In adults with new nephrotic syndrome, biopsy is the standard — the histological pattern determines the cause and the immunosuppression. The exceptions are the child with classic steroid-responsive MCD (treat empirically) and the adult with classic diabetic nephropathy (no biopsy if retinopathy and slowly progressive albuminuria). Biopsy provides light microscopy (architecture), immunofluorescence (immune complex pattern — "full house" for lupus, granular capillary-loop IgG for membranous, mesangial IgA for IgA nephropathy), and electron microscopy (location of deposits and foot process effacement). [1]
Imaging
- Renal ultrasound — kidney size, cortical thickness, exclude obstruction. Large kidneys suggest amyloidosis, HIVAN, diabetic nephropathy; small kidneys suggest chronic irreversible disease (and may make biopsy risky).
- CT or MR renal venography — if renal vein thrombosis suspected (acute flank pain, sudden GFR fall, membranous nephropathy).
- CT chest / age-appropriate malignancy screen — if malignancy-associated membranous or MCD suspected. [1]
DWE exam trap: A positive anti-PLA2R antibody is highly specific for primary membranous nephropathy and is absent in secondary membranous nephropathy (malignancy, infection, drugs). A negative anti-PLA2R does not exclude primary membranous — about 20 to 30 per cent are PLA2R-negative (some are THSD7A-positive). If PLA2R is negative, biopsy and search hard for secondary causes. [1]
Management — the integrated plan
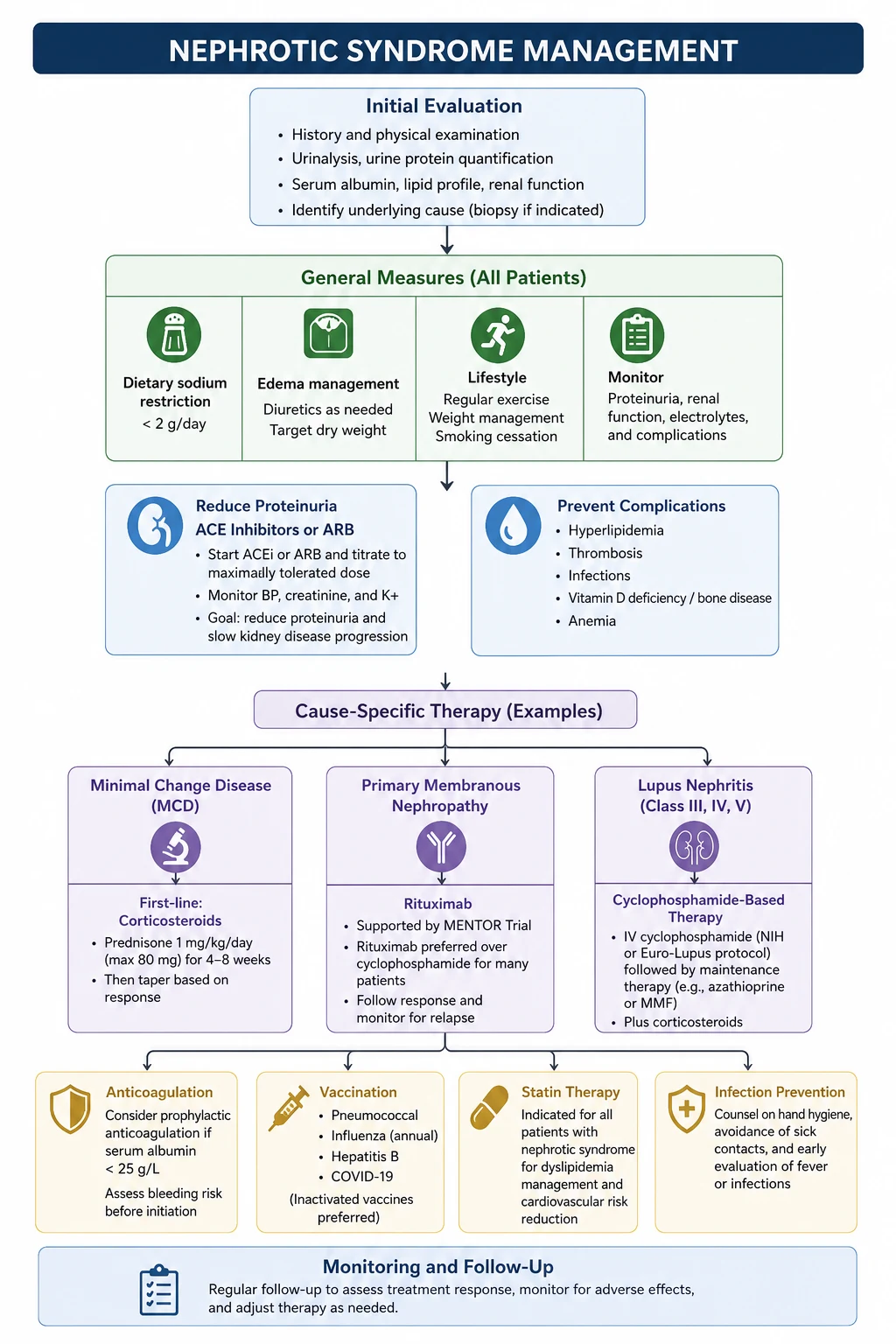
Management has four limbs that run in parallel: (1) cause-specific treatment, (2) anti-proteinuric therapy, (3) oedema and volume control, (4) complication prevention. [1]
Limb 1 — Cause-specific treatment
The cause determines the immunosuppression. Treat empirically only where the evidence supports it (childhood MCD); otherwise biopsy and tailor. [1]
| Cause | First-line immunosuppression | Key evidence |
|---|---|---|
| Minimal change disease | Oral prednisolone 1 mg/kg/day (60 mg/m² in children, max 80 mg) for 4 to 8 weeks, then taper | Steroid response over 90 per cent in children |
| FSGS (primary) | Prednisolone 1 mg/kg/day for up to 16 weeks; calcineurin inhibitor (cyclosporin or tacrolimus) if steroid-resistant | KDIGO 2021 [1] |
| Membranous nephropathy (primary) | Rituximab is preferred first-line immunosuppression in most patients; cyclophosphamide plus corticosteroids (modified Ponticelli) and calcineurin inhibitors are alternatives | MENTOR [2]; GEMRITUX [3]; STARMEN [5] |
| MPGN | Treat the underlying cause (HCV with DAAs, lupus with mycophenolate/cyclophosphamide); complement-mediated disease with eculizumab | KDIGO 2021 [1] |
| Diabetic nephropathy | Glycaemic control, ACEi/ARB, SGLT2 inhibitor; no immunosuppression | KDIGO 2024 [9] |
| Amyloidosis (AL) | Treat the plasma-cell clone (bortezomib-based, daratumumab) | Haematology-guided |
| Lupus nephritis (class V) | Mycophenolate mofetil or calcineurin inhibitor with corticosteroids for induction; mycophenolate for maintenance | KDIGO 2024 lupus update |
| HIVAN | Antiretroviral therapy plus ACE inhibitor | ART era |
Membranous nephropathy immunosuppression in depth
Membranous nephropathy is the highest-yield immunosuppression topic for the exam. Three randomised trials define the modern landscape: [1]
- GEMRITUX (Dahan 2017) — rituximab (375 mg/m² on days 1 and 8) versus non-immunosuppressive antiproteinuric treatment. The primary endpoint at 6 months was not met, but extended follow-up showed significantly higher remission with rituximab, with anti-PLA2R antibody depletion preceding remission [3].
- MENTOR (Fervenza 2019) — rituximab (1 g IV on days 1 and 15, with a second course at 6 months if needed) versus cyclosporine. Rituximab was non-inferior at 6 months and superior at 24 months, with far fewer relapses and a better safety profile than cyclosporine [2]. This established rituximab as the preferred first-line immunosuppression in most patients with primary membranous nephropathy and nephrotic syndrome.
- STARMEN (2020) — cyclical corticosteroid-cyclophosphamide (modified Ponticelli) versus sequential tacrolimus-rituximab. The cyclophosphamide regimen was superior in remission rate, but at the cost of more toxicity [5].
Who to immunosuppress? KDIGO 2021 recommends immunosuppression for patients at moderate-to-high risk of progression (persistent nephrotic-range proteinuria despite 6 months of conservative therapy, anti-PLA2R-positive, normal or near-normal GFR). Low-risk patients (sub-nephrotic proteinuria, normal GFR, falling anti-PLA2R titre) are managed conservatively because one-third undergo spontaneous remission. High-risk patients (rapidly falling GFR, very high anti-PLA2R) may need combination therapy. [1]
Limb 2 — Anti-proteinuric therapy for every patient
Every nephrotic patient, regardless of cause, should be on an ACE inhibitor or ARB at maximum tolerated dose [9]. These reduce proteinuria by lowering intraglomerular pressure and stabilising the slit diaphragm. The effect is independent of blood pressure. Monitor creatinine (a rise of up to 30 per cent is acceptable) and potassium (use potassium-binding resins, dietary counselling, or dose reduction before abandoning the agent). Dual ACEi and ARB blockade is harmful (ONTARGET) and is not recommended.
SGLT2 inhibitors reduce proteinuria and slow progression in proteinuric CKD across aetiologies, including glomerular disease — KDIGO 2024 endorses their use in proteinuric CKD [9]. The role in the acute nephrotic phase is evolving, but they are a strong candidate for the proteinuric patient who is not yet at kidney failure.
Limb 3 — Oedema and volume control
- Salt restriction (less than 2 g sodium per day). [1]- Loop diuretic — frusemide is first-line. Nephrotic patients are diuretic-resistant because frusemide is albumin-bound and the intravascular volume is contracted; doses are often high (80 to 120 mg or more). Adding a thiazide (sequential nephron blockade) or a potassium-sparing diuretic (amiloride, spironolactone) overcomes resistance.
- Albumin with frusemide — in diuretic-resistant anasarca, IV albumin followed by frusemide can mobilise fluid, but evidence is limited and the approach is reserved for severe cases.
- Avoid over-diuresis — a nephrotic patient may look oedematous but be intravascularly depleted; over-diuresis precipitates AKI. Aim for a weight loss of 0.5 to 1 kg per day. [1]
Limb 4 — Complication prevention
| Complication | Intervention |
|---|---|
| Thromboembolism | Prophylactic anticoagulation (warfarin or DOAC) when albumin less than 25 to 30 g/L, especially in membranous nephropathy; treat RVT with full anticoagulation |
| Hyperlipidaemia | Statin per cardiovascular risk |
| Hypertension | ACEi/ARB first-line (dual purpose); add calcium channel blocker or diuretic |
| Vitamin D deficiency | Cholecalciferol supplementation |
| Bone health (on steroids) | Calcium and vitamin D; bisphosphonate if high fracture risk |
| Vaccination in immunosuppression | Avoid live vaccines during active immunosuppression |
Prognosis and follow-up
Prognosis is cause-specific and proteinuria-driven: [1]
- Minimal change disease — excellent; over 90 per cent achieve remission, kidney survival is near-universal in children. Adults do slightly less well. Relapse is common and usually steroid-responsive.
- FSGS — guarded; remission predicts good outcome, but steroid-resistant FSGS progresses to kidney failure in 5 to 10 years. Collapsing variant is the most aggressive.
- Membranous nephropathy — one-third spontaneous remission, one-third persistent, one-third progress to kidney failure over 10 to 15 years. Anti-PLA2R titre and proteinuria response to immunosuppression stratify prognosis.
- MPGN — guarded; complement-mediated disease may respond to eculizumab; immune-complex disease depends on the cause.
- Diabetic nephropathy — progressive without intervention; SGLT2 inhibitors and RAAS blockade have transformed prognosis.
- Amyloidosis — historically poor, but modern haematology regimens have markedly improved kidney and overall survival. [1]
Follow-up is lifelong for most causes. Monitor proteinuria (PCR), renal function, albumin, blood pressure, lipids, and (in membranous nephropathy) anti-PLA2R titre. Reassess immunosuppression, anticoagulation, and vaccination status at each visit. Plan for kidney replacement therapy early in progressive disease. [1]
Guideline anchoring and regional variation
- ANZ primary: KDIGO 2021 Glomerular Diseases [1]; KDIGO 2024 CKD [9]; CARI Guidelines; Kidney Health Australia. Rituximab is PBS-authorised for membranous nephropathy in many indications.
- UK secondary: NICE NG203 (CKD/proteinuria); UKKA (UK Kidney Association) glomerulonephritis guidelines; rituximab is NICE-endorsed for membranous nephropathy.
- US tertiary: KDIGO; ACR; the MENTOR and STARMEN trials are the US-defining evidence.
Controversies:
- Rituximab versus cyclophosphamide in membranous nephropathy — MENTOR favoured rituximab; STARMEN favoured cyclophosphamide over a tacrolimus-rituximab sequence. The international trend is to rituximab first for toxicity reasons, reserving cyclophosphamide for aggressive or rituximab-resistant disease.
- Prophylactic anticoagulation threshold — albumin less than 25 g/L (most aggressive) versus less than 30 g/L, with risk models individualising the decision; no single trial definitively answers this.
- Steroid duration in MCD — KDIGO 2021 shortened the recommended course in children to 8 to 12 weeks (from the historical 6 months) to reduce steroid toxicity without losing efficacy. [1]
Communication and shared decision-making
A new diagnosis of nephrotic syndrome is frightening. The patient sees anasarca, learns about a kidney biopsy, immunosuppression, anticoagulation, and the word "transplant" or "dialysis" surfaces in their reading. Communicate: [1]
- The cause is identifiable and often treatable. Nephrotic syndrome is a pattern, not a destiny.
- The immunosuppression plan is cause-specific and evidence-based. Explain the agent, the duration, the monitoring, and the side effects (steroid toxicity, infection risk, cyclophosphamide fertility and malignancy, rituximab infusion reactions and reactivation of hepatitis B). [1]3. The complications are preventable. Anticoagulation when albumin is low; vaccination; oedema control; cardiovascular risk reduction.
- The long-term plan. Most patients achieve remission or stabilisation. Plan surveillance; discuss the realistic probability of kidney failure; involve the kidney transplant service early if progression is likely.
- Screen hepatitis B before rituximab — rituximab can reactivate hepatitis B and cause fatal fulminant hepatitis; check HBsAg, anti-HBc, anti-HBs, and give antiviral prophylaxis (entecavir or tenofovir) if there is evidence of past infection. [1]
DWE high-yield discriminators
- Child with first nephrotic episode: minimal change disease; treat empirically with steroids.
- Adult with new nephrotic syndrome and insidious onset: membranous nephropathy; check anti-PLA2R.
- African ancestry, HIV, rapid progression: collapsing FSGS / HIVAN; urgent ART and ACEi.
- Older adult with massive proteinuria, macroglossia, cardiomyopathy: AL amyloid; free light chains and Congo red.
- Young woman with nephrotic syndrome, malar rash, arthralgia: lupus nephritis class V; ANA, anti-dsDNA, complement, biopsy.
- Nephrotic with acute flank pain and haematuria: renal vein thrombosis; CT renal venography; anticoagulate.
- Nephrotic with low C3 and low C4: MPGN with cryoglobulinaemia, hepatitis C; cryoglobulins and HCV RNA.
- Membranous nephropathy refractory to rituximab: cyclophosphamide-corticosteroid (modified Ponticelli) per STARMEN.
- Hypercoagulability mechanism: antithrombin loss in urine, hepatic upregulation of fibrinogen and factor VIII.
- Encapsulated-organism infection mechanism: urinary loss of IgG and alternative complement pathway factors. [1]
References and guideline anchoring
KDIGO 2021 Glomerular Diseases [1]; KDIGO 2024 CKD [9]; MENTOR (rituximab versus cyclosporine in membranous nephropathy) [2]; GEMRITUX (rituximab for severe membranous nephropathy) [3]; Beck — PLA2R as target antigen in idiopathic membranous nephropathy [4]; STARMEN (cyclophosphamide versus tacrolimus-rituximab) [5]; Iijima — rituximab in frequently relapsing childhood nephrotic syndrome [6]; Llach — hypercoagulability and renal vein thrombosis in nephrotic syndrome [7]; Kerlin — antithrombin in nephrotic hypercoagulopathy [8].
References
- [1]Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases Kidney Int, 2021.PMID 34556256
- [2]Fervenza FC, Appel GB, Barbour SJ, et al. Rituximab or Cyclosporine in the Treatment of Membranous Nephropathy N Engl J Med, 2019.PMID 31269364
- [3]Dahan K, Debiec H, Plaisier E, et al. Rituximab for Severe Membranous Nephropathy: A 6-Month Trial with Extended Follow-Up J Am Soc Nephrol, 2017.PMID 27352623
- [4]Beck LH Jr, Bonegio RGB, Lambeau G, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy N Engl J Med, 2009.PMID 19571279
- [5]Fernández-Juárez G, Rojas-Rivera J, van de Logt AE, et al. The STARMEN trial indicates that alternating treatment with corticosteroids and cyclophosphamide is superior to sequential treatment with tacrolimus and rituximab in primary membranous nephropathy Kidney Int, 2021.PMID 33166580
- [6]Iijima K, Sako M, Nozu K, et al. Rituximab for childhood-onset, complicated, frequently relapsing nephrotic syndrome or steroid-dependent nephrotic syndrome: a multicentre, double-blind, randomised, placebo-controlled trial Lancet, 2014.PMID 24965823
- [7]Llach F Hypercoagulability, renal vein thrombosis, and other thrombotic complications of nephrotic syndrome Kidney Int, 1985.PMID 3906225
- [8]Kerlin BA, Iorember FM, Callerame KL, et al. Exploring the Role of Antithrombin in Nephrotic Syndrome-Associated Hypercoagulopathy: A Multi-Cohort Study and Meta-Analysis Clin J Am Soc Nephrol, 2023.PMID 36754010
- [9]Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease Kidney Int, 2024.PMID 38490803