Phys · renal
Tubulointerstitial Disease
Also known as acute interstitial nephritis · AIN · acute tubulointerstitial nephritis · ATIN · chronic interstitial nephritis · chronic tubulointerstitial nephritis · drug-induced interstitial nephritis · allergic interstitial nephritis · TINU · analgesic nephropathy · lithium nephropathy · aristolochic acid nephropathy · Balkan endemic nephropathy
Consultant-physician-depth guide to acute and chronic interstitial nephritis — AIN as the great AKI mimic, the unreliability of the allergic triad, culprit drug classes (antibiotics, PPIs, NSAIDs, checkpoint inhibitors), urine clues and the limits of eosinophiluria, biopsy triggers, the observational evidence for corticosteroids, checkpoint-inhibitor AIN, TINU, chronic TIN (analgesic, lithium, aristolochic acid), recovery and CKD risk — structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Tubulointerstitial Disease
The answer first
Acute interstitial nephritis (AIN) is the great mimic of acute kidney injury — an immune-mediated inflammation of the renal interstitium that presents as a quiet, subacute creatinine drift with unimpressive urine, and that is caused by a drug until proven otherwise [1]. Three rules carry you through the DWE and the long case [1] [5]:
- Suspect it in every unexplained AKI. The textbook allergic syndrome — fever, rash, eosinophilia — is the exception, not the rule. Modern drug-induced AIN is usually just a creatinine that will not stop rising, and the triad appears together in only a small minority of biopsy-proven cases [3] [4].
- Stop the culprit drug immediately. Withdrawal of the offending agent is the one intervention with consistent outcome data, and delay in withdrawal is associated with worse renal recovery — the steroid question always comes second [2].
- Steroids are a decision, not a default. The evidence is entirely retrospective; benefit concentrates in patients treated early after biopsy confirmation with no contraindication. In an elderly polypharmacy patient with modest AKI, watchful waiting after drug withdrawal is a defensible, examinable position [2] [13].
AIN — the great AKI mimic
The typical modern case is unimpressive to look at, which is exactly why it is missed. A patient started on a drug weeks ago — a proton pump inhibitor, an antibiotic, an NSAID — has a creatinine that has drifted upward over days to weeks. There is no oliguria drama, no nephritic sediment, no nephrotic picture. The urine shows modest abnormalities at most, the kidneys are normal-sized on ultrasound, and the only real clue is the timeline [3] [6]. In biopsy series performed for unexplained AKI, AIN accounts for roughly 15–20% of diagnoses — common enough that "we never biopsied, so we never saw it" is a real phenomenon on every renal unit [1] [3].
The classic teaching — fever, rash, eosinophilia — comes from the methicillin era. In contemporary series each individual feature appears in only a minority of patients, and the full triad is present in about 10% or fewer of biopsy-proven drug-induced cases [4] [3]. Peripheral eosinophilia is present in a minority. What remains as the consistent signal is the drug exposure and the tempo: a subacute, non-oliguric creatinine rise days to weeks after the culprit began [6].
Causes — the drug chart first
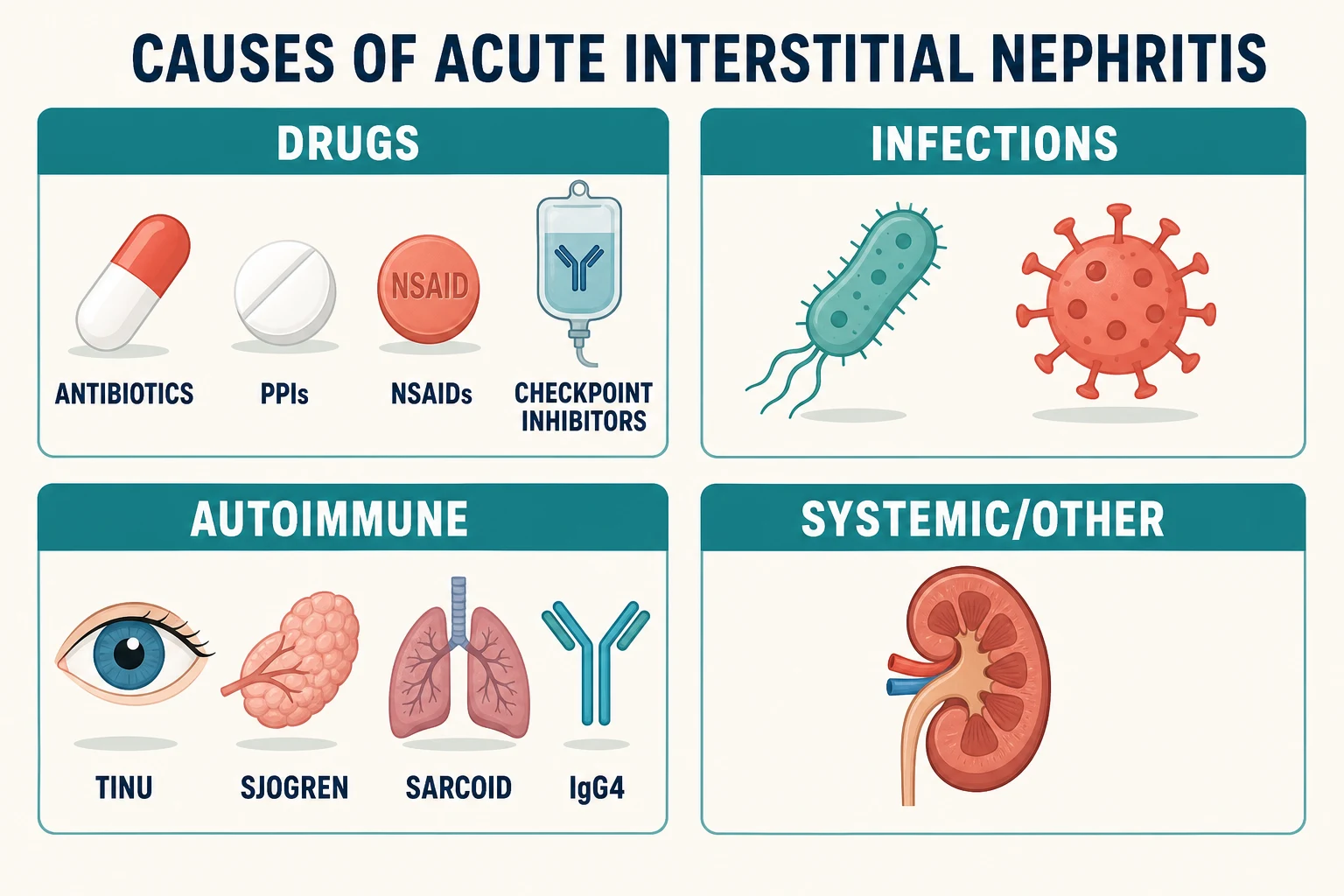
Drugs now cause the large majority of AIN in modern series, displacing the infections that dominated older textbooks [3]. The classes worth knowing individually, because examiners test their peculiarities:
| Culprit class | Latency and pattern | Features that distinguish it |
|---|---|---|
| Beta-lactams (penicillins, cephalosporins) | Days to weeks after starting | The historical prototype of allergic AIN; systemic features more common here than with other classes [1] |
| Fluoroquinolones | Days to weeks | Increasingly recognised; same subacute drift pattern [5] |
| Rifampicin | Classically on intermittent or restarted dosing | Can be abrupt, with flank pain and systemic upset after re-exposure [1] |
| Proton pump inhibitors | Often months of uneventful use first | The silent one — no allergic features, discovered late, a leading cause in elderly polypharmacy [9] [10] |
| NSAIDs | Weeks to months | The outlier: often accompanied by nephrotic-range proteinuria from a concurrent minimal-change-like glomerular lesion [19] |
| Immune checkpoint inhibitors | Weeks to months into therapy | AIN is the dominant biopsy finding in ICI-associated AKI; management is a joint decision with oncology [11] [12] |
| Allopurinol | Weeks, within a DRESS picture | Severe systemic hypersensitivity with rash, eosinophilia, hepatitis and AIN [5] |
| Others (sulfonamides, diuretics, phenytoin, mesalazine) | Variable | The principle matters more than the list: any drug can do it [1] |
Proton pump inhibitors deserve their own paragraph because they dominate the modern long case. A nationwide nested case-control study found current PPI use carried a severalfold increased risk of AIN, and a systematic review of reported cases describes the typical patient: older, months into treatment, with none of the allergic features, presenting as unexplained CKD drift [9] [10]. Because PPIs are started casually and almost never reviewed, the drug is still on the chart when the biopsy comes back — which is why deprescribing is the centrepiece of the DCE defence [9].
Infections are the second category: AIN accompanies leptospirosis, tuberculosis, EBV and a range of bacterial and viral illnesses, usually as part of the systemic infection rather than an isolated renal event [1]. Tuberculosis also appears on the granulomatous interstitial nephritis list, where it competes with drugs and sarcoidosis [20].
Autoimmune and systemic causes complete the map: TINU syndrome (tubulointerstitial nephritis with uveitis), Sjögren syndrome, sarcoidosis, IgG4-related disease, and lupus [22] [21]. These matter for two reasons: they change management (the underlying disease dictates treatment), and they are the reason "no culprit drug" does not end the workup [20].
Pathophysiology — why the tubules fail quietly
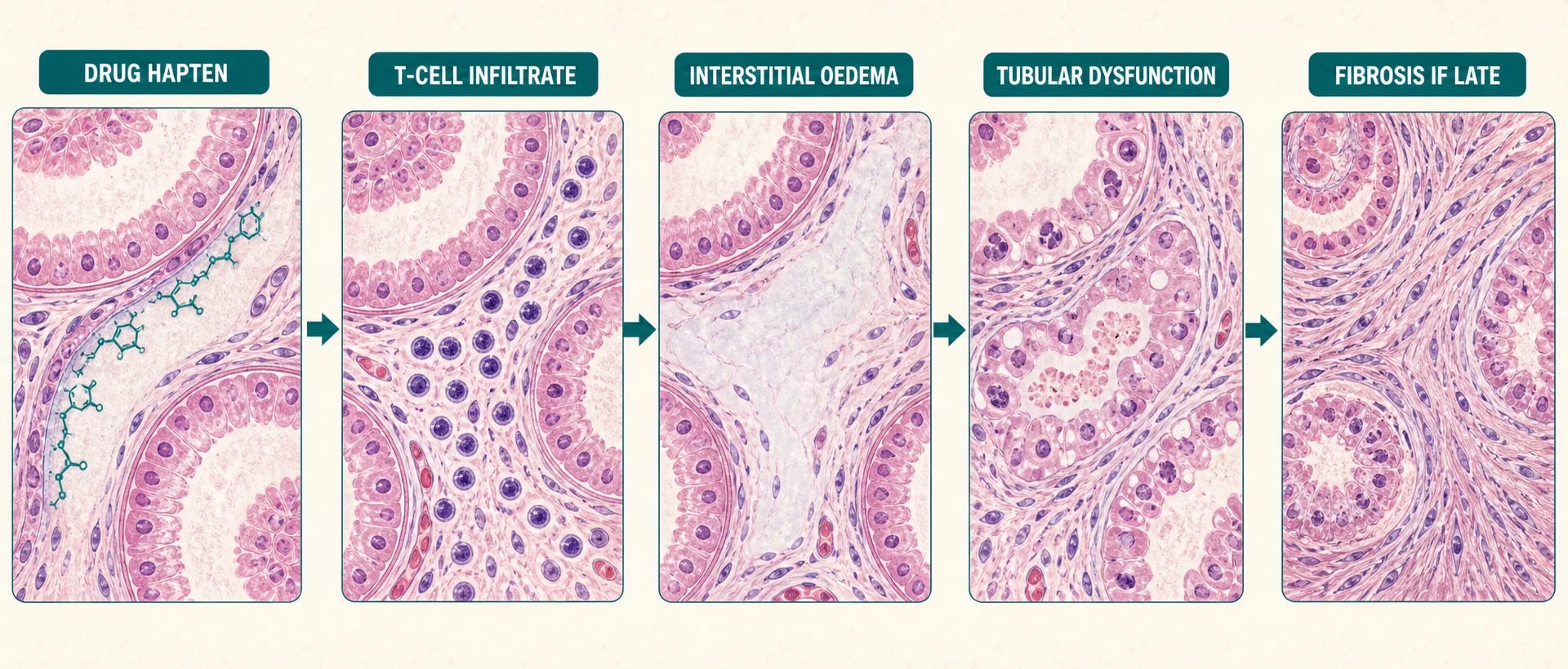
Drug-induced AIN is an idiosyncratic, delayed-type (type IV) T-cell hypersensitivity — not a dose-dependent toxicity and not IgE-mediated, which is why it can appear after months of uneventful use and why prior tolerance means nothing [1] [5]. The drug (or its metabolite) acts as a hapten, binding tubular basement membrane or interstitial components, and the resulting lymphocytic response does the damage [1].
The injurious cascade
Drug hapten
Culprit drug or metabolite seeds the interstitium or tubular basement membrane and is seen as foreign
T-cell infiltrate
Lymphocytes (with monocytes, plasma cells, eosinophils) invade the interstitium; tubulitis follows
Interstitial oedema
Inflammatory swelling expands the interstitial space and compresses tubules and peritubular capillaries
Tubular dysfunction
Sodium handling, concentrating ability and secretion suffer first; GFR falls as tubuloglomerular feedback engages
Fibrosis if late
Persistent inflammation lays down interstitial fibrosis and tubular atrophy — the point at which recovery becomes incomplete
The cascade explains the clinical signature. Inflammation sits in the interstitium, not the glomerulus, so there is no heavy proteinuria (NSAIDs excepted), no dysmorphic haematuria, and no nephritic sediment — just a GFR that slides as tubular function and peritubular blood flow deteriorate [1]. It also explains the two prognostic facts examiners care about: recovery tracks the amount of fibrosis already laid down at biopsy, and both delayed drug withdrawal and delayed treatment allow more of it to accumulate [2] [4].
Diagnosis — urine clues, then the biopsy question
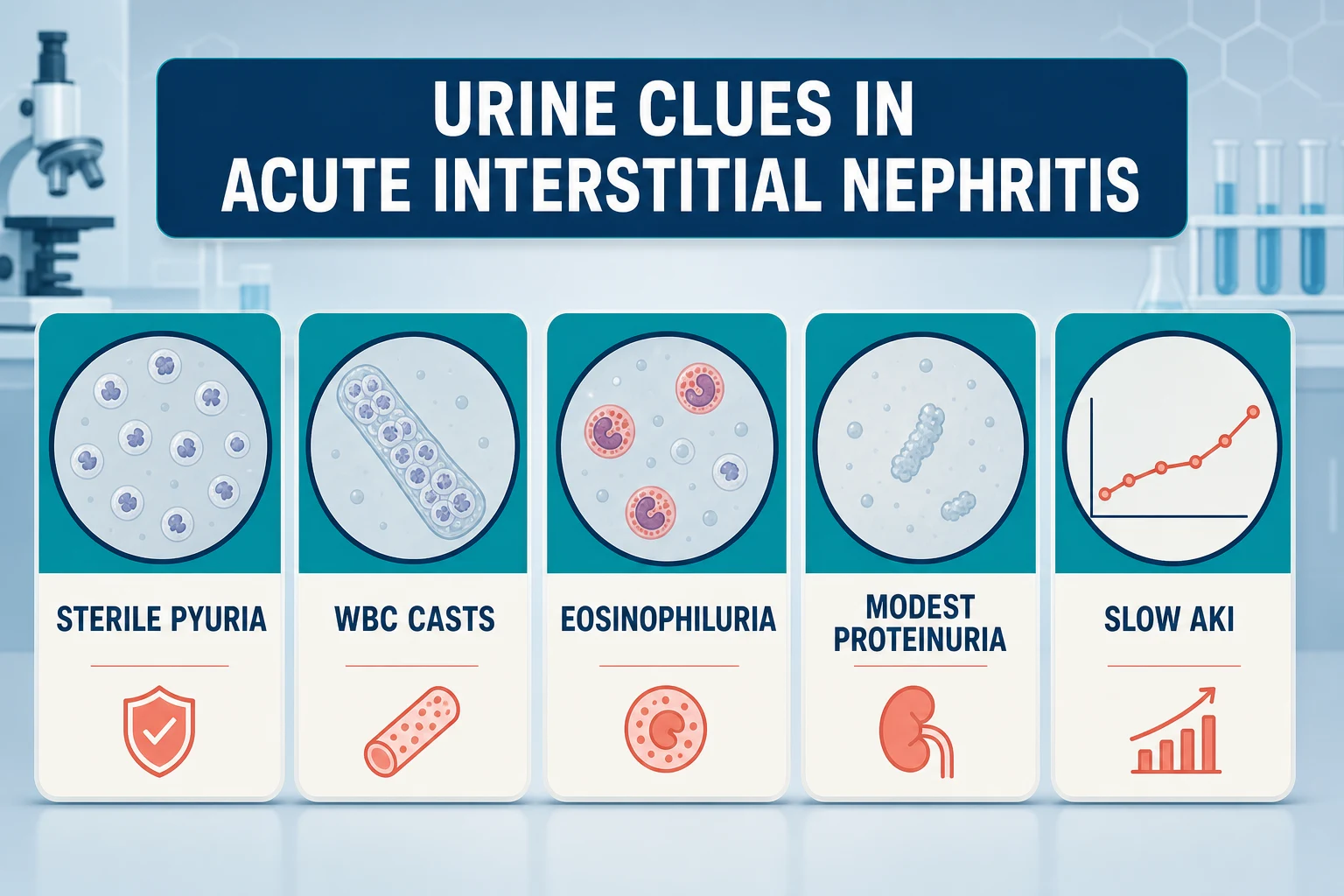
The urine gives you probability, not proof. The findings to name aloud, in the order examiners want them [6] [7]:
| Finding | What it tells you | The catch |
|---|---|---|
| Sterile pyuria | White cells without bacteriuria — inflammation in the kidney, not the bladder | Non-specific: also seen in TB, stones, papillary necrosis, glomerulonephritis [6] |
| White-cell casts | The most localising urine clue — points the inflammation at the tubulointerstitium | Often absent; absence excludes nothing [1] |
| Eosinophiluria | Historically celebrated; Hansel stain improved detection over Wright stain | Poor sensitivity and specificity — present in cystitis, pyelonephritis, prostatitis and other AKI causes; a weak test that should change no decision on its own [8] [7] |
| Modest proteinuria | Usually sub-nephrotic, consistent with tubular rather than glomerular pathology | Nephrotic-range proteinuria with AIN points at an NSAID culprit [19] |
| Eosinophilia (blood) | Supportive when present | Present in a minority — a normal count excludes nothing [4] |
| Renal ultrasound | Normal-sized kidneys support an acute process | Supportive only; never diagnostic [1] |
The pivotal modern data on urinary eosinophils comes from a biopsy-proven case series: urinary eosinophils performed poorly as a discriminator between AIN and other causes of kidney disease, in line with the original description that defined eosinophiluria across a broad clinical spectrum beyond AIN [7] [8]. The exam translation is blunt: eosinophiluria neither rules AIN in nor rules it out, and a question that hinges on it is testing whether you know its limits [7].
Management — stop the drug, then the steroid conversation
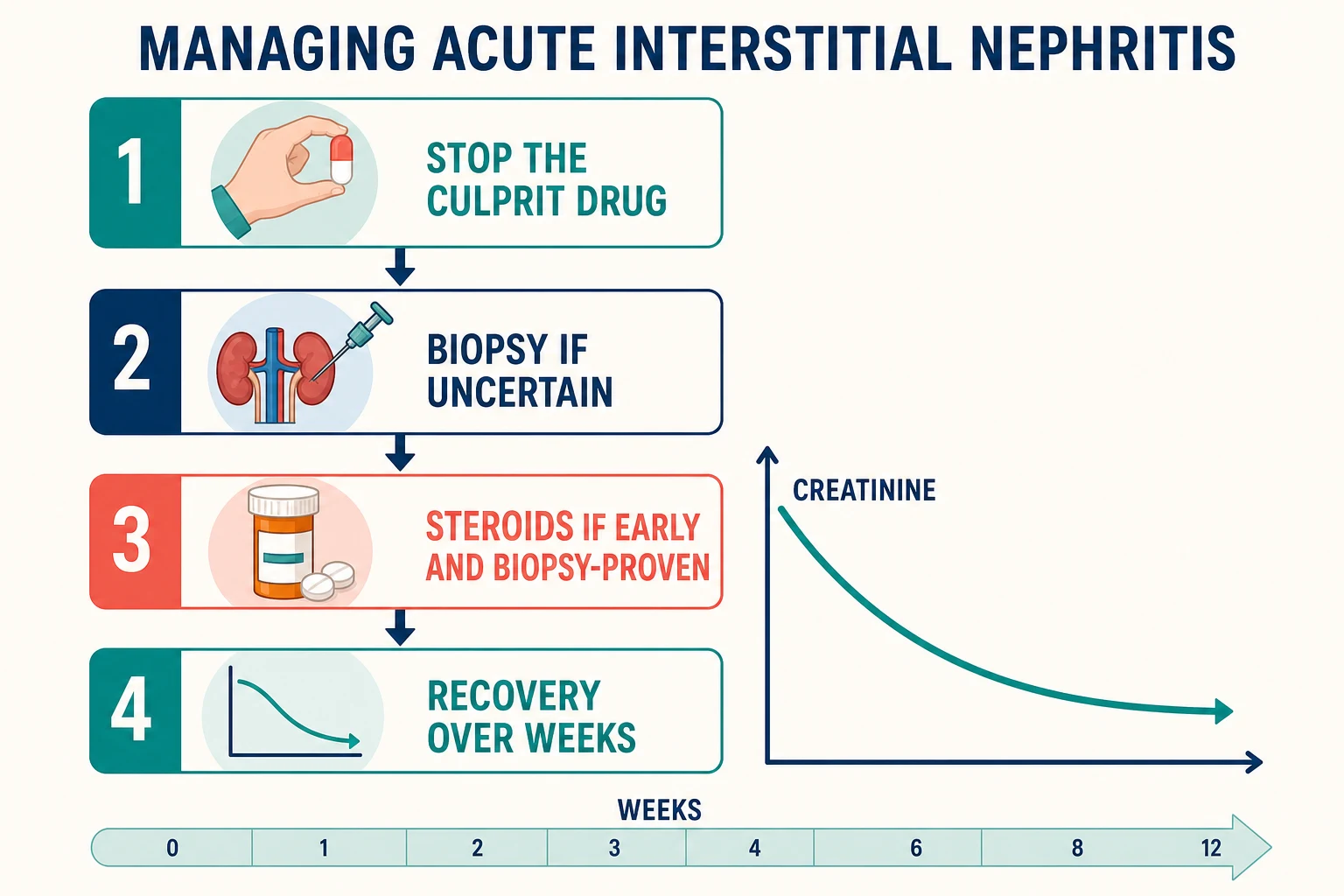
The management sequence
Stop the culprit drug now
Immediate withdrawal of the offending agent — and document it as an adverse drug reaction so it is never re-prescribed
Reconcile the whole chart
Deprescribe non-essential agents, avoid further nephrotoxins, and review herbal and over-the-counter products
Support the AKI
Volume status, potassium and acidosis surveillance; renal replacement therapy if the usual indications arise
Biopsy if uncertain or not recovering
Confirm the diagnosis and grade chronicity before committing to immunosuppression
Decide on steroids explicitly
Early, biopsy-proven, significant AKI, no contraindication — and frame it as a shared decision because the evidence is observational
Follow the recovery for months
Creatinine can improve over weeks; arrange CKD follow-up because incomplete recovery is common
Step one is not steroids — it is the drug chart. Withdrawal of the culprit agent is the one intervention every study and every reviewer agrees on, and the retrospective data tie delay in withdrawal to worse recovery [2]. In the González series of drug-induced AIN, both delayed withdrawal and delayed steroid initiation were associated with worse final renal function — the message that timing, not just treatment, is the lever [2].
The steroid evidence — know it by name
There has never been an adequate randomised trial of corticosteroids in AIN; the entire evidence base is retrospective and confounded by indication. The DWE expects you to hold that uncertainty honestly and still have a working position [13] [14].
| Study | Design | What it contributes |
|---|---|---|
| Clarkson 2004 | Retrospective cohort with corticosteroid response data | Clinical features catalogued; the allergic triad rare; steroid-treated patients tended to recover better [4] |
| González 2008 | Retrospective, drug-induced AIN | Early steroid treatment improved recovery of renal function; delays in withdrawal and in steroids both worsened outcomes [2] |
| Prendecki 2017 | Large multicentre retrospective series with long-term follow-up | Benefit concentrated in those treated early; little signal for late treatment [13] |
| Fernandez-Juarez 2018 | Retrospective comparison of steroid duration | No additional recovery benefit from prolonged courses over shorter tapers [14] |
The defensible synthesis: offer corticosteroids to the patient who is early (within days of withdrawal, before fibrosis accumulates), biopsy-proven, has significant AKI, and has no contraindication — and make it a shared decision, because the evidence is observational and the harms (hyperglycaemia, infection, delirium, myopathy) are real, especially in the elderly polypharmacy patient who typifies PPI-induced disease [2] [13]. There is no mandate to treat every AIN with steroids; a modest AKI already recovering after withdrawal is legitimately watched [14].
When steroids are given, the commonly used regimen is prednisone about 1 mg/kg daily (up to roughly 60–80 mg) with a taper over about 8–12 weeks — intravenous methylprednisolone first in severe cases — acknowledging that the duration data argue against dragging the taper out [1] [2] [14].
AIN management anchors
Checkpoint-inhibitor AIN — the oncology handshake
Immune checkpoint inhibitors (anti-PD-1, anti-PD-L1, anti-CTLA-4) have added a new, examinable chapter. AIN is the dominant lesion on biopsy in ICI-associated AKI, typically appearing weeks to months into therapy, often alongside other immune-related adverse events [12]. The mechanism fits the drug class: releasing T-cell brakes loses peripheral tolerance, and the kidney is one of the organs that pays for it [11].
Management is a negotiation, not a solo renal decision: hold the checkpoint inhibitor, treat with corticosteroids (most patients recover at least partial renal function), and decide on rechallenge jointly with oncology once renal function has stabilised — multicentre data show recurrent AKI occurs in a minority of rechallenged patients, so rechallenge is a real option when the cancer indication is strong, with close creatinine surveillance [11]. The viva-ready nuance: this is the one AIN setting where "stop the culprit drug forever" is explicitly negotiable, because the culprit may be the patient's only effective cancer therapy [11] [12].
The named syndromes — TINU, granulomatous, IgG4
TINU syndrome — tubulointerstitial nephritis with uveitis — is the trap with a physical sign. A patient (classically an adolescent or young adult, though it occurs at any age) presents with AIN, and the uveitis may precede, accompany or follow the nephritis by months; it is frequently relapsing [22]. The practical rules: ask about eye symptoms in every AIN, examine the eyes, get ophthalmology to slit-lamp them, and warn the patient that the uveitis can return after the kidneys have recovered [22].
Granulomatous interstitial nephritis has its own short differential: drugs and sarcoidosis are the commonest causes, with tuberculosis and other infections behind them — so granulomas on biopsy redirect the workup toward chest imaging, calcium, ACE and a TB assessment rather than simply lengthening the drug-stop list [20].
IgG4-related tubulointerstitial nephritis typically affects older men, often with other organ involvement (pancreas, salivary glands, retroperitoneum), a high serum IgG4, characteristic imaging lesions, and a plasma-cell-rich infiltrate with IgG4 staining on biopsy; it is notably steroid-responsive, which makes it the AIN variant you least want to miss [21].
Chronic tubulointerstitial nephritis — the slow burns
Chronic TIN presents differently: years of slowly progressive CKD, tubular dysfunction out of proportion to GFR (concentrating defects, distal acidification problems), modest proteinuria, bland sediment, and kidneys that may be small or scarred on imaging. The causes are the exam's favourites because each carries a story [17] [18]:
| Cause | The story to tell | Key clinical points |
|---|---|---|
| Analgesic nephropathy | Chronic daily combination-analgesic use (historically phenacetin-containing), often for headache or back pain | Renal papillary necrosis with chronic TIN; calcifications at the papillae on CT; stop the analgesics and progression slows [17] |
| Lithium nephropathy | Decades of lithium for bipolar disorder | Chronic TIN with tubular microcysts; nephrogenic diabetes insipidus; can progress despite withdrawal — the hardest deprescribing conversation in nephrology, taken with psychiatry [18] |
| Aristolochic acid nephropathy | Chinese-herb slimming remedies (the Belgian outbreak) and Balkan endemic nephropathy along the Danube | Rapidly progressive interstitial fibrosis; characteristic DNA adducts and mutational signature; lifelong urothelial cancer surveillance — many patients undergo prophylactic upper-tract surgery [15] [16] |
| Sarcoidosis | Granulomatous TIN with hypercalcaemia | Steroid-responsive; look for pulmonary and other organ disease [20] |
| Other (reflux nephropathy, obstruction, hypokalaemic nephropathy, lead) | Mechanical and metabolic chronic interstitial injury | The biopsy theme is shared: fibrosis and tubular atrophy dominate [6] |
Recovery and prognosis
Recovery from AIN is slow and often incomplete, and saying so early is part of good consulting. Creatinine typically begins improving within days to weeks of drug withdrawal (faster with early steroids in the responders), but the nadir-to-plateau journey runs over weeks, and a substantial fraction of biopsy-proven patients are left with CKD — the series that track long-term function after steroids make this plain [2] [13]. The predictors of incomplete recovery are the same in every series: delayed withdrawal, delayed treatment, and interstitial fibrosis already present on biopsy [2] [4].
The follow-up plan is therefore part of the management, not an afterthought: repeat creatinine through the recovery arc, review blood pressure and proteinuria, avoid re-exposure to the culprit class, document the adverse drug reaction, and refer back to nephrology if function plateaus at a reduced eGFR — because "recovered from AIN" and "back at baseline" are not synonyms [1] [13].
The exam angles — DCE long case and data station
The long case is the biopsy-proven PPI AIN in a polypharmacy patient: a 74-year-old whose creatinine drifted for six weeks on omeprazole, no rash, no fever, eosinophils normal, biopsied when function failed to recover. The examiner's interest is never the histology — it is your medication reasoning: how you reconciled the chart, what you deprescribed and why, how you defended (or declined) steroids with observational evidence, how you counselled about incomplete recovery, and how you stopped the next doctor restarting a PPI "for reflux" without a plan [9] [2]. Bring a deprescribing framework (indication, benefit, harm, burden, patient priorities) and the open-disclosure obligation that comes with an iatrogenic injury [5].
The short case is a data station, not a bedside: urine microscopy showing sterile pyuria with white-cell casts, a drug chart containing a recently started agent, and a creatinine trend. The task is to integrate them — name AIN as the unifying diagnosis, state what you would stop, state when you would biopsy, and resist every invitation to lean on eosinophiluria or the absent rash [7] [8].
Exam traps, collected
References
- [1]Praga M, González E. Acute interstitial nephritis Kidney Int, 2010.PMID 20336051
- [2]González E, Gutiérrez E, Galeano C, et al. Early steroid treatment improves the recovery of renal function in patients with drug-induced acute interstitial nephritis Kidney Int, 2008.PMID 18185501
- [3]Baker RJ, Pusey CD. The changing profile of acute tubulointerstitial nephritis Nephrol Dial Transplant, 2004.PMID 14671029
- [4]Clarkson MR, Giblin L, O'Connell FP, et al. Acute interstitial nephritis: clinical features and response to corticosteroid therapy Nephrol Dial Transplant, 2004.PMID 15340098
- [5]Moledina DG, Perazella MA. Drug-Induced Acute Interstitial Nephritis Clin J Am Soc Nephrol, 2017.PMID 28893923
- [6]Raghavan R, Eknoyan G. Acute interstitial nephritis - a reappraisal and update Clin Nephrol, 2014.PMID 25079860
- [7]Muriithi AK, Leung N, Valeri AM, et al. Biopsy-proven acute interstitial nephritis, 1993-2011: a case series Am J Kidney Dis, 2014.PMID 24927897
- [8]Nolan CR 3rd, Anger MS, Kelleher SP. Eosinophiluria--a new method of detection and definition of the clinical spectrum N Engl J Med, 1986.PMID 2431314
- [9]Blank ML, Parkin L, Paul C, Herbison P. A nationwide nested case-control study indicates an increased risk of acute interstitial nephritis with proton pump inhibitor use Kidney Int, 2014.PMID 24646856
- [10]Sierra F, Suarez M, Rey M, Vela MF. Systematic review: Proton pump inhibitor-associated acute interstitial nephritis Aliment Pharmacol Ther, 2007.PMID 17661758
- [11]Cortazar FB, Kibbelaar ZA, Glezerman IG, et al. Clinical Features and Outcomes of Immune Checkpoint Inhibitor-Associated AKI: A Multicenter Study J Am Soc Nephrol, 2020.PMID 31896554
- [12]Cortazar FB, Marrone KA, Troxell ML, et al. Clinicopathological features of acute kidney injury associated with immune checkpoint inhibitors Kidney Int, 2016.PMID 27282937
- [13]Prendecki M, Tanna A, Salama AD, et al. Long-term outcome in biopsy-proven acute interstitial nephritis treated with steroids Clin Kidney J, 2017.PMID 28396740
- [14]Fernandez-Juarez G, Perez JV, Caravaca-Fontán F, et al. Duration of Treatment with Corticosteroids and Recovery of Kidney Function in Acute Interstitial Nephritis Clin J Am Soc Nephrol, 2018.PMID 30397027
- [15]Vanherweghem JL, Depierreux M, Tielemans C, et al. Rapidly progressive interstitial renal fibrosis in young women: association with slimming regimen including Chinese herbs Lancet, 1993.PMID 8094166
- [16]Grollman AP, Shibutani S, Moriya M, et al. Aristolochic acid and the etiology of endemic (Balkan) nephropathy Proc Natl Acad Sci U S A, 2007.PMID 17620607
- [17]De Broe ME, Elseviers MM. Analgesic nephropathy N Engl J Med, 1998.PMID 9459649
- [18]Markowitz GS, Radhakrishnan J, Kambham N, et al. Lithium nephrotoxicity: a progressive combined glomerular and tubulointerstitial nephropathy J Am Soc Nephrol, 2000.PMID 10906157
- [19]Clive DM, Stoff JS. Renal syndromes associated with nonsteroidal antiinflammatory drugs N Engl J Med, 1984.PMID 6363936
- [20]Joss N, Morris S, Young B, Geddes C. Granulomatous interstitial nephritis Clin J Am Soc Nephrol, 2007.PMID 17699417
- [21]Raissian Y, Nasr SH, Larsen CP, et al. Diagnosis of IgG4-related tubulointerstitial nephritis J Am Soc Nephrol, 2011.PMID 21719792
- [22]Mackensen F, Billing H. Tubulointerstitial nephritis and uveitis syndrome Curr Opin Ophthalmol, 2009.PMID 19752730