Phys · respiratory
Diffuse Alveolar Haemorrhage and Pulmonary-Renal Syndromes
Also known as DAH · diffuse alveolar hemorrhage · alveolar haemorrhage · pulmonary-renal syndrome · pulmonary renal syndrome · lung-kidney syndrome · Goodpasture syndrome · anti-GBM disease · pulmonary capillaritis · idiopathic pulmonary haemosiderosis
Consultant-physician-depth guide to diffuse alveolar haemorrhage — the haemoptysis-absent trap, capillaritis versus bland versus diffuse alveolar damage mechanisms, serial BAL diagnosis, the ANCA/anti-GBM/SLE pulmonary-renal differential, and evidence-based severe DAH management from pulse methylprednisolone to plasma exchange — structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Diffuse Alveolar Haemorrhage and Pulmonary-Renal Syndromes
The answer first
Diffuse alveolar haemorrhage (DAH) is bleeding into the alveolar spaces from the pulmonary microcirculation — a syndrome, not a diagnosis. The job is never just to recognise it; it is to find which of the immune, cardiac, toxic or coagulopathic causes is bleeding, because the treatments are opposites [1] [2]. Three rules carry you through the DWE and the DCE:
- Haemoptysis is not required. Up to a third of DAH episodes present without any haemoptysis. The real triad is a falling haemoglobin, new diffuse pulmonary infiltrates, and worsening hypoxaemia — in a ventilated ICU patient that combination, unexplained by pneumonia or pulmonary oedema, is DAH until bronchoscopy proves otherwise [1] [2].
- Bronchoscopy with serial lavage is the diagnostic move. Not another CT, not empiric antibiotics: bronchoalveolar lavage aliquots that become progressively bloodier, later confirmed by haemosiderin-laden macrophages [1].
- The urine finishes the examination. DAH plus an active urinary sediment (haematuria, red-cell casts, rising creatinine) is the pulmonary-renal syndrome — and the shortlist is ANCA-associated vasculitis, anti-GBM disease and SLE. Same-day serology decides the treatment: plasma exchange is the cornerstone for anti-GBM, while rituximab or cyclophosphamide anchors ANCA vasculitis — and after PEXIVAS, plasma exchange is no longer routine for ANCA disease [7] [12] [16].
What DAH is — and the trap that fails candidates
DAH arises from disruption of the alveolar-capillary membrane, so blood floods the airspaces faster than macrophages can clear it. Any cause of diffuse lung bleeding can produce it, and the clinical signature is deceptively quiet: dyspnoea, cough, low-grade fever and fatigue are common, while the textbook symptom — haemoptysis — is absent in up to a third of presentations, because blood distributes through alveoli without reaching the central airways [1] [2].
The surrogate triad that examiners want you to verbalise is falling haemoglobin, new or changing bilateral infiltrates, and disproportionate hypoxaemia — especially when the trajectory is days, and especially in a patient with an autoimmune background, new haematuria, anticoagulation, or a cardiac valve lesion [1].
A useful physiological footnote for vivas: fresh intra-alveolar blood avidly binds inhaled carbon monoxide, so the DLCO (or KCO) is elevated in acute DAH — a raised or rising DLCO with new infiltrates is a subtle, stable-patient clue that what looks like infection is actually blood [1].
Severity has no formal score, but the working triage is pragmatic: mild DAH is managed on the ward with workup proceeding in parallel; severe DAH is any episode with hypoxaemic respiratory failure, a rapid haemoglobin fall, or haemodynamic compromise — and it belongs in the ICU with treatment started on suspicion rather than after the full serology panel returns. Quantifying the haemorrhage matters less than quantifying its trajectory: a Golde score or haemosiderin-macrophage percentage on cytology confirms that bleeding occurred, but the daily haemoglobin, the oxygen requirement and the repeat lavage appearance tell you whether it is still happening [1] [2].
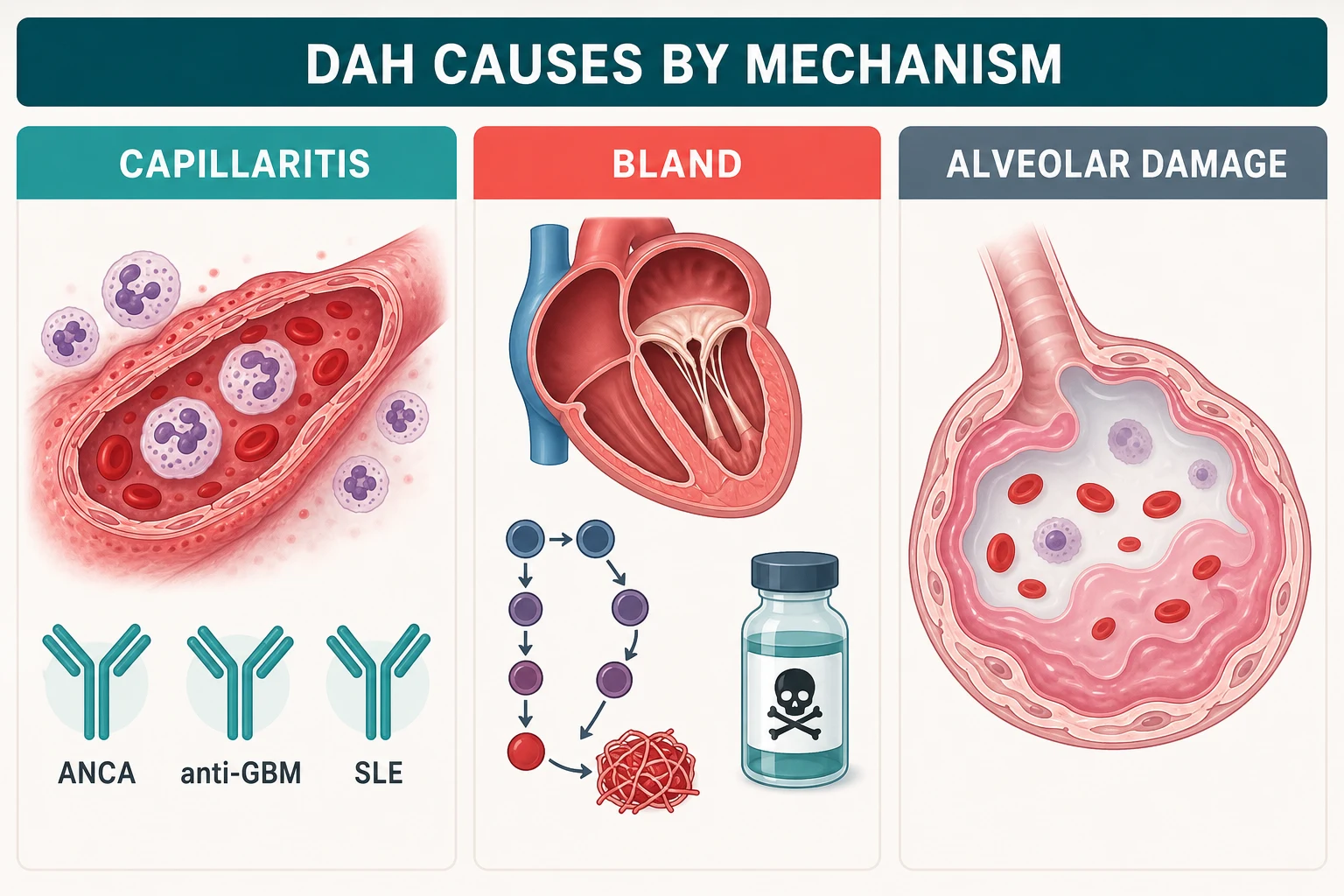
Three mechanisms — read the biopsy without a biopsy
Histology sorts DAH into three patterns, and each pattern carries its own cause list. You rarely see the lung tissue, so you infer the mechanism from the story, the serology and the kidney [2].
| Mechanism | What is happening | Classic causes | Exam anchor |
|---|---|---|---|
| Pulmonary capillaritis | Neutrophilic infiltration of alveolar septal capillaries with fibrinoid necrosis — vessel walls destroyed, blood leaks | ANCA vasculitis (GPA, MPA, eGPA), anti-GBM disease, SLE, antiphospholipid syndrome, drug-induced (propylthiouracil, hydralazine) | The immune causes — this is where immunosuppression works [2] |
| Bland alveolar haemorrhage | Blood leaks without inflammation — pressure or haemostasis failure | Mitral stenosis and severe left-heart failure, coagulopathy and anticoagulation, inhaled toxins such as crack cocaine | Treat the valve, reverse the anticoagulant, remove the toxin — NOT steroids [19] |
| Diffuse alveolar damage | Alveolar epithelial injury with hyaline membranes — the ARDS pattern bleeding | Bone-marrow transplant, cytotoxic drugs, radiation, severe sepsis and ARDS, idiopathic | Supportive care; the bleeding reflects lung injury itself [2] |
The causes map — who bleeds, and why
The cause list is long, but the exam shortlist is short. Learn it as layers: the autoimmune big three, the cardiac and haemostatic mimics, the toxins and drugs, and the diagnosis of exclusion [1] [2].
| Layer | Cause | The tell |
|---|---|---|
| Autoimmune | ANCA vasculitis — GPA (PR3), MPA (MPO), eGPA (MPO with asthma and eosinophilia) | Upper-airway disease, purpuric rash, mononeuritis, active urine sediment; DAH is the most feared AAV lung manifestation [3] |
| Autoimmune | Anti-GBM (Goodpasture) disease | Young male smokers; simultaneous lung and kidney; linear IgG on immunofluorescence [5] [7] |
| Autoimmune | SLE | Usually established lupus with nephritis; high dsDNA, low complement, thrombocytopenia; mortality is high [17] |
| Cardiac | Mitral stenosis, severe left-heart failure | Chronic pulmonary venous hypertension; miliary haemosiderosis pattern on CT; diastolic murmur and opening snap if you listen [19] |
| Haemostatic | Anticoagulation (warfarin, DOACs), thrombolytics, DIC, thrombocytopenia | The drug chart and the coagulation profile make the diagnosis [1] |
| Toxic and iatrogenic | Crack cocaine, hydrocarbon solvents; propylthiouracil and hydralazine (drug-induced ANCA); checkpoint inhibitors; bone-marrow transplant | Exposure history; PTU-ANCA is classically MPO and can cause true capillaritis [1] [2] |
| Idiopathic | Idiopathic pulmonary haemosiderosis | Mostly children and young adults; recurrent DAH with iron-deficiency anaemia, negative serology, bland histology with haemosiderin — a diagnosis of exclusion [18] |
Workup — CT, then the bronchoscope, then the serology, then the kidney
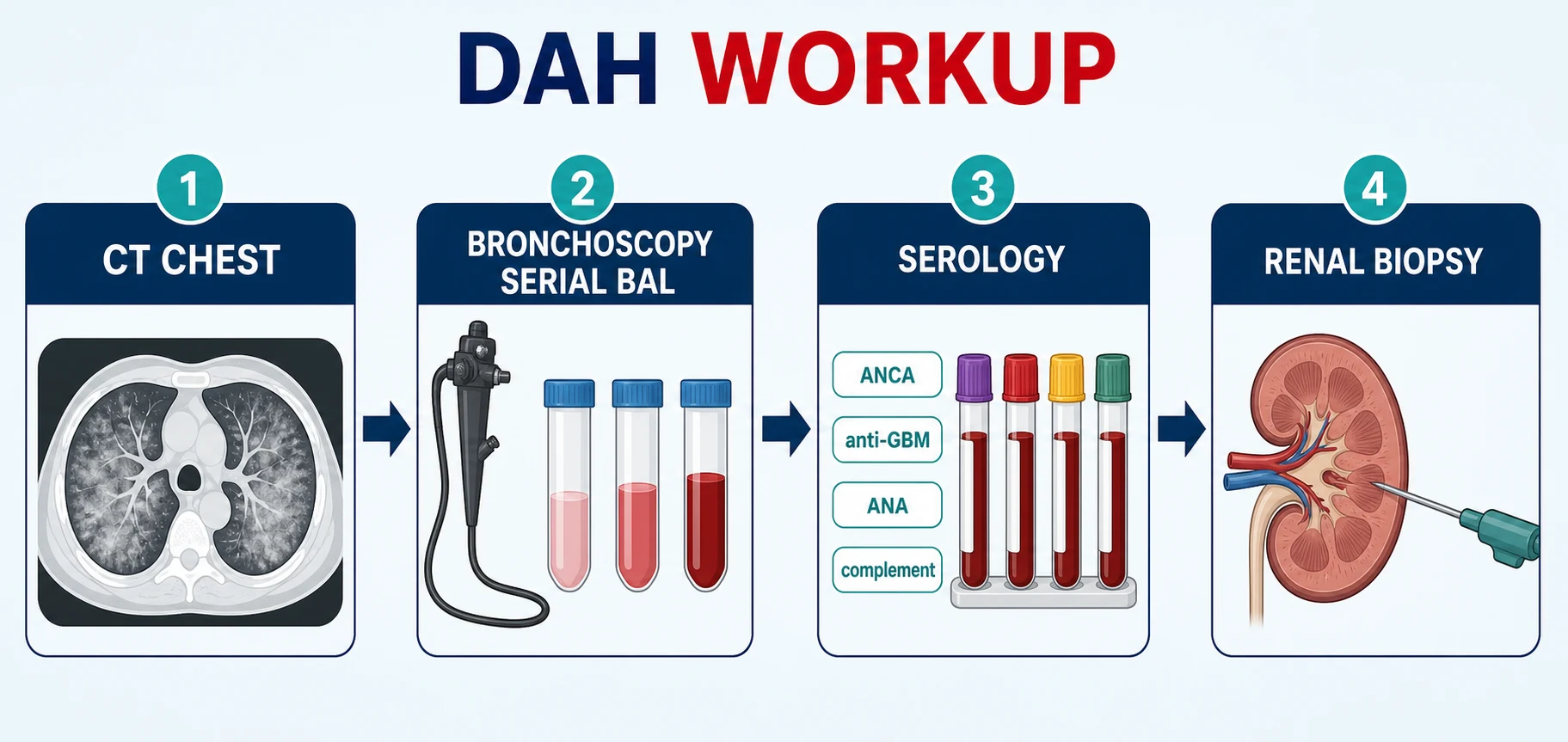
The workup has a rhythm that examiners reward: confirm the bleeding, then hunt the cause in parallel — and do not let any single negative test slow the sequence when the patient is deteriorating [1].
The DAH workup sequence
CT chest (non-contrast HRCT)
Diffuse or patchy ground-glass opacities and consolidation, typically mid and lower zones; chronic haemosiderosis gives a miliary or interstitial pattern; CT localises but does not prove blood
Bronchoscopy with serial BAL
Three aliquots from the same segment becoming progressively bloodier confirms DAH; send cytology for haemosiderin-laden macrophages (Prussian blue), plus cultures to exclude infection
Serology panel, same day
ANCA by ELISA with MPO and PR3 specificity, anti-GBM antibody, ANA and dsDNA, complement C3/C4, antiphospholipid antibodies if suggested; HIV and hepatitis serologies complete the renal-safe panel
The urine
Dipstick for blood and protein; microscopy for dysmorphic red cells and red-cell casts; protein:creatinine ratio; creatinine trend — the kidney tells you whether this is pulmonary-renal syndrome
Renal biopsy when the kidney is involved
Pauci-immune necrotising crescentic GN (ANCA), linear IgG (anti-GBM), or immune-complex GN (lupus) — the biopsy drives prognosis and treatment intensity
Echocardiogram
Exclude mitral stenosis and estimate pulmonary pressures in every unexplained or recurrent case before labelling anything idiopathic
Serology interpretation is where the DWE lives. MPO-ANCA points to microscopic polyangiitis (and eGPA in the right story), PR3-ANCA to granulomatosis with polyangiitis, anti-GBM antibody to Goodpasture disease — send it by immunoassay, and remember up to 10% of anti-GBM patients are double-positive for ANCA, usually MPO, which changes their relapse risk to the ANCA pattern [4] [7]. ANA with high dsDNA and low complement points to SLE; isolated low complement with immune deposits suggests lupus or cryoglobulinaemia rather than the pauci-immune vasculitides [17].
When the kidney is involved, the biopsy is the decision-maker: pauci-immune necrotising crescentic GN (little immunoglobulin — the ANCA pattern), linear IgG along the glomerular basement membrane (the anti-GBM pattern, mirrored in the lung), or full-house immune-complex GN (lupus). A patient too sick or too anticoagulated for biopsy is treated on the serology — do not let tissue become the bottleneck for plasma exchange or induction therapy [6] [16].
Lung biopsy is the opposite case: rarely required and usually avoided acutely. The combination of serial BAL, serology and renal tissue answers the management question in almost every patient, and surgical lung biopsy in a hypoxic, bleeding lung carries real morbidity. Reserve it for the patient with negative serology, bland urine and an unclear mechanism — the suspected idiopathic pulmonary haemosiderosis or isolated capillaritis — where the histology (capillaritis versus bland haemorrhage with haemosiderin) is the only way forward [2] [18].
The pulmonary-renal differential — the table examiners test
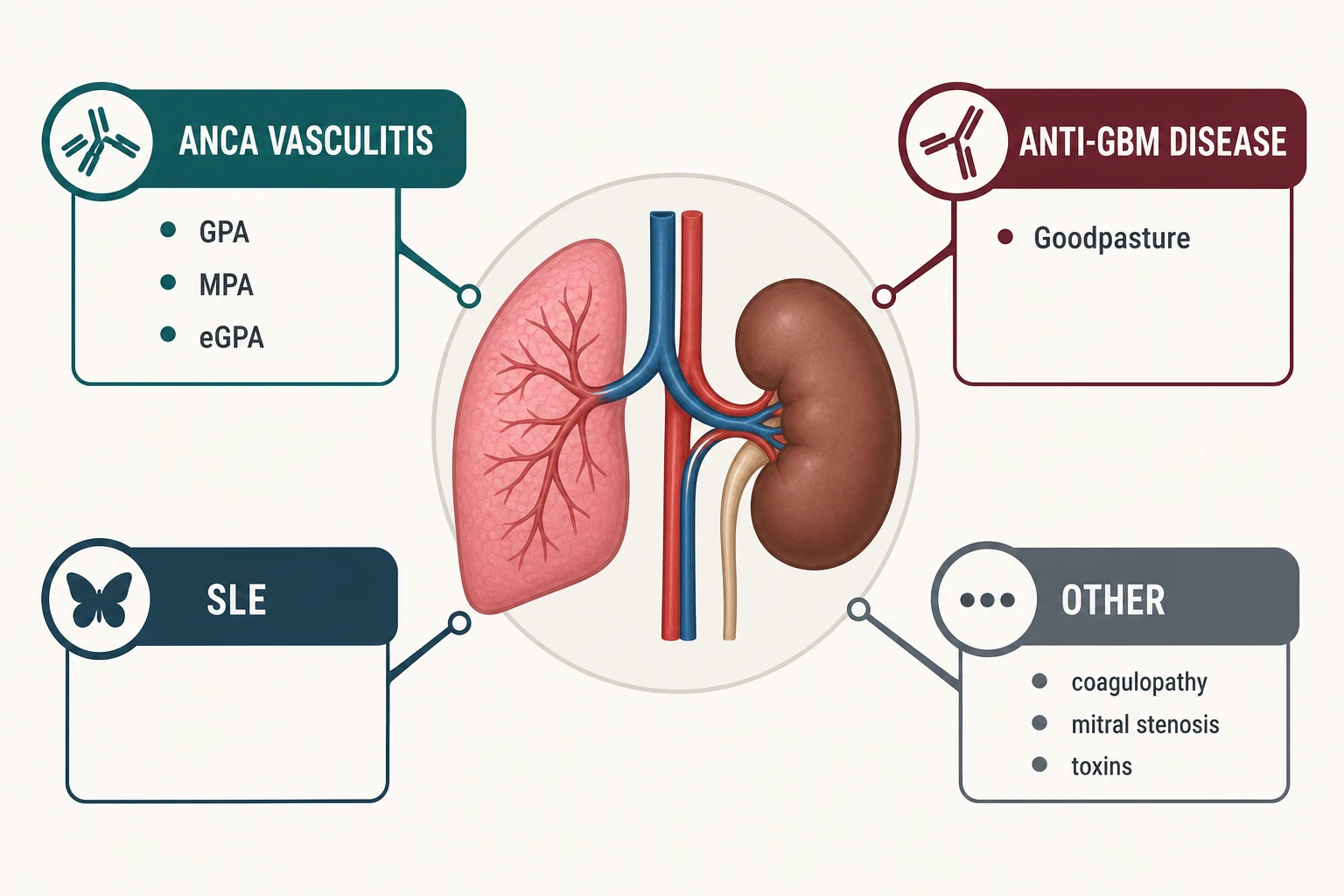
Pulmonary-renal syndrome has a short differential with long consequences. The pattern-recognition table below is the high-yield core of this topic [1] [7].
| Feature | Anti-GBM disease | GPA (PR3-ANCA) | MPA (MPO-ANCA) | SLE |
|---|---|---|---|---|
| Typical patient | Young male smoker | 50s–60s, either sex | 60s–70s, either sex | Young woman, often known SLE |
| Lung | DAH, often severe; trigger-linked (smoking, hydrocarbons) [5] | DAH plus nodules, cavitation, endobronchial disease | DAH plus pulmonary fibrosis background | DAH with pleuritis and effusions [17] |
| Kidney | Crescentic GN, linear IgG; rapid progression | Pauci-immune crescentic GN | Pauci-immune crescentic GN | Immune-complex (full-house) GN |
| Serology | Anti-GBM positive; up to 10% also MPO-ANCA [4] | PR3-ANCA positive | MPO-ANCA positive | ANA, high dsDNA, LOW complement |
| Extra clues | No systemic features beyond lung and kidney | Saddle-nose, nasal crusting, sinusitis, subglottic stenosis | Neuropathy, rash; less upper-airway disease | Rash, arthritis, serositis, cytopenias |
| Cornerstone treatment | Plasma exchange + steroids + cyclophosphamide [7] | Rituximab or cyclophosphamide + steroids [8] | Same — rituximab or cyclophosphamide | Steroids + cyclophosphamide or rituximab |
| Relapse pattern | Rare once antibody cleared — unless double ANCA-positive | Relapses — needs maintenance | Relapses — needs maintenance | Flares with lupus activity [14] |
Two additions round out the differential honestly: eGPA (asthma, eosinophilia, MPO-ANCA in about a third — renal disease is usually milder than in GPA/MPA, but DAH occurs), and the infective mimics (endocarditis with septic emboli and immune-complex GN, severe leptospirosis) which must be excluded before committing a patient to months of immunosuppression [1] [16].
Drug-induced vasculitis and antiphospholipid disease deserve explicit mention because both are treatable by subtraction. Propylthiouracil and hydralazine induce MPO-ANCA antibodies that can produce true pulmonary capillaritis with DAH — the treatment is stopping the drug, with immunosuppression reserved for organ-threatening disease [1] [2]. Antiphospholipid syndrome bleeds through a different door: microvascular thrombosis plus anticoagulation, so the management balances continued anticoagulation against the bleeding — a genuinely difficult call that examiners reward you for naming rather than solving glibly [2].
Anti-GBM disease — the smoking gun diagnosis
Anti-GBM disease is autoimmunity against the alpha-3 chain of type IV collagen, the same basement-membrane antigen in glomerulus and alveolus. The epitope is normally cryptic; alveolar injury exposes it, which is why the lung disease clusters in smokers and those with hydrocarbon or solvent exposure — the classic 1983 observation that cigarette smoking tracks with lung haemorrhage in anti-GBM nephritis remains the best exam answer to 'why do only some patients bleed from the lung?' [5] [7]. Systematic-review data confirm a bimodal young-male predominance, simultaneous lung and kidney involvement in most presentations, and a strong association between pulmonary haemorrhage and worse early mortality [4].
The diagnostic signature is linear IgG deposition along the glomerular basement membrane on immunofluorescence — smooth and continuous, unlike the granular deposits of immune-complex disease. Serum anti-GBM immunoassay is fast and reliable enough that treatment starts on the serology, with the biopsy confirming and prognosticating (proportion of crescents, degree of sclerosis) [7].
Treatment is a race to clear the antibody before the kidney scars [6] [7]:
- Plasma exchange — daily exchanges (typically for 10–14 days or until the antibody is cleared) physically remove anti-GBM IgG; it is the cornerstone and the one pulmonary-renal indication where apheresis is uncontested [6] [7].
- Glucocorticoids — pulse methylprednisolone then oral taper, as the anti-inflammatory base [6].
- Cyclophosphamide — classically added to stop new antibody synthesis; rituximab and the IgG-cleaving enzyme imlifidase (IdeS) are the modern salvage and bridging options discussed at viva level [7].
The double-positive patient (anti-GBM plus MPO-ANCA) deserves its own sentence in any viva: they present like anti-GBM but relapse like ANCA vasculitis, so they need both the acute plasma exchange and the longer-term maintenance thinking of AAV [4] [7].
ANCA vasculitis with DAH — the induction evidence, in order
DAH is the most dangerous pulmonary manifestation of ANCA-associated vasculitis: cohort data show severe alveolar haemorrhage marks a subgroup with high early mortality and a heavy relapse burden, which is why it defines 'severe disease' at induction [3] [16]. The trials, in the order examiners expect:
| Trial | Population | Comparison | Result | What it changed |
|---|---|---|---|---|
| RAVE (2010) | Severe AAV, new or relapsing | Rituximab (375 mg/m² weekly × 4) vs cyclophosphamide→azathioprine, both with steroids | Rituximab non-inferior for remission at 6 months; trend to superiority in relapsing disease | Rituximab became a first-line induction agent [8] |
| RITUXVAS (2010) | Newly diagnosed AAV with renal involvement | Rituximab + 2 cyclophosphamide pulses vs standard cyclophosphamide | Non-inferior remission, similar adverse events | Rituximab works in severe renal disease [9] |
| CYCLOPS (2009) | Generalised AAV | Pulse IV cyclophosphamide vs daily oral | Equal remission rates, lower cumulative dose with pulses — at the cost of more relapse | Pulse dosing is standard; relapse risk is the price [10] |
| MEPEX (2007) | Severe renal vasculitis (creatinine above about 500 micromol/L) | Plasma exchange vs pulse IV methylprednisolone, added to oral cyclophosphamide | PLEX improved dialysis independence at 3–12 months | The historical basis for PLEX in severe renal AAV [11] |
| PEXIVAS (2020) | Severe AAV (eGFR below 50 mL/min or DAH) | PLEX vs no PLEX; standard-dose vs reduced-dose steroids, all with rituximab or cyclophosphamide | No PLEX benefit on death or ESKD; reduced-dose steroids non-inferior with fewer infections | PLEX is no longer routine in AAV; steroid minimisation is [12] |
| ADVOCATE (2021) | AAV on rituximab or cyclophosphamide | Avacopan (oral C5a-receptor antagonist) vs prednisone taper | Avacopan non-inferior at week 26, superior at week 52, steroid-sparing | A licensed steroid-sparing adjunct [13] |
Rituximab versus cyclophosphamide is the other recurring viva fork. Choose rituximab for relapsing disease, PR3-ANCA/GPA phenotype, younger patients where fertility and bladder-cancer exposure matter, and where maintenance will also be rituximab. Choose cyclophosphamide when B-cell depletion is undesirable or rituximab has failed, acknowledging the fertility, bladder and marrow costs and keeping cumulative dose low with the pulse regimen [8] [10] [16].
A note on eGPA, because examiners probe its difference: it is the ANCA disease of asthma and eosinophilia, and its guideline pathway diverges for non-severe disease — mepolizumab and other steroid-sparing biologics feature where GPA and MPA do not. But severe eGPA with DAH or threatening renal disease is treated like any other severe AAV: pulse steroids plus rituximab or cyclophosphamide, with the same PEXIVAS restraint on apheresis [16].
Severe DAH — the ICU bundle
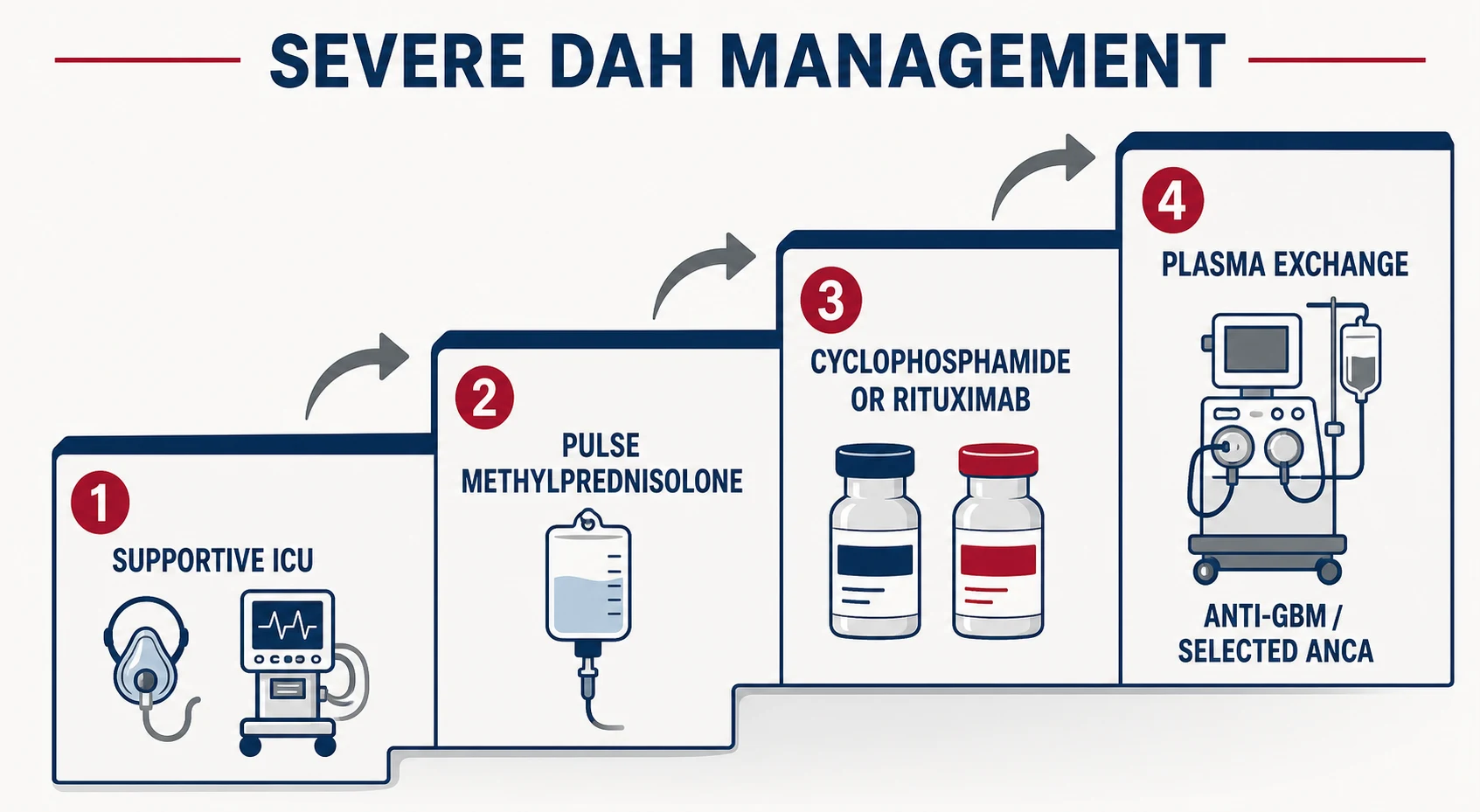
Severe DAH management runs on two rails simultaneously: keep the patient oxygenated while the cause-specific therapy lands [1] [16].
The severe-DAH pharmacology card (all with cause-specific framing)
The first 24 hours of severe DAH
ICU and airway
Early ICU involvement; intubate for refractory hypoxaemia; lung-protective ventilation with PEEP — the bleeding lung still obeys ARDS physiology
Stop the bleeding diathesis
Reverse anticoagulation, correct coagulopathy and thrombocytopenia, transfuse as required; hold the toxin or drug trigger (crack cocaine, PTU, checkpoint inhibitor)
Pulse methylprednisolone
500–1000 mg IV daily for 3 days on suspicion of immune DAH — do not wait for serology when capillaritis is likely
Cause-specific induction
Rituximab or cyclophosphamide for ANCA vasculitis; add daily plasma exchange NOW for anti-GBM disease; cyclophosphamide or rituximab for lupus DAH
Infection cover and prophylaxis
BAL cultures guide antibiotics; start PJP prophylaxis with combination immunosuppression; screen hepatitis B/C and HIV before rituximab or cyclophosphamide
Taper and plan maintenance
Switch to oral prednisolone taper after the pulse; book the maintenance decision (rituximab vs azathioprine) before discharge, not after relapse
Supportive details that separate consultant answers from registrar ones: expect a transient radiographic worsening as blood organises; re-bleeding in the first week usually means the trigger is still present or the diagnosis is wrong, so revisit anticoagulation, infection and the serology; and in the ventilated patient, daily haemoglobin trends plus BAL appearance are your monitoring, because CT cannot distinguish re-bleed from organising blood [1] [3].
For refractory hypoxaemia, the frame is bridge-to-therapy: immunosuppression takes days to land, so ventilation strategy buys time — lung-protective tidal volumes, PEEP titrated to oxygenation, proning for the ARDS physiology, and in selected quaternary centres veno-venous extracorporeal support while the capillaritis comes under control. Rescue haemostatics (intrapulmonary or systemic recombinant factor VIIa) appear in the literature only as case-level salvage — name them as such if asked, and never as standard care [1] [3].
Maintenance and relapse — what happens after the ICU
Induction wins the month; maintenance wins the years. The evidence anchors [14] [15] [16]:
- MAINRITSAN (2014): after cyclophosphamide induction, maintenance rituximab 500 mg every 6 months was superior to azathioprine for sustained remission — this established rituximab as the preferred maintenance for severe AAV [14].
- RITAZAREM (2023): in patients with relapsing AAV re-induced with rituximab, rituximab maintenance again beat azathioprine for preventing further relapse — so the relapsing phenotype gets rituximab at both steps [15].
- Azathioprine and mycophenolate remain the alternatives when rituximab is unsuitable; methotrexate is reserved for non-severe disease without renal involvement. The 2021 ACR/VF guideline prefers rituximab maintenance for severe AAV, particularly PR3 disease [16].
Working doses for the viva: rituximab 500 mg every 6 months (the MAINRITSAN schedule) or re-dose by CD19 count/ANCA titre where a tailored protocol is used; azathioprine about 2 mg/kg/day with TPMT-aware prescribing and blood-count surveillance; mycophenolate 2–3 g/day in divided doses as the azathioprine-alternative; methotrexate up to 20–25 mg weekly only in non-severe disease with preserved renal function [14] [16]. State a duration: at least 18–24 months of maintenance for severe AAV, longer for relapsing PR3 disease, and re-evaluate rather than stop by default [15] [16].
Monitoring is a system, not a blood test: scheduled clinical review with urinalysis, creatinine, CRP and full blood count; ANCA titres as an adjunct — a rising titre supports clinical suspicion but is not, alone, a reason to treat; haemoglobin trends and a low threshold for repeat CT or BAL when DAH recurrence is possible; and for anti-GBM disease, antibody clearance is the treatment endpoint, after which maintenance immunosuppression is usually unnecessary unless the patient is ANCA double-positive [7] [16].
Do not forget the unglamorous chronic care: smoking cessation (therapeutic in anti-GBM and GPA), vaccination before B-cell depletion, bone protection through the steroid tapers, fertility preservation before cyclophosphamide, and cumulative-dose bookkeeping across relapses [5] [10] [16].
Special situations the examiners reach for
Pregnancy. Active AAV or lupus DAH in pregnancy forces the safest-effective subset: glucocorticoids and azathioprine are the compatible backbone; cyclophosphamide is teratogenic and avoided, particularly in the first trimester; rituximab crosses the placenta in later pregnancy and causes neonatal B-cell depletion, so it is reserved for when the alternative is losing the mother. Say explicitly that severe DAH is a treat-first situation in which maternal stabilisation is the fetal intervention [16] [17].
The transplant and oncology ward. DAH after haematopoietic stem-cell transplantation and with cytotoxic or checkpoint-inhibitor therapy sits in the diffuse-alveolar-damage mechanism: management is supportive — lung protection, platelet and coagulation support, steroids in selected peri-engraftment or immune-related cases — and the prognosis tracks the underlying lung injury rather than any immune target [2].
The child with recurrent 'anaemia and pneumonia'. Recurrent DAH with iron-deficiency anaemia, negative serology and bland haemosiderin-laden histology is idiopathic pulmonary haemosiderosis — but only after the full exclusion hunt (serology, echo, coagulation, toxin history), because the label commits a child to long-term immunosuppression whose evidence base is case series [18].
DCE angles — how this topic is examined live
The long case is ANCA vasculitis with DAH and dialysis-dependent renal disease. The examiner's arc: presentation (did you pick up DAH without haemoptysis?), induction (rituximab vs cyclophosphamide — defend your choice), the PEXIVAS question (did plasma exchange belong in this patient?), maintenance (rituximab, and for how long?), and the systems layer (prophylaxis, vaccination, fertility, smoking, relapse plan). Have numbers: remission rates, the PEXIVAS composite, MAINRITSAN's relapse reduction [8] [12] [14].
The short case is the systemic vasculitis examination: palpable purpura on the legs, nasal crusting or a saddle-nose bridge, crusted bloody nasal mucosa, diffuse fine crackles, hypoxaemia at rest — and then the move that passes the station: ask for the urine dipstick and microscopy before you summarise, because the kidney completes the syndrome. Offer the serology panel you would send and the one treatment you would start tonight [1] [7].
Exam traps, collected
References
- [1]Ioachimescu OC, Stoller JK Diffuse alveolar hemorrhage: diagnosing it and finding the cause Cleve Clin J Med, 2008.PMID 18491433
- [2]Lara AR, Schwarz MI Diffuse alveolar hemorrhage Chest, 2010.PMID 20442117
- [3]Hruskova Z, Casian AL, Konopasek P, et al. Long-term outcome of severe alveolar haemorrhage in ANCA-associated vasculitis: a retrospective cohort study Scand J Rheumatol, 2013.PMID 23374071
- [4]Kuang H, Jiang N, Jia XY, et al. Epidemiology, clinical features, risk factors, and outcomes in anti-glomerular basement membrane disease: A systematic review and meta-analysis Autoimmun Rev, 2024.PMID 38493958
- [5]Donaghy M, Rees AJ Cigarette smoking and lung haemorrhage in glomerulonephritis caused by autoantibodies to glomerular basement membrane Lancet, 1983.PMID 6140495
- [6]Johnson JP, Moore J Jr, Austin HA 3rd, et al. Therapy of anti-glomerular basement membrane antibody disease: analysis of prognostic significance of clinical, pathologic and treatment factors Medicine (Baltimore), 1985.PMID 3892220
- [7]Shin JI, Geetha D, Szpirt WM, et al. Anti-glomerular basement membrane disease (Goodpasture disease): From pathogenesis to plasma exchange to IdeS Ther Apher Dial, 2022.PMID 34339589
- [8]Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis N Engl J Med, 2010.PMID 20647199
- [9]Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis N Engl J Med, 2010.PMID 20647198
- [10]de Groot K, Harper L, Jayne DR, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial Ann Intern Med, 2009.PMID 19451574
- [11]Jayne DR, Gaskin G, Rasmussen N, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis J Am Soc Nephrol, 2007.PMID 17582159
- [12]Walsh M, Merkel PA, Peh CA, et al. Plasma Exchange and Glucocorticoids in Severe ANCA-Associated Vasculitis N Engl J Med, 2020.PMID 32053298
- [13]Jayne DRW, Merkel PA, Schall TJ, et al. Avacopan for the Treatment of ANCA-Associated Vasculitis N Engl J Med, 2021.PMID 33596356
- [14]Guillevin L, Pagnoux C, Karras A, et al. Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis N Engl J Med, 2014.PMID 25372085
- [15]Smith RM, Jones RB, Specks U, et al. Rituximab versus azathioprine for maintenance of remission for patients with ANCA-associated vasculitis and relapsing disease: an international randomised controlled trial Ann Rheum Dis, 2023.PMID 36958796
- [16]Chung SA, Langford CA, Maz M, et al. 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the Management of Antineutrophil Cytoplasmic Antibody-Associated Vasculitis Arthritis Rheumatol, 2021.PMID 34235894
- [17]Zamora MR, Warner ML, Tuder R, et al. Diffuse alveolar hemorrhage and systemic lupus erythematosus. Clinical presentation, histology, survival, and outcome Medicine (Baltimore), 1997.PMID 9193454
- [18]Ioachimescu OC, Sieber S, Kotch A Idiopathic pulmonary haemosiderosis revisited Eur Respir J, 2004.PMID 15293620
- [19]Agrawal G, Agarwal R, Rohit MK, et al. Miliary nodules due to secondary pulmonary hemosiderosis in rheumatic heart disease World J Radiol, 2011.PMID 21390194