Phys · renal
Polycystic Kidney Disease (ADPKD)
Also known as ADPKD · autosomal dominant polycystic kidney disease · polycystic kidneys · adult polycystic kidney disease · PKD1 · PKD2 · polycystin · polycystic liver disease · tolvaptan · Mayo Imaging Classification · htTKV
Consultant-physician-depth guide to autosomal dominant polycystic kidney disease — PKD1 versus PKD2 genetics and natural history, ultrasound and genetic diagnosis, progression assessment with htTKV and the Mayo Imaging Classification, HALT-PKD blood-pressure targets, tolvaptan selection and its liver monitoring, extrarenal disease with selective aneurysm screening, cyst complications, ESKD and transplantation, and family counselling — structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Polycystic Kidney Disease (ADPKD)
The answer first
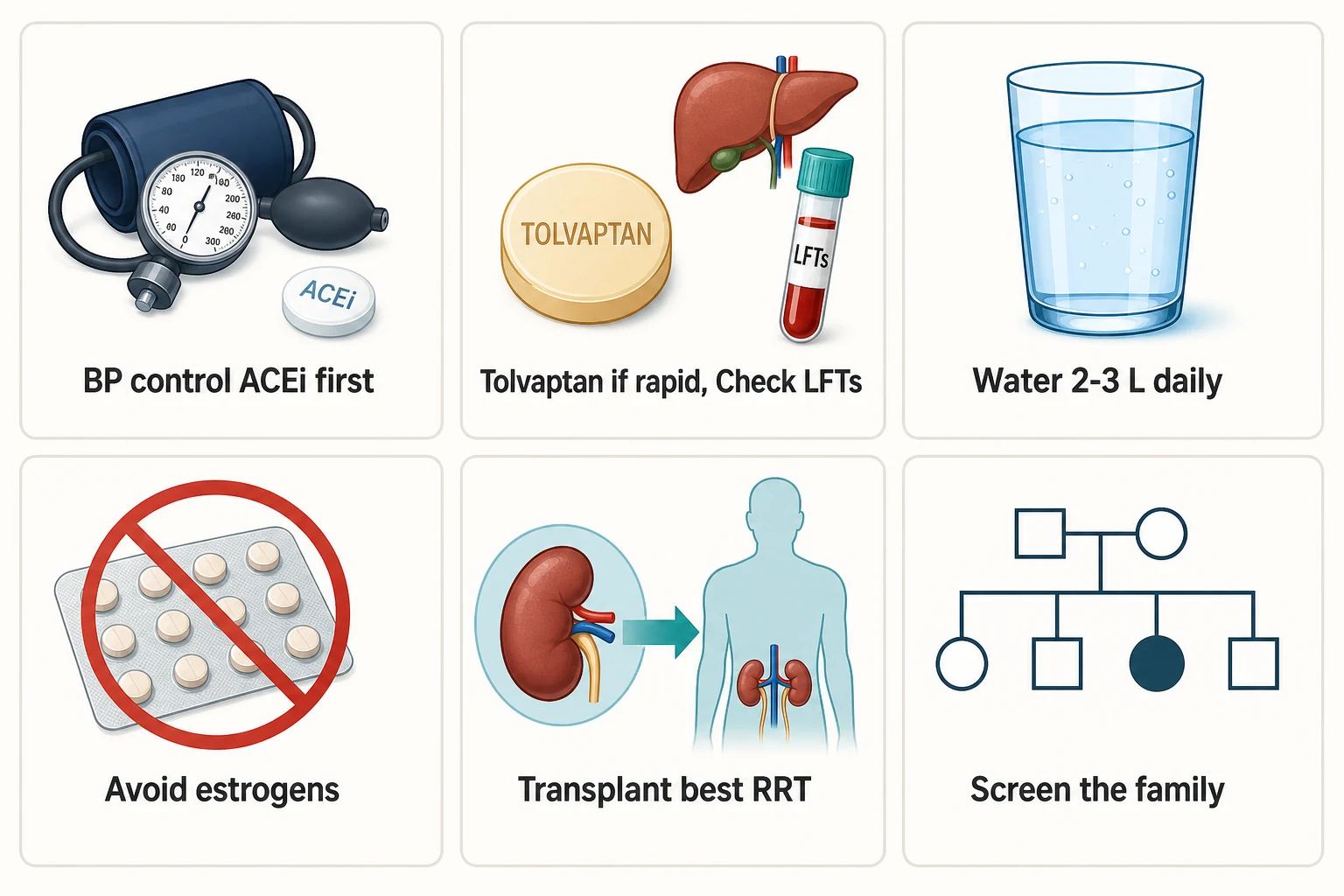
ADPKD is the commonest monogenic kidney disease — an autosomal dominant disorder that replaces both kidneys with cysts over decades, and the one inherited nephropathy where you now have a disease-modifying drug to offer. Five rules carry the DWE and the long-case defence [7] [20]:
- The gene sets the tempo. PKD1 families (about 85%) reach end-stage kidney disease around two decades earlier than PKD2 families (about 15%) — median ESKD in the late 50s versus the late 70s. Genotype is destiny on average, but within a family the course still varies, so never quote a fixed sentence to a patient [6] [7].
- No family history does not exclude it. Up to about one in five genotyped patients reports no affected parent — de novo variants, parental mosaicism, hypomorphic alleles and quietly mild PKD2 ancestors all break the "dominant, so someone must have it" reflex [11].
- Measure progression before you treat progression. Height-adjusted total kidney volume plotted against age (the Mayo Imaging Classification) separates typical classes 1A to 1E; classes 1C to 1E are the rapid progressors who benefit from tolvaptan. eGFR alone lies for years because hyperfiltration masks cyst growth — kidneys grow before function falls [4] [8].
- Tolvaptan is for rapid progressors, not everyone. TEMPO 3:4 and REPRISE established slower kidney growth and slower eGFR decline, at the price of aquaresis and scheduled liver-function monitoring for idiosyncratic hepatotoxicity. Selection, consent and monitoring are the exam — not the drug name [1] [2] [19].
- Think outside the kidney. Polycystic liver disease, intracranial aneurysms, valvular disease, hernias and diverticulosis travel with the genotype. Screen for aneurysms selectively — family history of aneurysm or subarachnoid haemorrhage, prior rupture, high-risk occupation, pre-transplant — not universally [12] [19].
Genetics — why the gene is the prognosis
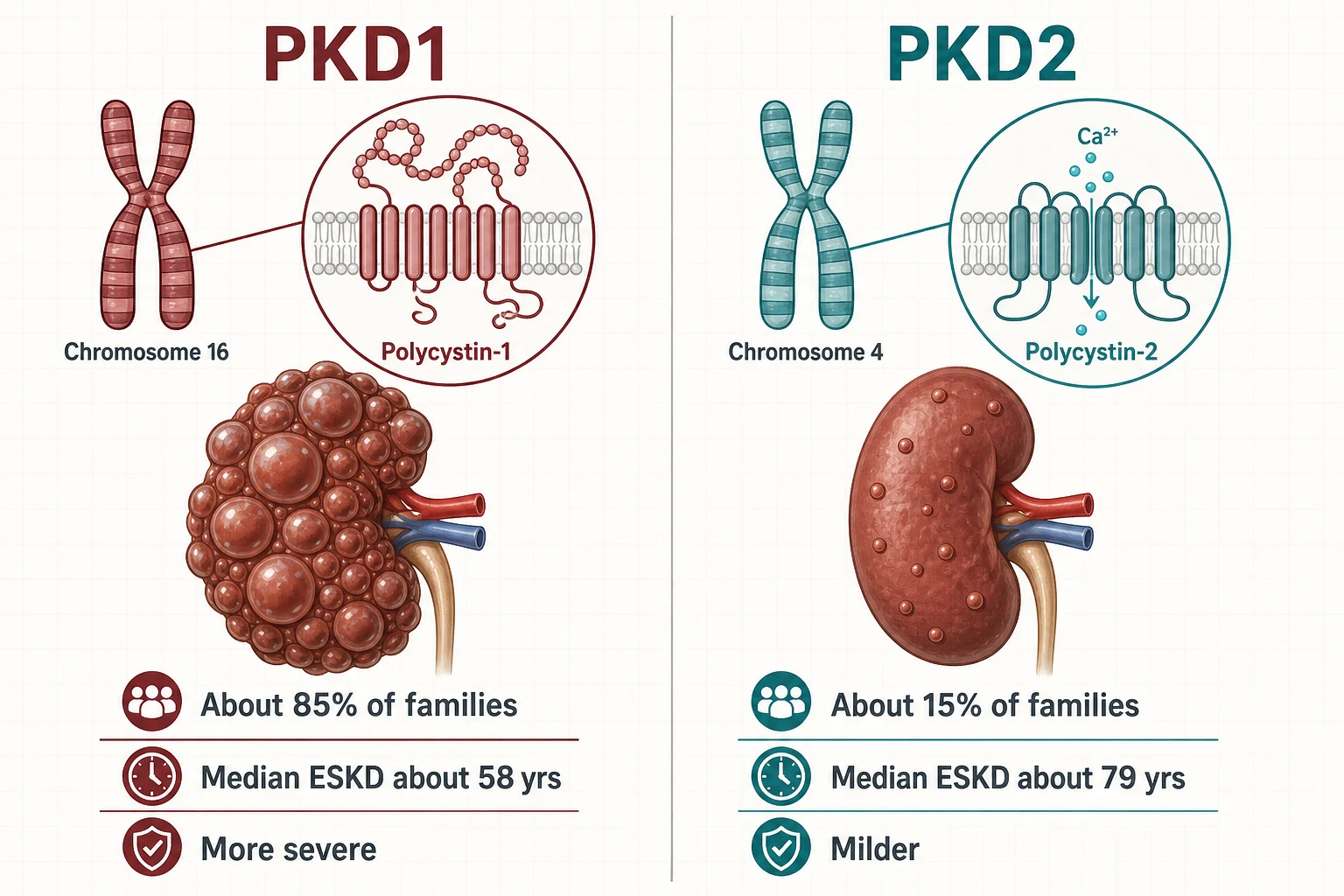
ADPKD affects roughly one in several hundred to one in a thousand people and accounts for about one in ten patients on renal replacement therapy in European registry data — making it the most common monogenic cause of kidney failure [21] [7]. Two genes produce nearly all of it [6]:
| Feature | PKD1 | PKD2 |
|---|---|---|
| Chromosome / protein | 16p13.3 — polycystin-1, a large mechanosensory membrane protein | 4q21 — polycystin-2, a calcium-permeable channel (TRPP2) [7] |
| Proportion of families | About 85% | About 15% [6] |
| Median age at ESKD | Late 50s (about 58) | Late 70s (about 79) — roughly two decades later [6] |
| Cyst burden | More cysts, earlier — kidneys enlarge sooner | Fewer cysts, later [6] |
| Extrarenal disease | Liver cysts in the great majority by middle age | Liver cysts still occur; aneurysm risk accompanies both [7] |
Both proteins localise to the primary cilium of tubular epithelial cells, where loss of polycystin function lowers intracellular calcium, raises cyclic AMP, and drives chloride-rich fluid secretion and tubular-cell proliferation — the cyst engine. This is why the vasopressin V2-receptor–cAMP axis became the therapeutic target: block the signal and you slow the fluid secretion that inflates every cyst [7] [20].
Within PKD1, allele type refines the prognosis further: truncating variants run faster than non-truncating ones, and hypomorphic (partial-function) alleles produce milder or atypical disease — including some of the "early severe child in a mild family" and "no family history" presentations. The PRO-PKD score packages this biology into a bedside number: points for a truncating PKD1 variant (most), non-truncating PKD1, PKD2 (least), male sex, and hypertension or a urological event (haematuria, cyst infection) before age 35 — a score above 6 flags rapid progression [18].
How it presents — the clinical shape
ADPKD is a decades-long disease that most often surfaces in the third to fifth decade, but the presenting doorways are remarkably consistent [20]:
| Doorway | Typical story | What to know |
|---|---|---|
| Hypertension | Found in a 30-year-old at an insurance medical | The commonest early sign — precedes any GFR loss, driven by intrarenal RAAS activation from cyst compression [3] |
| Flank and abdominal pain | Dull ache from stretched capsules, or acute severe pain | Acute pain means haemorrhage, infection or stone until proven otherwise [16] |
| Haematuria | Gross, sometimes with clots | Usually cyst haemorrhage — self-limiting; persistent or first-episode-later-in-life haematuria still warrants exclusion of tumour [20] |
| Stones | Colic in a known ADPKD patient | Uric acid and calcium oxalate; metabolic risk factors (low urine pH, hypocitraturia) travel with the disease [20] |
| Infection | UTI, pyelonephritis, or the dangerous one — an infected cyst | Fever with flank pain and high CRP; blood and urine cultures can be negative in true cyst infection [16] |
| Incidental imaging | CT for something else shows two huge cystic kidneys | Check the family history, the blood pressure, the creatinine — and look at the liver on the same scan [5] |
| Family screening | Asymptomatic adult child of an affected parent | The counselling conversation before the scan matters as much as the scan [19] |
Diagnosis — ultrasound first, genetics when it changes something
The diagnosis in a patient with a positive family history is made on the unified ultrasound criteria — cyst counts calibrated to age, because simple cysts accumulate with age in everyone and the threshold must outrun them [5]:
| Age of at-risk individual | Ultrasound criterion for ADPKD diagnosis | Note |
|---|---|---|
| 15–39 years | At least 3 unilateral or bilateral renal cysts | Sensitivity high for PKD1, lower for PKD2 in the young [5] |
| 40–59 years | At least 2 cysts in each kidney | [5] |
| 60 years and older | At least 4 cysts in each kidney | [5] |
| Exclusion, age 40 and over | Fewer than 2 cysts | Effectively excludes PKD1 disease; slightly weaker against PKD2 — an important caveat when a potential kidney donor is being cleared [5] |
Genetic testing earns its place in defined situations, and the viva answer is a list [19]:
- No family history with a compatible imaging phenotype — confirm or refute, and look for mosaicism and hypomorphic alleles [11].
- Atypical imaging — asymmetric, segmental or unilateral disease, or marked discordance between kidneys.
- A young potential living related donor whose ultrasound is clean but who is too young for the exclusion criteria to be reliable.
- Family planning — prenatal or preimplantation genetic testing (PGT) conversations need a confirmed familial variant [19].
- Early severe or syndromic disease — massive cystic kidneys in a child, or features suggesting a contiguous gene syndrome or a phenocopy (tuberous sclerosis, HNF1B disease, von Hippel–Lindau) [7].
Progression — measure the volume, not just the creatinine
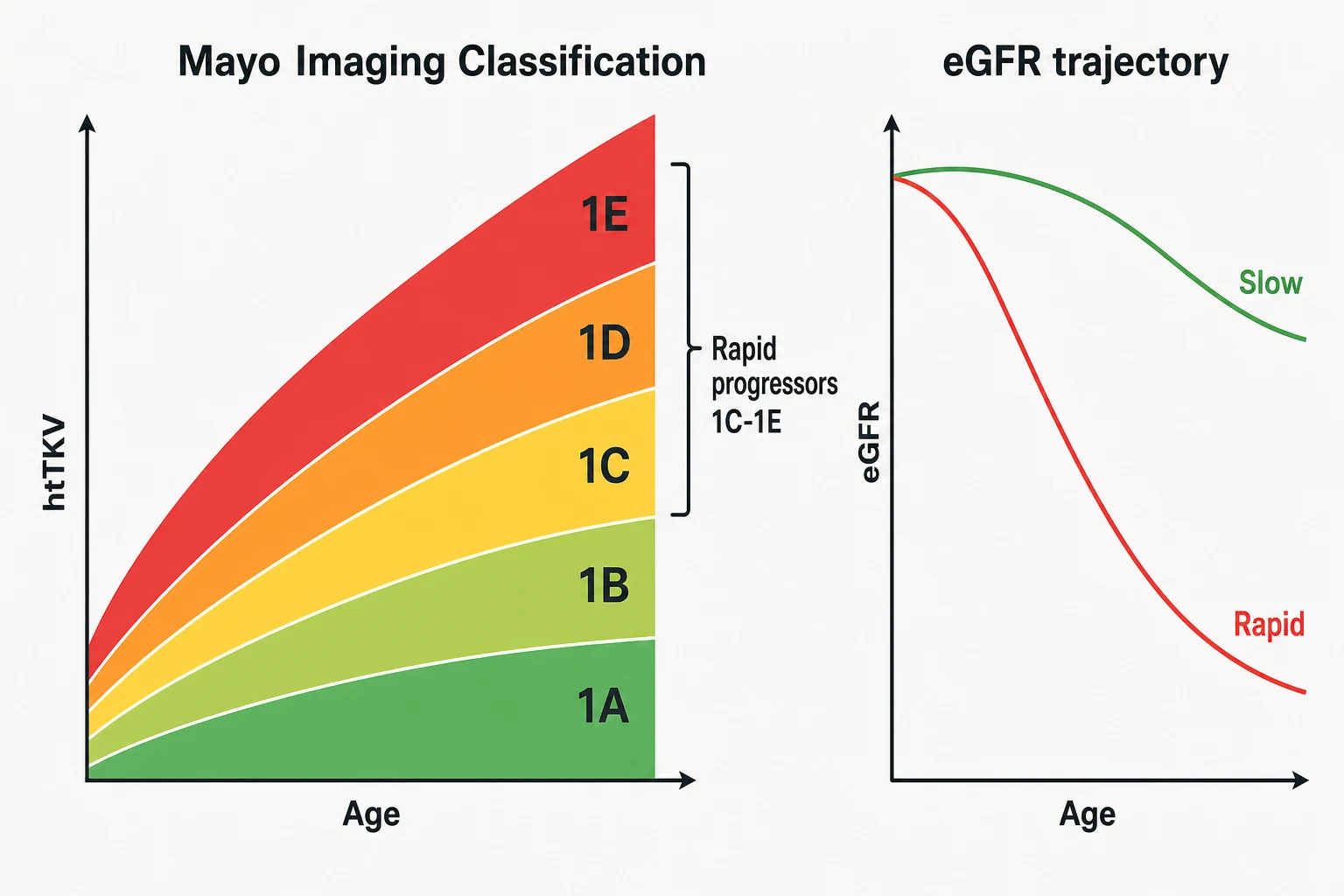
The central biological fact of ADPKD is that the kidneys grow for decades before the GFR falls — hyperfiltration and compensatory hypertrophy of surviving nephrons hold the creatinine normal while total kidney volume climbs at about 5% per year. CRISP proved the correlation: baseline volume and volume growth rate predict subsequent GFR decline, which is why volume, not creatinine, is the progression biomarker of choice in early disease [8].
The Mayo Imaging Classification operationalises this. Take height-adjusted total kidney volume (htTKV, from CT or MRI), plot it against age, and the patient lands in a class — for the typical diffuse bilateral phenotype, classes 1A through 1E, where 1A is indolent and 1E is explosive. The practical line the guideline draws: classes 1C, 1D and 1E are rapid progressors — the patients in whom you should expect future GFR loss and in whom disease-modifying therapy pays. Class 2 (atypical — asymmetric, segmental, unilateral, or atrophic patterns) sits outside the model and needs genotype-level thinking [4] [19].
How a consultant stages ADPKD progression
The eGFR trajectory still matters — it is the check on the imaging call and the metric trials were powered on. A patient losing eGFR at 3 mL/min/1.73 m² per year or faster is a rapid progressor by any definition; once CKD is established, the slope becomes the working measure because volume growth plateaus as fibrosis replaces expansion [2] [8].
The PRO-PKD score adds the genotype and early clinical events to the picture: truncating PKD1 scores highest, then non-truncating PKD1, then PKD2; male sex and hypertension or a urological event before age 35 add points. Above 6 predicts rapid progression, and the score is most useful where imaging is equivocal or discordant with the clinical story [18].
Extrarenal disease — the genotype travels
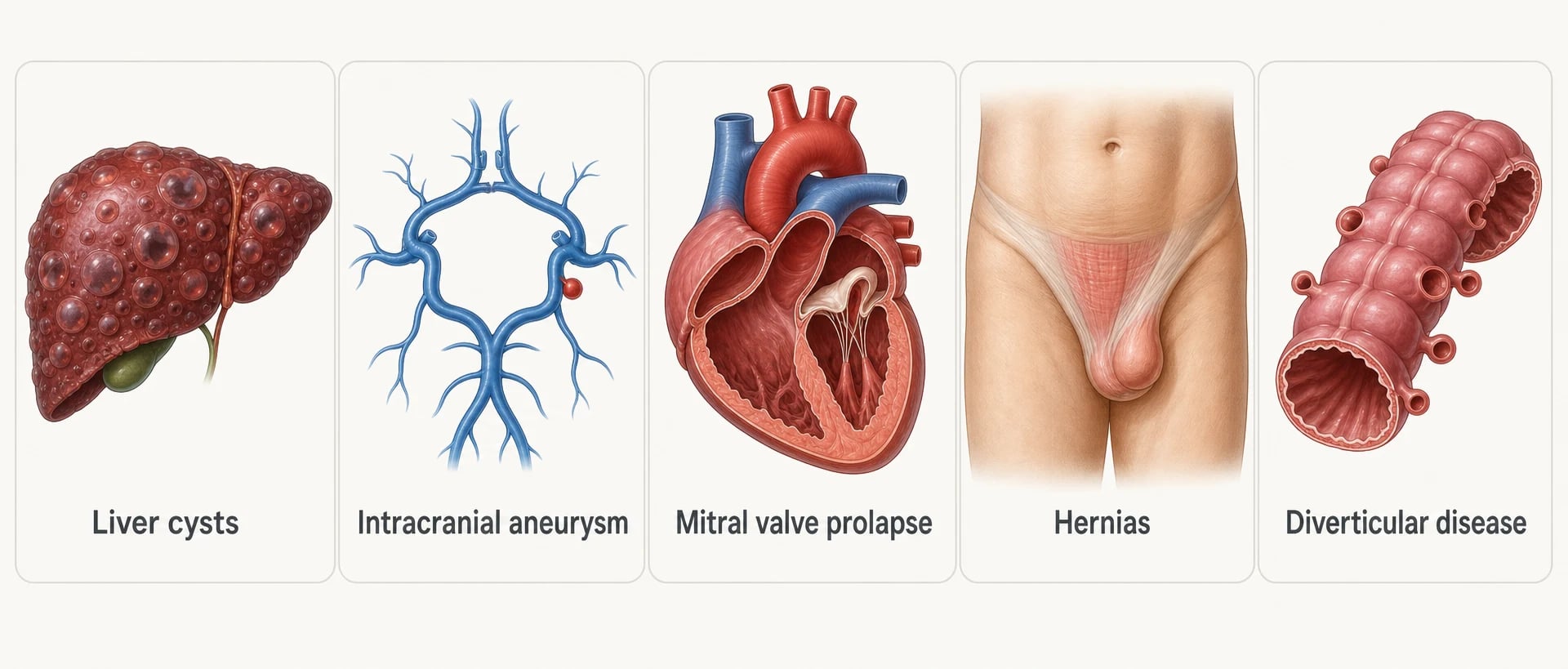
ADPKD is a systemic connective-tissue-flavoured disease; the kidneys simply fail first. The extrarenal survey is a core part of every long-case defence [7] [20].
Polycystic liver disease is the commonest extrarenal manifestation — cysts in the great majority of patients by middle age, and almost always clinically benign: liver synthetic function is preserved because the parenchyma between cysts is normal [7]. The important subset is massive hepatomegaly, overwhelmingly a disease of women and of estrogen exposure — postmenopausal estrogen therapy selectively stimulates hepatic cyst growth, which is why estrogen-containing contraception and hormone replacement are used cautiously or avoided in significant liver disease [10]. Symptoms are mechanical: early satiety, reflux, pain, abdominal wall distortion. For severe symptomatic volume disease, somatostatin analogues reduce liver volume modestly in randomised data, and the surgical options (fenestration, resection, occasionally transplant) belong to specialist centres [17].
Intracranial aneurysms are the manifestation that kills young. Prevalence is several-fold higher than in the general population, and it clusters — a family history of aneurysm or subarachnoid haemorrhage raises an individual's risk substantially [12]. The discipline is selective, not universal, screening with MR angiography: screen patients with a personal or family history of aneurysm or subarachnoid haemorrhage, those in high-risk occupations or hobbies where sudden incapacity endangers others (pilots, professional drivers, divers), those being prepared for transplant or major anticoagulation, and those whose anxiety after counselling still demands an answer. Repeat imaging on an interval for small aneurysms; extended follow-up of screen-detected ADPKD aneurysms shows growth and rupture are uncommon in small anterior-circulation lesions, which is what makes conservative surveillance defensible [12] [19].
Valvular disease — the original echocardiographic series found mitral valve prolapse in about a quarter of ADPKD patients, with increased mitral and aortic regurgitation. Listen for the mid-systolic click and the murmurs; image on clinical grounds, not by reflex annual echo [14].
Hernias and diverticular disease complete the connective-tissue picture: inguinal and incisional hernias are commoner (and matter when you are planning peritoneal dialysis), and diverticulosis is commoner still — diverticulitis in the dialysis and transplant population runs a more complicated course in ADPKD, with higher rates of perforation reported in series of kidney failure patients [15] [20].
And do not forget the cardiovascular core: early hypertension drives left ventricular hypertrophy and faster progression, which is why the blood-pressure section below is treatment, not housekeeping [3].
Hypertension — treat it early, treat it hard in the young
Hypertension arrives before any loss of GFR in most ADPKD patients, and the mechanism is renal: expanding cysts compress and stretch intrarenal vessels, producing patchy ischaemia that switches on the renin–angiotensin system. That physiology dictates the drug class — ACE inhibitors or ARBs are first-line [3] [20].
The HALT-PKD trial gives the exam its numbers. In hypertensive ADPKD patients aged 15 to 49 with preserved GFR (above 60 mL/min/1.73 m²), a low blood-pressure target — 95/60 to 110/75 mmHg on home measurement — against standard control (120/70 to 130/85) produced slower growth of total kidney volume, a greater fall in left ventricular mass and lower albuminuria, at the cost of more dizziness. The eGFR difference between arms did not reach significance over the trial's timeframe — so quote the target as organ-protective and progression-modifying, not as proven to delay ESKD [3].
The practical synthesis the guideline supports: in a young ADPKD patient with preserved GFR, aim below 110/75 mmHg as tolerated; in older patients or established CKD, individualise toward standard CKD targets, using RAAS blockade first and adding a calcium-channel blocker or diuretic as needed [3] [19]. And hold the line on combination RAAS blockade — the companion HALT-PKD comparison found adding an ARB to an ACE inhibitor conferred no advantage over ACE inhibition alone [3].
Tolvaptan — the disease-modifying decision
Tolvaptan is a vasopressin V2-receptor antagonist: it blocks the cAMP signal that drives cyst fluid secretion, and it is the only therapy proven to slow ADPKD progression. The two trials are the exam [1] [2]:
| Trial | Population | Result | What it established |
|---|---|---|---|
| TEMPO 3:4 (2012) | 1,445 adults, 18–50 years, early CKD with large kidneys (TKV at least 750 mL) | Tolvaptan roughly halved kidney-volume growth and slowed eGFR decline by about a quarter over 3 years | Proof of concept in early, rapidly progressing disease [1] |
| REPRISE (2017) | 1,370 adults, 18–65 years, later CKD (eGFR 25–65) | Slower eGFR decline by about 1.3 mL/min/1.73 m² per year versus placebo | Benefit persists into established CKD — the drug is not just for the early [2] |
Who should be offered it. The guideline position: adults with ADPKD and evidence of rapid progression — practically, Mayo classes 1C to 1E with typical disease, supported where helpful by eGFR slope or PRO-PKD score — with enough GFR left to protect and an age range in which benefit accrues. The ideal candidate is the younger patient with preserved GFR and a 1C–1E kidney; REPRISE extends the offer into later CKD for documented rapid progressors. It is explicitly not for everyone with ADPKD — indolent Mayo 1A/1B disease gains little and pays the full burden [19] [4].
The burden you must name in the consent conversation [1]:
- Aquaresis — free-water diuresis of several litres daily: polyuria, nocturia, thirst. It is the price of the mechanism, the commonest reason for stopping, and the reason you warn about occupations where toilet access is poor [1].
- Idiosyncratic hepatocellular injury — a small excess of significant transaminase elevations in the trials, reversible on stopping, with no requirement for pre-existing liver disease. This is why scheduled liver-function testing is mandatory: baseline, then monthly for the first 18 months and every 3 months thereafter, and the drug stops for significant enzyme elevation [1] [19].
- Practicalities — split daily dosing titrated as tolerated (starting at 60 mg per day as 45/15 mg, toward 90/30 mg), drug interactions (strong CYP3A inhibitors), and cost and access through local funding criteria [19].
Starting and monitoring tolvaptan
Confirm rapid progression
Mayo class on htTKV (1C-1E rapid), eGFR slope, PRO-PKD where helpful — do not treat indolent disease
Baseline checks
Liver enzymes, sodium, renal function, pregnancy status and contraception discussion; screen for contraindications including significant liver disease
Consent conversation
Benefit in slowed decline versus daily aquaresis, monitoring burden, interactions and cost — a shared decision, documented
Start low, titrate
Split-dose regimen (45/15 mg) titrated as tolerated toward 90/30 mg; morning dose early to spare the night
Scheduled LFTs
Monthly for 18 months, then every 3 months; stop for significant transaminase elevation and do not rechallenge casually
Sick-day and surgery rules
Pause for dehydration, vomiting, fasting or major surgery — aquaresis plus hypovolaemia risks hypernatraemia and AKI
Cyst complications — infection, haemorrhage, stones, pain
Cyst infection is the dangerous mimic. The picture is fever, flank pain and high inflammatory markers in a patient whose urine culture may be negative because the infected cyst no longer communicates with the collecting system; blood cultures are positive in only a minority. Distinguishing an infected cyst from ordinary pyelonephritis changes the antibiotic: the drug must penetrate the cyst wall, which means lipophilic agents — fluoroquinolones or trimethoprim–sulfamethoxazole — while beta-lactams penetrate cysts poorly as monotherapy [16]. Treat for weeks, not days; image for an abscessed cyst (CT or MRI, sometimes labelled white-cell scanning when the picture is obscure); and aspirate or drain a large infected cyst that fails antibiotics — persistent sepsis with a dominant cyst on imaging is the drainage indication [16].
Cyst haemorrhage produces sudden flank pain and gross haematuria, sometimes with clot colic. It is overwhelmingly self-limiting: rest, hydration, analgesia avoiding NSAIDs, and time — settle within days. The consultant's job is to know when not to be reassured: haemodynamic compromise, a dropping haemoglobin, pain with fever, or haematuria that persists — especially a first episode in an older patient, where tumour exclusion with contrast imaging is mandatory [20].
Stones complicate ADPKD more often than the general population — uric acid and calcium oxalate, fostered by low urine pH, hypocitraturia and the urinary stasis of distorted calyces. Management follows standard stone principles with two caveats: imaging interpretation is harder (a stone hides well inside a cyst wall on plain films — CT is the test), and drainage procedures must navigate the cystic anatomy [20].
Chronic pain is the undertreated complication — capsular stretch and mass effect. Escalate through simple analgesia and physical measures, avoid chronic NSAIDs (they accelerate GFR loss), and reserve cyst-decompressing procedures (aspiration-sclerosis, fenestration) for dominant-cyst pain that fails conservative management [20].
ESKD, dialysis and transplantation
ADPKD supplies about one in ten European renal-replacement patients, and — a consistent registry finding — they do better on RRT than age-matched patients with other kidney diseases, with the survival advantage most marked after transplantation [21].
Transplantation is the renal replacement therapy of choice, and the exam answer is that ADPKD patients are often excellent recipients: they are typically less anaemic than other dialysis patients (preserved erythropoietin production from the cystic kidneys), carry less cardiac disease than the diabetic ESKD population, and tolerate surgery well [21] [20]. Native nephrectomy is the exception, not the routine — reserve it for defined problems: kidneys so massive there is no room for the graft, recurrent cyst infection, recurrent significant haemorrhage, intractable pain, or suspicion of malignancy. It carries real operative morbidity and can be timed pre-, peri- or post-transplant; do not offer it for size alone [20]. Pre-transplant work-up in ADPKD also includes the extrarenal survey — aneurysm screening for the indicated patients, and attention to diverticular disease and hernias [19].
Dialysis when needed. Haemodialysis is conventional; note the haemoglobin often runs higher than expected for the CKD stage — do not chase it with erythropoietin reflexively. Peritoneal dialysis is feasible in many but deserves a thought about massive kidneys, massive livers and hernias before committing [20].
And a therapeutic negative worth quoting: mTOR inhibition failed — sirolimus did not slow kidney growth in a randomised trial despite compelling preclinical biology. It is the humility footnote for the viva: not every pathway drug works, which is exactly why tolvaptan needed TEMPO and REPRISE before it earned a guideline place [9] [1].
Lifestyle and the long game
Three everyday levers have real mechanistic or trial support [13]:
- Water intake. Vasopressin is the agonist of the cyst engine, so sustained dilute urine is a rational brake. Pilot data show a prescribed water intake — typically around 2 to 3 litres daily, titrated to keep urine dilute — is achievable and safe in patients with preserved GFR; it is adjunct, not drug, and it is inappropriate once GFR is very low or sodium handling is impaired [13].
- Salt and weight. Salt restriction potentiates RAAS blockade and blood-pressure control; weight management reduces the cardiometabolic load on a cardiovascularly vulnerable population [3] [19].
- Avoiding accelerants. Chronic NSAIDs, unnecessary estrogen exposure with significant liver disease, and smoking all push the wrong way [10] [20].
Structure surveillance to the stage: blood pressure and renal function at least annually, htTKV re-measurement when it would change a decision (tolvaptan candidacy, prognosis), liver enzymes on tolvaptan per schedule, and interval imaging of known aneurysms per neurosurgical advice [19].
Family counselling — the conversation that defines the specialty
An ADPKD diagnosis is a family diagnosis. Each child of an affected parent has a 50% chance of inheriting the variant, and the question "should I be tested, and how?" arrives in every clinic [7].
The consultant framework [19]:
- Counsel before screening. An ultrasound that finds cysts changes insurance, family planning and self-image; an adult child is entitled to decline testing, and the right not to know is respected. Screening is offered, never imposed [19].
- Imaging is the usual first test — the age-calibrated ultrasound criteria, remembering that a clean scan under age 40 does not fully exclude PKD2 disease [5].
- Genetic testing has defined jobs — no-family-history cases, atypical imaging, young potential living related donors, and family planning [11] [19].
- Reproductive options belong on the table: conceive naturally and test children in adulthood, prenatal testing, or preimplantation genetic testing (PGT) to select unaffected embryos — a conversation for clinical genetics, offered early rather than discovered late [19].
DCE angles — how this topic is examined
The long case is the 40-year-old with ADPKD whose GFR is declining — Mayo class 1D, eGFR in the 50s, and a family history that includes an aneurysm. The examiner's arc runs: stage the progression (htTKV, Mayo class, eGFR slope), defend or decline tolvaptan (selection, consent, aquaresis, scheduled LFTs), run the extrarenal survey (MRA for her sister's-aneurysm indication, liver, valves), manage the blood pressure to target, and close with the family — her children's screening options and the PGT conversation. Every limb of that arc has evidence to quote, which is why this case is a DCE favourite [4] [1] [12].
The short case is the abdominal examination. Bilateral ballotable renal masses that move with respiration and are bimanually palpable; a palpable, sometimes nodular liver edge; hernial orifices worth examining; blood pressure measured properly; fundi for hypertensive change; and the synthesis sentence — "bilateral enlarged ballotable kidneys with hepatomegaly and hypertension: ADPKD until proven otherwise, and I would assess severity, complications and family implications" [20].
Exam traps, collected
References
- [1]Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease N Engl J Med, 2012.PMID 23121377
- [2]Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in Later-Stage Autosomal Dominant Polycystic Kidney Disease N Engl J Med, 2017.PMID 29105594
- [3]Schrier RW, Abebe KZ, Perrone RD, et al. Blood pressure in early autosomal dominant polycystic kidney disease N Engl J Med, 2014.PMID 25399733
- [4]Irazabal MV, Rangel LJ, Bergstralh EJ, et al. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials J Am Soc Nephrol, 2015.PMID 24904092
- [5]Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD J Am Soc Nephrol, 2009.PMID 18945943
- [6]Hateboer N, v Dijk MA, Bogdanova N, et al. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group Lancet, 1999.PMID 10023895
- [7]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease Nat Rev Dis Primers, 2018.PMID 30523303
- [8]Grantham JJ, Torres VE, Chapman AB, et al. Volume progression in polycystic kidney disease N Engl J Med, 2006.PMID 16707749
- [9]Serra AL, Poster D, Kistler AD, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease N Engl J Med, 2010.PMID 20581391
- [10]Sherstha R, McKinley C, Russ P, et al. Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease Hepatology, 1997.PMID 9362373
- [11]Iliuta IA, Kalatharan V, Wang K, et al. Polycystic Kidney Disease without an Apparent Family History J Am Soc Nephrol, 2017.PMID 28522688
- [12]Irazabal MV, Huston J 3rd, Kubly V, et al. Extended follow-up of unruptured intracranial aneurysms detected by presymptomatic screening in patients with autosomal dominant polycystic kidney disease Clin J Am Soc Nephrol, 2011.PMID 21551026
- [13]Wang CJ, Creed C, Winklhofer FT, et al. Water prescription in autosomal dominant polycystic kidney disease: a pilot study Clin J Am Soc Nephrol, 2011.PMID 20876670
- [14]Hossack KF, Leddy CL, Johnson AM, et al. Echocardiographic findings in autosomal dominant polycystic kidney disease N Engl J Med, 1988.PMID 3419455
- [15]Lederman ED, McCoy G, Conti DJ, et al. Diverticulitis and polycystic kidney disease Am Surg, 2000.PMID 10695753
- [16]Sallée M, Rafat C, Zahar JR, et al. Cyst infections in patients with autosomal dominant polycystic kidney disease Clin J Am Soc Nephrol, 2009.PMID 19470662
- [17]Hogan MC, Masyuk TV, Page LJ, et al. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease J Am Soc Nephrol, 2010.PMID 20431041
- [18]Cornec-Le Gall E, Audrézet MP, Rousseau A, et al. The PROPKD Score: A New Algorithm to Predict Renal Survival in Autosomal Dominant Polycystic Kidney Disease J Am Soc Nephrol, 2016.PMID 26150605
- [19]Kidney Disease: Improving Global Outcomes (KDIGO) ADPKD Work Group KDIGO 2025 Clinical Practice Guideline for the Evaluation, Management, and Treatment of Autosomal Dominant Polycystic Kidney Disease (ADPKD) Kidney Int, 2025.PMID 39848759
- [20]Ong AC, Devuyst O, Knebelmann B, et al. Autosomal dominant polycystic kidney disease: the changing face of clinical management Lancet, 2015.PMID 26090645
- [21]Spithoven EM, Kramer A, Meijer E, et al. Renal replacement therapy for autosomal dominant polycystic kidney disease (ADPKD) in Europe: prevalence and survival--an analysis of data from the ERA-EDTA Registry Nephrol Dial Transplant, 2014.PMID 25165182