Phys · respiratory
Interstitial Lung Disease
Also known as diffuse parenchymal lung disease · DPLD · interstitial lung disease · ILD · idiopathic pulmonary fibrosis · IPF · usual interstitial pneumonia · UIP · fibrosing lung disease · pulmonary fibrosis · connective tissue disease-associated ILD · CTD-ILD · hypersensitivity pneumonitis · HP · sarcoidosis
Consultant-physician-depth guide to the interstitial lung diseases — classification, HRCT pattern recognition, diagnostic approach, and evidence-based management of IPF and the progressive fibrosing ILDs — structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Interstitial Lung Disease
The answer first
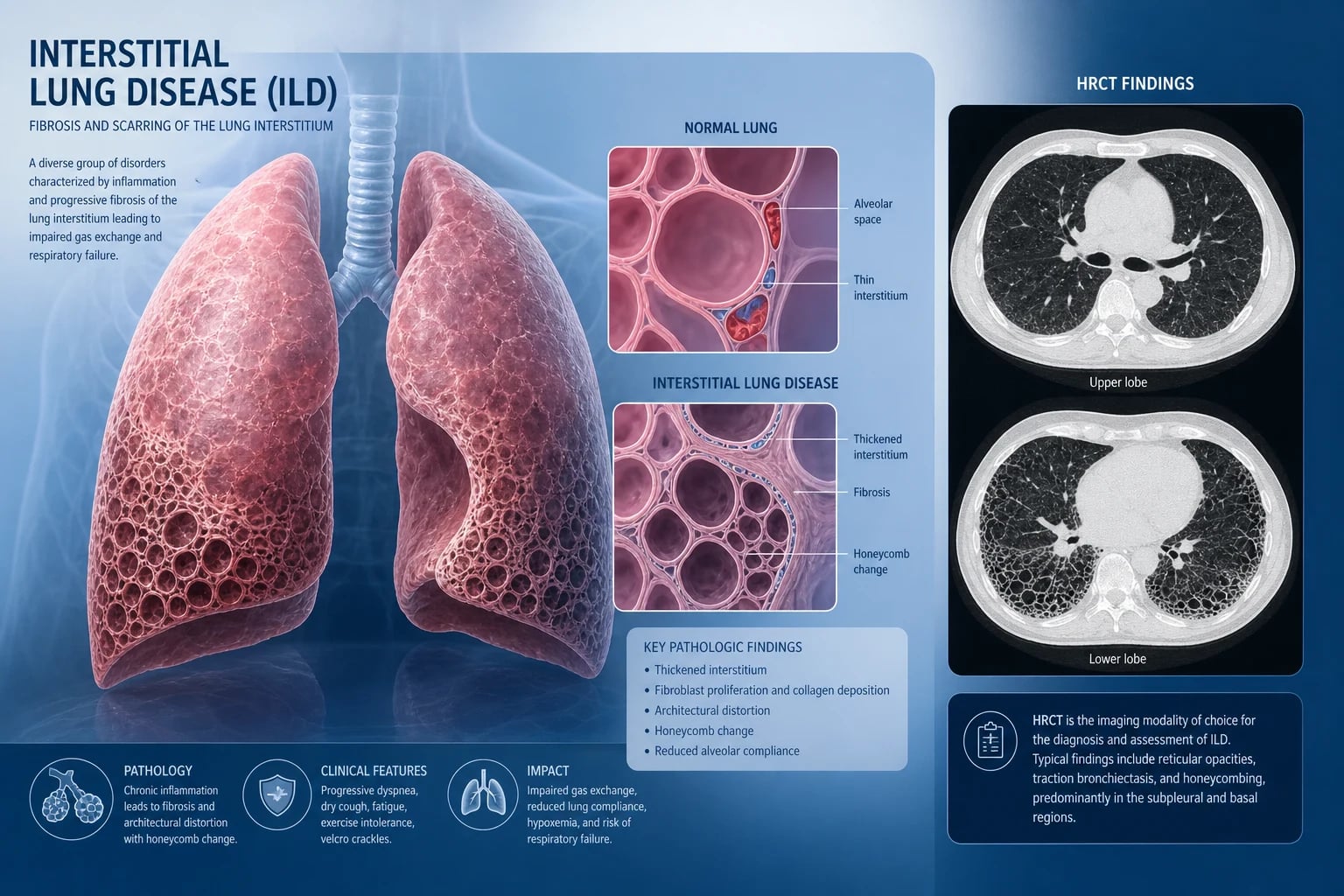
Interstitial lung disease (ILD) is a heterogenous group of over 200 disorders that share inflammation and fibrosis of the lung interstitium and alveolar walls. The term diffuse parenchymal lung disease (DPLD) is preferred by many because the disease is not confined to the interstitium — it involves alveoli, airways, and vasculature. [1]
Three facts must land before everything else: [1]
- A confident diagnosis requires a multidisciplinary team (MDT) discussion — a respiratory physician, thoracic radiologist, and pathologist reviewing clinical, radiological, and pathological data together. No single piece of evidence stands alone. The MDT is the new gold standard.
- The HRCT pattern is the single most powerful diagnostic tool. A definite usual interstitial pneumonia (UIP) pattern on HRCT is diagnostic of IPF in the right clinical context, without biopsy. Master HRCT pattern recognition — it earns marks in every exam format.
- Two antifibrotics — nintedanib and pirfenidone — slow FVC decline in IPF. Neither reverses fibrosis. Neither cures the disease. Both reduce the rate of decline by approximately 50 percent and are now standard of care for IPF and other progressive fibrosing ILDs. [1]
The clinical question is always: Is this IPF, or is it something else? The distinction matters because IPF is managed with antifibrotics while many other ILDs respond to immunosuppression. Treating IPF with corticosteroids is harmful; treating hypersensitivity pneumonitis with antifibrotics alone without antigen avoidance misses the primary therapy. [1]
Classification
The clinically useful framework divides ILD into four major categories. The first diagnostic question is: does this patient have a known cause? [1]
Category 1: ILD with a known cause
| Subcategory | Key examples | Distinguishing clue |
|---|---|---|
| Connective tissue disease (CTD-ILD) | Systemic sclerosis (SSc), rheumatoid arthritis (RA), Sjogren syndrome, inflammatory myopathies (anti-synthetase syndrome), mixed connective tissue disease (MCTD) | Raynaud phenomenon, skin changes, arthralgia, sicca, muscle weakness, specific autoantibodies |
| Hypersensitivity pneumonitis (HP) | Bird fancier's lung, farmer's lung, hot tub lung, metal-working fluid exposure | Temporal relationship to antigen exposure (birds, hay, mouldy organic dust), occupational or avocational history |
| Occupational / inhalational | Asbestosis, silicosis, coal worker's pneumoconiosis, berylliosis, hard metal disease | Exposure history (mining, construction, sandblasting), latency from exposure to disease, characteristic radiological features (pleural plaques in asbestos, eggshell calcification in silicosis) |
| Drug-induced ILD | Amiodarone, methotrexate, nitrofurantoin, bleomycin, checkpoint inhibitors, radiotherapy, gold, sulfasalazine, hydralazine, chemotherapy agents | Temporal relationship to drug commencement, dose dependence for some agents (amiodarone, bleomycin), specific radiological patterns |
| Radiation-induced | Post-radiotherapy fibrosis | Confined to radiation field, temporal relationship to radiotherapy |
Category 2: Granulomatous ILD
| Disease | Distinguishing features |
|---|---|
| Sarcoidosis | Non-caseating granulomas, bilateral hilar lymphadenopathy, perilymphatic nodules, multi-organ involvement (skin, eye, cardiac, neurological), non-steroidal features including erythema nodosum and uveitis |
| Hypersensitivity pneumonitis (granulomatous variant) | Poorly formed granulomas, centrilobular nodules, air trapping, antigen exposure |
| Berylliosis | Indistinguishable from sarcoidosis histologically; beryllium lymphocyte proliferation test positive |
Category 3: Idiopathic interstitial pneumonias (IIPs)
The IIPs are ILDs of unknown cause. The 2013 ATS/ERS classification divides them into chronic fibrosing, acute/subacute, and smoking-related: [1]
| IIP | HRCT pattern | Clinical context | Prognosis |
|---|---|---|---|
| Idiopathic pulmonary fibrosis (IPF) | UIP — basal, subpleural reticulation, honeycombing, traction bronchiectasis | Older male (over 60), smoker or former smoker | Median survival 3-5 years untreated |
| Idiopathic NSIP | Uniform ground-glass and reticulation, basal and symmetrical, little or no honeycombing | Middle-aged adults, often female, better prognosis than IPF | 5-year survival approximately 80 percent |
| Cryptogenic organising pneumonia (COP) | Patchy consolidation (often peripheral or peribronchial), reverse halo sign | Middle-aged adults, flu-like illness, productive cough | Usually steroid-responsive, excellent prognosis |
| Respiratory bronchiolitis-ILD (RB-ILD) | Centrilobular nodules, ground-glass, upper lobe predominance | Heavy smokers, 40-50 years | Smoking cessation is primary therapy |
| Desquamative interstitial pneumonia (DIP) | Diffuse ground-glass, basal predominance | Smokers | Usually steroid-responsive, good prognosis |
| Lymphoid interstitial pneumonia (LIP) | Diffuse ground-glass, septal thickening, cysts | Associated with Sjogren, HIV, common variable immunodeficiency | Variable; treat underlying condition |
Category 4: Rare ILDs
- Lymphangioleiomyomatosis (LAM) — cystic lung disease in women of reproductive age, chylous effusions, pneumothorax, associated with tuberous sclerosis complex. Sirolimus stabilises lung function.
- Pulmonary Langerhans cell histiocytosis (PLCH) — upper lobe cysts and nodules in young smokers. Smoking cessation is critical; may progress to fibrosis.
- Pulmonary alveolar proteinosis — alveolar filling with lipoproteinaceous material, crazy-paving pattern on HRCT. Whole lung lavage or GM-CSF therapy.
- Eosinophilic pneumonias — blood or lavage eosinophilia, peripheral infiltrates. Often steroid-responsive. [1]
DWE trap: The classification is not academic — it drives treatment. IPF gets antifibrotics. CTD-ILD and COP get immunosuppression. HP gets antigen avoidance plus immunosuppression. Sarcoidosis gets corticosteroids for significant organ involvement. Getting the classification wrong leads to harmful therapy. [1]
The diagnostic approach
The diagnostic pathway follows a structured sequence. Every ILD patient gets the same workup. [1]
Step 1: History and examination
History must include: [1]
- Temporal profile — months of progressive dyspnoea and dry cough are typical of fibrosing ILD; acute onset over days suggests infection, diffuse alveolar haemorrhage, or acute interstitial pneumonia
- Occupational and avocational history — every patient gets asked about birds, farming, mouldy environments, construction, mining, sandblasting, and dental work. This is non-negotiable
- Drug history — specifically ask about amiodarone, methotrexate, nitrofurantoin, bleomycin, checkpoint inhibitors, and immunotherapy. Review all medications started within 6-12 months of symptom onset
- Connective tissue disease symptoms — Raynaud phenomenon, arthralgia, sicca (dry eyes and mouth), skin thickening, muscle weakness, proximal myopathy, dysphagia, oesophageal reflux
- Smoking history — pack-years, current or former smoker, never-smoker
- Family history — familial pulmonary fibrosis (mutations in TERT, TERC, surfactant protein genes)
- Aspiration risk — GERD symptoms, neurological dysphagia [1]
Examination: [1]
- Respiratory — fine bilateral basal Velcro-like crackles (the hallmark sign), tachypnoea, reduced chest expansion. In advanced disease: signs of pulmonary hypertension (loud P2, RV heave, raised JVP, peripheral oedema)
- Hands — digital clubbing (present in up to 50 percent of IPF patients; its absence does not exclude IPF). Hypertrophic pulmonary osteoarthropathy is possible
- Skin and joints — sclerodactyly, digital pitting scars or telangiectasia (systemic sclerosis), Gottron papules and mechanic's hands (dermatomyositis/anti-synthetase), rheumatoid nodules, active synovitis, Raynaud phenomenon [1]
DCE short-case discriminator: The crackles of ILD are described as "Velcro-like" — fine, dry, end-inspiratory, bilateral basal, and they do not clear with coughing. This distinguishes them from the coarse, mid-inspiratory crackles of bronchiectasis (which may clear with coughing) and the fine basal crackles of pulmonary oedema (which shift with position). Master this distinction for your short case. [1]
Step 2: Pulmonary function tests
PFTs in ILD show a restrictive pattern: [1]
- Reduced TLC (total lung capacity) — the defining abnormality
- Reduced FVC and RV (forced vital capacity, residual volume)
- Preserved or increased FEV1/FVC ratio (no airflow obstruction, unless there is coexisting COPD)
- Reduced DLCO (diffusing capacity) — often disproportionately reduced relative to the degree of restriction. A markedly reduced DLCO with preserved lung volumes suggests pulmonary hypertension or early CTD-ILD [1]
DWE point: The DLCO pattern matters. In IPF, DLCO falls as the disease progresses. A DLCO less than 40 percent predicted correlates with poor prognosis. In systemic sclerosis-ILD, an isolated low DLCO with relatively preserved volumes suggests pulmonary arterial hypertension overlapping with ILD — this changes management and requires echocardiography and right heart catheterisation. [1]
Step 3: High-resolution CT (HRCT)
HRCT is the single most important diagnostic test. Master the major patterns: [1]
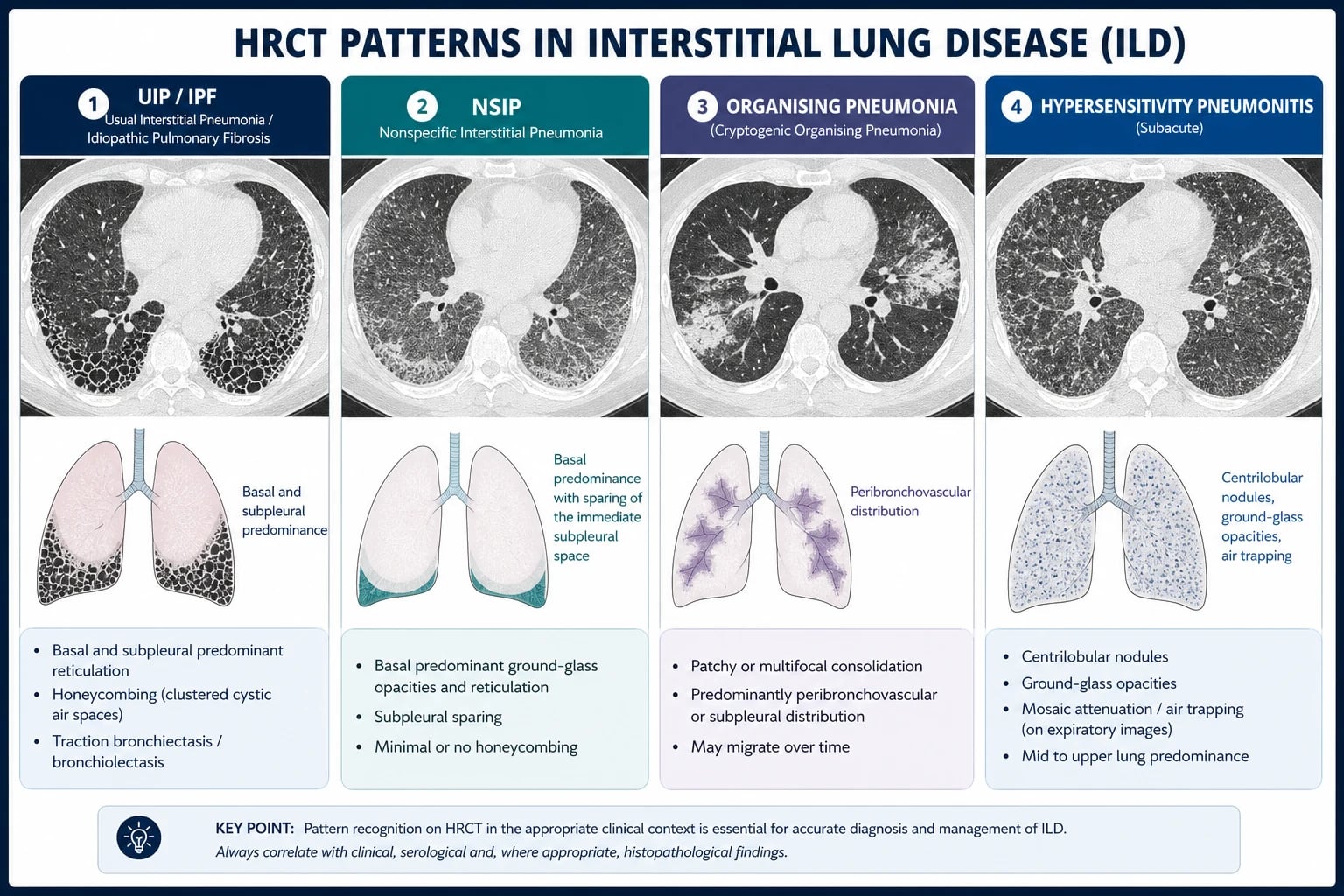
Usual interstitial pneumonia (UIP) pattern — the hallmark of IPF: [1]
- Basal and subpleural predominant reticulation and honeycombing
- Traction bronchiectasis and bronchiolectasis
- Heterogeneous distribution — areas of normal lung adjacent to fibrotic lung
- No significant ground-glass opacity (if present, suggests an alternative or superimposed process)
- A definite UIP pattern on HRCT is diagnostic of IPF without biopsy when infection, CTD, and drug causes are excluded [1]
NSIP pattern: [1]
- Symmetrical, basal ground-glass opacity and fine reticulation
- Uniform distribution (contrast with UIP's heterogeneity)
- Little or no honeycombing
- Traction bronchiectasis may be present
- Most common IIP in CTD-ILD (especially systemic sclerosis) [1]
Organising pneumonia pattern: [1]
- Patchy consolidation — often peripheral or peribronchial
- Reverse halo sign (atoll sign) — central ground-glass surrounded by consolidation
- May show nodular opacities [1]
Hypersensitivity pneumonitis pattern: [1]
- Centrilobular nodules (poorly defined)
- Ground-glass opacity
- Air trapping on expiratory images (mosaic attenuation)
- Upper or mid-zone predominance (contrast with IPF's basal pattern)
- Chronic HP may evolve to a UIP-like fibrotic pattern — careful MDT review needed [1]
Sarcoidosis pattern: [1]
- Perilymphatic nodules (along bronchovascular bundles, fissures, pleura)
- Bilateral hilar and mediastinal lymphadenopathy
- Upper and mid-zone predominance
- May show galaxy sign (clustered nodules with satellite lesions) [1]
Examiner point: When presented with an HRCT in a DCE or viva, describe the pattern systematically: distribution (upper vs lower, central vs peripheral), predominant abnormality (reticulation, ground-glass, consolidation, nodules, cysts), and specific features (honeycombing, traction bronchiectasis, lymphadenopathy, air trapping). Then give your pattern diagnosis (UIP, NSIP, organising pneumonia, HP, sarcoidosis) and your differential diagnosis. This demonstrates structured radiological reasoning. [1]
Step 4: Serological workup
Every ILD patient gets a connective tissue disease screen: [1]
| Test | What it detects |
|---|---|
| ANA (antinuclear antibody) | Systemic sclerosis, SLE, MCTD, Sjogren syndrome |
| Rheumatoid factor and anti-CCP | Rheumatoid arthritis-associated ILD |
| Anti-Scl-70 (anti-topoisomerase I) | Diffuse systemic sclerosis |
| Anti-centromere | Limited systemic sclerosis (CREST) |
| Anti-Ro52 | SSc-ILD, inflammatory myopathy, interstitial pneumonia with autoimmune features (IPAF) |
| Anti-Jo-1 and anti-synthetase panel (PL-7, PL-12, EJ, OJ, KS) | Anti-synthetase syndrome — characteristic ILD |
| Anti-MDA5 | Clinically amyopathic dermatomyositis with rapidly progressive ILD |
| Anti-Ro/La (SS-A/SS-B) | Sjogren syndrome-associated ILD |
| CK (creatine kinase) and aldolase | Inflammatory myopathy |
| HIV serology | HIV-associated LIP and opportunistic ILDs |
DCE long-case trap: Missing occult CTD-ILD is the most common error. In a patient with "IPF," the presence of any CTD feature — Raynaud phenomenon, sicca symptoms, an abnormal nailfold capillaroscopy — demands a full autoimmune panel. Anti-synthetase syndrome is the single most commonly missed diagnosis in ILD clinics. An anti-Jo-1 or anti-PL-7 antibody changes management from antifibrotic-only to immunosuppression plus antifibrotic. [1]
Step 5: Bronchoalveolar lavage (BAL)
BAL is useful for excluding infection and narrowing the differential: [1]
- Lymphocytosis (lymphocytes greater than 15 percent) — sarcoidosis, hypersensitivity pneumonitis, CTD-ILD, drug-induced ILD. In sarcoidosis, CD4/CD8 ratio greater than 3.5 is suggestive
- Neutrophilia (greater than 3-5 percent) — IPF, fibrotic NSIP, asbestosis
- Eosinophilia (greater than 2-5 percent) — eosinophilic pneumonia, drug-induced, Churg-Strauss. Greater than 25 percent supports eosinophilic pneumonia
- Increased CD1a-positive Langerhans cells (greater than 5 percent) — PLCH [1]
BAL is not diagnostic of IPF — it is used to exclude alternative diagnoses. A normal BAL cellular profile does not exclude UIP/IPF. [1]
Step 6: Surgical lung biopsy
A surgical lung biopsy (via video-assisted thoracoscopic surgery, VATS) is indicated when: [1]
- The HRCT pattern is indeterminate (possible UIP or inconsistent with UIP)
- The clinical context is ambiguous (overlap features, possible CTD)
- The MDT needs histopathological confirmation before committing to therapy [1]
A transbronchial lung cryobiopsy (TBLC) is emerging as an alternative to VATS with lower morbidity, providing larger samples than conventional transbronchial biopsy. [1]
Biopsy is contraindicated in patients with severe physiological impairment (FVC less than 50 percent or DLCO less than 30 percent predicted), where perioperative risk of acute exacerbation or mortality is unacceptably high. [1]
Examiner trap: The histopathological correlate of the UIP pattern on HRCT is spatial and temporal heterogeneity — areas of normal lung, fibrosis, honeycomb change, and fibroblastic foci coexisting in the same biopsy. This heterogeneity is the defining feature. NSIP shows uniformity. Understanding this pathophysiological distinction earns marks in viva. [1]
Step 7: Multidisciplinary team (MDT) discussion
The MDT integrates clinical, radiological, and pathological data. It is the gold standard for ILD diagnosis and outperforms individual clinician assessment. The MDT includes: [1]
- Respiratory physician — clinical context, PFT trajectory, treatment decisions
- Thoracic radiologist — HRCT pattern interpretation
- Pathologist — histopathological diagnosis (if biopsied)
- Rheumatologist — CTD assessment (if autoimmune features present)
- ILD nurse coordinator — patient education, adherence, symptom monitoring
- Transplant team — early referral for progressive disease [1]
Idiopathic pulmonary fibrosis (IPF)
IPF is the most common and most important IIP. Master it thoroughly. [1]
Diagnosis
Per the ATS/ERS/JRS/ALAT 2018 diagnostic guideline, the diagnosis of IPF requires: [1]
- A UIP pattern on HRCT (or a combination of UIP pattern on HRCT and surgical lung biopsy)
- Exclusion of known causes of ILD (CTD, HP, occupational, drug-induced)
- Multidisciplinary discussion confirming the diagnosis [1]
The HRCT UIP pattern is classified as definite UIP, probable UIP, indeterminate for UIP, or alternative diagnosis: [1]
| HRCT category | Key features |
|---|---|
| Definite UIP | Basal and subpleural reticulation with honeycombing; absence of features suggesting alternative diagnosis |
| Probable UIP | Basal and subpleural reticulation without honeycombing but with other UIP features |
| Indeterminate | Reticulation that is subtly or not clearly UIP; no features of alternative diagnosis |
| Alternative diagnosis | Features suggesting non-IPF diagnosis (ground-glass predominant, perilymphatic nodules, consolidation, mid/upper zone predominance) |
A definite UIP on HRCT in a patient with exclusion of known causes is diagnostic of IPF — no biopsy needed. A probable UIP or indeterminate pattern requires MDT discussion and possibly biopsy. [1]
Pathophysiology
IPF results from recurrent micro-injury to the alveolar epithelium in a susceptible host (ageing lung, genetic predisposition). The injured epithelium fails to regenerate properly. Instead, aberrant wound healing follows: fibroblasts proliferate and differentiate into myofibroblasts, depositing excessive extracellular matrix. Fibroblastic foci — collections of activated myofibroblasts — are the histopathological hallmark and the leading edge of fibrosis. [1]
Key signalling pathways driving fibrosis:
- TGF-beta — the master profibrotic cytokine; drives myofibroblast differentiation and collagen deposition
- PDGF (platelet-derived growth factor) — fibroblast proliferation and migration
- FGF (fibroblast growth factor) — angiogenesis and fibroblast activation [1]
Nintedanib inhibits multiple receptor tyrosine kinases (PDGFR, FGFR, VEGFR), and pirfenidone has anti-TGF-beta and anti-PDGF activity. Both reduce the rate of fibrosis — the rationale for calling them antifibrotics. [1]
Antifibrotic therapy
Two antifibrotics are approved for IPF. Both slow FVC decline by approximately 50 percent. Neither reverses established fibrosis. [1]
Nintedanib (Ofev): [1]
- Dose: 150 mg orally twice daily, with food. May reduce to 100 mg BID for tolerability
- Mechanism: Multi-target tyrosine kinase inhibitor (PDGFR alpha/beta, FGFR 1-3, VEGFR 1-3)
- Evidence (INPULSIS, PMID 24836310): Two replicate Phase 3 trials in 1066 patients. Reduced annual FVC decline by approximately 110 mL/year (placebo: -239 mL/year; nintedanib: -114 mL/year)
- Adverse effects: Diarrhoea (over 60 percent, leading to discontinuation in approximately 5 percent), nausea, abdominal pain, liver enzyme elevation. Manage with loperamide, taking with food, and dose reduction
- Monitoring: LFTs at baseline, then monthly for 3 months, then every 3 months [1]
Pirfenidone (Esbriet): [1]
- Dose: Titrate to 801 mg orally three times daily (2403 mg/day total) with food. Titration: Week 1: 267 mg TID; Week 2: 534 mg TID; Week 3 and beyond: 801 mg TID
- Mechanism: Anti-fibrotic and anti-inflammatory; inhibits TGF-beta production and collagen synthesis
- Evidence (CAPACITY, PMID 21571362; ASCEND, PMID 24836312): ASCEND confirmed FVC decline reduction (47.9 percent relative reduction in proportion of patients with 10 percent or greater FVC decline). Pooled analysis of ASCEND and CAPACITY showed significant mortality reduction
- Adverse effects: Photosensitivity rash, nausea, fatigue, dyspepsia, liver enzyme elevation. Manage with sunscreen, taking with food, antiemetics, and dose reduction
- Monitoring: LFTs at baseline, then monthly for 3 months, then every 3 months [1]
DWE key point: Both antifibrotics are equivalent in efficacy — there is no head-to-head superiority trial. The choice is driven by tolerability and patient preference. The most common DWE question tests whether you know that these drugs slow decline, not reverse it, and that they are indicated for progressive fibrosing ILD, not just IPF. [1]
What NOT to prescribe in IPF
- Corticosteroid monotherapy is harmful in IPF — no benefit, increased infection risk
- Triple therapy (prednisone + azathioprine + N-acetylcysteine) was evaluated in the PANTHER-IPF trial and stopped early for harm — increased mortality, more hospitalisations, and more adverse events versus placebo. This combination is contraindicated in IPF
- Anticoagulation is not indicated in the absence of a specific thromboembolic indication
- AmbriSENTAN (an endothelin receptor antagonist) showed no benefit in IPF [1]
Acute exacerbation of IPF
An acute exacerbation is an acute, clinically significant respiratory deterioration characterised by new widespread alveolar abnormality (ground-glass or consolidation) not fully explained by cardiac failure or fluid overload. [1]
Diagnostic criteria (Collard 2016 International Working Group, PMID 27299520): [1]
- Previous or concurrent diagnosis of IPF
- Acute worsening or development of dyspnoea typically of less than 1 month duration
- CT with new bilateral ground-glass opacity and/or consolidation superimposed on a background pattern consistent with UIP
- Deterioration not fully explained by cardiac failure or fluid overload [1]
Triggers include infection (viral and bacterial), aspiration, post-procedural (bronchoscopy, surgery, particularly thoracic), drug toxicity, and a substantial proportion are idiopathic. [1]
Management: [1]
- Broad-spectrum antibiotics to cover infection (the most common mimic and trigger)
- Corticosteroids — high-dose IV methylprednisolone (0.5-1 g daily for 3 days) then tapering oral prednisolone. Evidence is weak; many experts use it empirically given the lack of alternatives [1]- Oxygen supplementation for hypoxaemia
- Consider NIV for moderate hypoxaemic respiratory failure, primarily as a palliative or bridge measure
- Mechanical ventilation is generally not recommended in end-stage IPF — outcomes are poor (hospital mortality exceeds 50 percent, approaching 90 percent in severe cases). Discuss goals of care
- Consider antifibrotic continuation if the patient was on therapy; do not stop during an acute exacerbation unless drug toxicity is suspected [1]
Exam trap: Acute exacerbation of IPF is defined by Collard 2016 criteria. The key exclusion is cardiac failure — you must exclude pulmonary oedema as the cause of new bilateral infiltrates. Echocardiography and clinical assessment of volume status are mandatory. [1]
Prognosis
Median survival of untreated IPF is 3-5 years from diagnosis. Antifibrotics slow decline but do not restore normal life expectancy. The GAP model (Gender, Age, Physiology; PMID 22586007) is a validated staging system: [1]
| Component | Score 0 | Score 1 | Score 2 |
|---|---|---|---|
| Gender | Female | Male | — |
| Age | 60 or less | 61-65 | Over 65 |
| FVC (% predicted) | 75 or more | 50-75 | Less than 50 |
| DLCO (% predicted) | 55 or more | 36-55 | 35 or less |
GAP Stage I (score 0-3): 1-year mortality 6 percent, 3-year mortality 16 percent GAP Stage II (score 4-5): 1-year mortality 11 percent, 3-year mortality 29 percent GAP Stage III (score 6-10): 1-year mortality 39 percent, 3-year mortality 62 percent [1]
Progressive pulmonary fibrosis (PPF)
The 2022 ATS/ERS/JRS/ALAT guideline introduced the concept of progressive pulmonary fibrosis (PPF) — a clinical phenotype of worsening seen in ILDs other than IPF. [1]
PPF criteria (applied after at least 12 months of observation): [1]
- Worsening respiratory symptoms
- Worsening of at least one of the following:
- FVC decline of 5 percent or more (absolute) within 12 months
- DLCO decline (corrected for haemoglobin) of 10 percent or more within 12 months
- Worsening on HRCT (increased extent of fibrosis)
- No alternative explanation (infection, pulmonary embolism, cardiac failure) [1]
The INBUILD trial (PMID 31566307) demonstrated that nintedanib reduced the rate of FVC decline in patients with progressive fibrosing ILDs other than IPF (including CTD-ILD, chronic HP, unclassifiable ILD, and others). This led to the approval of nintedanib for progressive fibrosing ILD. [1]
Clinical impact: A patient with systemic sclerosis-ILD whose FVC is falling by 10 percent per year despite mycophenolate should have nintedanib added. The progressive fibrotic phenotype is treatable regardless of the underlying aetiology. [1]
Connective tissue disease-associated ILD (CTD-ILD)
CTD-ILD is the most common group of ILDs with a known cause. ILD may be the presenting feature of a CTD — the lung disease precedes the systemic manifestations by months or years. [1]
Systemic sclerosis (SSc-ILD)
- ILD occurs in over 50 percent of SSc patients; it is the leading cause of death
- NSIP is the most common pattern (contrast with IPF's UIP)
- Screening: All SSc patients should have baseline HRCT and PFTs (including DLCO) at diagnosis, then annually for the first 5 years
- Treatment: Mycophenolate mofetil (first-line immunosuppressant), cyclophosphamide (Scleroderma Lung Study, PMID 16790698 — modest benefit), or rituximab for refractory disease. Add nintedanib if fibrosis is progressive
- Scleroderma renal crisis is a critical consideration — corticosteroids at doses above 15 mg/day prednisolone equivalent increase risk. ACE inhibitors (not ARBs) are first-line for scleroderma renal crisis [1]
Rheumatoid arthritis (RA-ILD)
- ILD occurs in approximately 10-20 percent of RA patients (more if subclinical disease is counted)
- UIP is the most common pattern (unlike SSc), and the prognosis approaches that of IPF
- Methotrexate may rarely cause drug-induced pneumonitis — distinguish from RA-ILD progression by HRCT pattern and temporal relationship
- Treatment: Mycophenolate or rituximab for inflammatory disease; antifibrotics for progressive fibrosis [1]
Inflammatory myopathy and anti-synthetase syndrome
- Anti-synthetase syndrome is defined by the presence of anti-aminoacyl-tRNA synthetase antibodies (anti-Jo-1, anti-PL-7, anti-PL-12, anti-EJ, anti-OJ, anti-KS) plus ILD, fever, mechanic's hands, Raynaud phenomenon, arthritis, and non-erosive arthritis
- Anti-MDA5 antibody is critical to identify — associated with rapidly progressive ILD in clinically amyopathic dermatomyositis. High early mortality if untreated
- Treatment: Aggressive immunosuppression — high-dose corticosteroids, plus mycophenolate, calcineurin inhibitors, rituximab, or IV cyclophosphamide for severe disease. Anti-MDA5 ILD may require triple immunosuppression from the outset [1]
Sjogren syndrome
- ILD occurs in approximately 10-20 percent; NSIP and LIP are the most common patterns
- Treatment targets the sicca syndrome and lung inflammation [1]
DCE long-case insight: In a patient with SSc-ILD, the long case demands integration: ILD management (immunosuppression plus antifibrotic for progressive fibrosis), pulmonary hypertension screening (echo then right heart catheter), GERD management (PPI, prokinetics), digital ulcer prevention (calcium channel blockers, bosentan), and renal crisis surveillance (BP monitoring, ACE inhibitors). This is a classic complex multisystem case. [1]
Hypersensitivity pneumonitis (HP)
HP is an immune-mediated alveolitis triggered by repeated inhalation of antigens. The antigen list is vast — the common ones are: [1]
| Antigen | Source | Disease name |
|---|---|---|
| Avian proteins | Pigeon, budgerigar, poultry droppings and feather dust | Bird fancier's lung |
| Thermophilic actinomycetes | Mouldy hay, compost, grain | Farmer's lung |
| Mycobacterium avium complex (MAC) | Hot tubs, metal-working fluids | Hot tub lung |
| Fungi (Aspergillus) | Mouldy environments | Various |
| Isocyanates | Spray painting, polyurethane foam | Occupational HP |
Acute HP
- Develops 4-12 hours after exposure to a high dose of antigen
- Presents with fever, chills, cough, dyspnoea, and malaise — mimics an atypical pneumonia
- Resolves within 24-48 hours of removing the antigen
- Chest X-ray/HRCT: diffuse ground-glass, centrilobular nodules, air trapping
- PFTs: may show restriction, or mixed obstructive/restrictive pattern
- BAL: lymphocytosis (often over 50 percent, with CD8 predominance) [1]
Chronic HP
- Develops insidiously from ongoing low-level exposure
- Presents with chronic cough, progressive dyspnoea, weight loss
- May progress to fibrotic ILD indistinguishable from IPF on HRCT (basal reticulation, honeycombing in advanced cases)
- Distinguishing clues: upper/mid-zone predominance, air trapping, centrilobular nodules, exposure history
- Treatment: Antigen avoidance is the primary therapy — this is non-negotiable. Remove birds, change occupations, remediate mould. Corticosteroids (prednisolone 0.5-1 mg/kg/day, tapering over months) for significant disease. Mycophenolate or rituximab for steroid-dependent or refractory disease. Antifibrotics for progressive fibrosis despite antigen avoidance. [1]
DWE trap: The most commonly tested HP scenario is bird fancier's lung in a patient with pigeons or budgerigars at home. The key is recognising the antigen exposure and prioritising antigen removal above all other interventions. Pharmacotherapy is adjunctive. [1]
Sarcoidosis
Sarcoidosis is a multisystem granulomatous disorder of unknown cause. It is one of the most commonly tested ILDs in the FRACP exam. [1]
Clinical features
- Most commonly affects adults aged 20-40 years
- Pulmonary involvement (over 90 percent): bilateral hilar lymphadenopathy, pulmonary infiltrates, may progress to fibrosis (upper lobe predominant)
- Extra-pulmonary manifestations: skin (erythema nodosum, lupus pernio), eyes (anterior and posterior uveitis), cardiac (arrhythmias, heart block, cardiomyopathy — a leading cause of death), neurological (cranial nerve palsies, hypothalamic involvement), hepatic (granulomatous hepatitis), hypercalcaemia (from increased 1,25-dihydroxyvitamin D production by granulomas) [1]
Scadding chest radiograph staging
| Stage | CXR finding | Spontaneous remission rate |
|---|---|---|
| 0 | Normal CXR | — |
| 1 | Bilateral hilar lymphadenopathy (BHL) | 60-90 percent |
| 2 | BHL plus parenchymal infiltrates | 40-70 percent |
| 3 | Parenchymal infiltrates without BHL | 10-20 percent |
| 4 | Pulmonary fibrosis (upper lobe, volume loss, traction bronchiectasis) | Less than 5 percent |
Examiner point: The Scadding stage predicts spontaneous remission. Stages 0-2 often resolve without treatment. Stage 3 may need corticosteroids. Stage 4 is irreversible fibrosis — corticosteroids may slow progression but cannot reverse established fibrosis. [1]
Indications for corticosteroids in sarcoidosis
Not all sarcoidosis requires treatment. Indications for systemic corticosteroids: [1]
- Symptomatic stage 2 or 3 pulmonary disease (cough, dyspnoea) not improving after 3-6 months of observation
- Stage 4 with active inflammation (ground-glass on HRCT suggesting ongoing granulomatous inflammation)
- Cardiac sarcoidosis — always treated, potentially fatal
- Neurological sarcoidosis — always treated
- Ocular sarcoidosis (sight-threatening uveitis) — topical or systemic
- Hypercalcaemia with renal impairment
- Lofgren syndrome (erythema nodosum plus BHL plus fever/arthritis) — usually self-limiting; treat with NSAIDs, not corticosteroids [1]
Dose: Prednisolone 20-40 mg daily, tapering slowly over 6-24 months. Steroid-sparing agents (methotrexate, azathioprine, mycophenolate, or infliximab for refractory disease) are used for prolonged courses or corticosteroid-resistant disease. [1]
When not to treat
- Asymptomatic bilateral hilar lymphadenopathy (Stage 1) — observe; the majority resolve spontaneously
- Isolated erythema nodosum without systemic involvement (Lofgren syndrome) — NSAIDs and observation
- Normal lung function and no organ-threatening disease — conservative management with surveillance [1]
Drug-induced ILD
Recognising drug-induced ILD requires a high index of suspicion. The temporal relationship to drug commencement or dose change is key. [1]
| Drug | Pattern | Mechanism | Key teaching point |
|---|---|---|---|
| Amiodarone | Chronic interstitial pneumonitis, organising pneumonia | Phospholipidosis — drug accumulates in lysosomes; accumulates in lung tissue; dose-dependent (greater risk above 400 mg/day or after more than 2 years) | Basal ground-glass and reticulation; high attenuation liver and lung on CT (iodine content) |
| Nitrofurantoin | Acute or chronic pneumonitis | Immune-mediated | Acute form within days to weeks (fever, eosinophilia, diffuse infiltrates); chronic form after months to years of continuous use (basal fibrosis) |
| Bleomycin | Pneumonitis progressing to fibrosis | Dose-dependent direct toxicity; oxygen worsens | Total cumulative dose risk; DLCO monitoring during chemotherapy; avoid supplemental oxygen |
| Checkpoint inhibitors (anti-PD-1/PD-L1) | Pneumonitis, organising pneumonia | Immune-mediated (immune-related adverse event) | Any pattern; treat by holding drug and corticosteroids; may be fatal |
| Radiotherapy | Fibrosis in radiation field | Direct tissue injury | Confined to radiation port; latency weeks to months |
DWE point: In any patient with ILD, review the medication list for amiodarone, methotrexate, nitrofurantoin, bleomycin, and immunotherapy. Stopping the offending drug is the primary therapy; corticosteroids are adjunctive. [1]
Multidisciplinary team approach and lung transplantation
The MDT
The MDT is the gold standard for ILD diagnosis. It achieves diagnostic confidence that exceeds that of any individual clinician. The MDT revisits the diagnosis at each visit — ILD diagnoses can change as new clinical information emerges. [1]
Lung transplantation
Lung transplantation is the only therapy proven to improve survival in progressive IPF. Timing of referral is critical — late referral is a common failure. [1]
Indications for transplant referral (early referral is always better): [1]
- IPF with FVC less than 80 percent predicted or DLCO less than 40 percent predicted — refer early
- Any ILD with progressive physiological decline (FVC decline 10 percent or more in 12 months, DLCO decline 15 percent or more)
- Pulmonary hypertension complicating ILD
- Functional decline despite optimal medical therapy
- Hospitalisation for respiratory deterioration or acute exacerbation [1]
Transplant considerations: [1]
- Bilateral sequential lung transplant is preferred for IPF
- 5-year post-transplant survival is approximately 50-60 percent
- Age limits have expanded — carefully selected patients up to age 70 may be considered
- Major contraindications: recent malignancy, uncontrolled infection, severe cardiac or hepatic dysfunction, obesity (BMI over 35), poor functional status, non-adherence [1]
DCE trap: The most common long-case error is late transplant referral. In a progressive ILD patient, refer to a transplant centre at the time of diagnosis, not when the patient is in end-stage respiratory failure. The workup takes months. Early referral preserves the option of transplant. [1]
Long-term follow-up and surveillance
Every ILD patient receives structured follow-up: [1]
- PFTs every 3-6 months (FVC trajectory is the primary disease activity marker)
- Annual HRCT to assess progression
- 6-minute walk test annually (desaturation, distance, Borg dyspnoea index)
- Echocardiography every 1-2 years for pulmonary hypertension screening
- Vaccinations — influenza annually, pneumococcal, COVID-19, and pertussis booster. Avoid live vaccines if on immunosuppression
- Pulmonary rehabilitation — improves exercise capacity and quality of life
- Oxygen therapy for PaO2 less than 55 mmHg at rest or exertional desaturation to 88 percent or below
- Advance care planning — early and ongoing; explore transplant candidacy, end-of-life preferences, and palliative care integration [1]
Key trial evidence summary
| Trial | Drug | Population | Key result | PMID |
|---|---|---|---|---|
| INPULSIS (2014) | Nintedanib 150 mg BID | IPF | Reduced annual FVC decline by approximately 110 mL/year | 24836310 |
| CAPACITY (2011) | Pirfenidone | IPF | Two trials; reduced FVC decline | 21571362 |
| INBUILD (2019) | Nintedanib | Progressive fibrosing ILD (non-IPF) | Reduced annual FVC decline by 107 mL/year | 31566307 |
| SLS (2006) | Cyclophosphamide | SSc-ILD | Modest FVC benefit at 12 months | 16790698 |
References
- [1]Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline Am J Respir Crit Care Med, 2022.PMID 35486072
- [2]Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis N Engl J Med, 2014.PMID 24836310
- [3]King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis N Engl J Med, 2014.PMID 24836312
- [4]Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases N Engl J Med, 2019.PMID 31566307
- [5]Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management Am J Respir Crit Care Med, 2011.PMID 21471066
- [6]Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis Ann Intern Med, 2012.PMID 22586007
- [7]Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials Lancet, 2011.PMID 21571362
- [8]Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease N Engl J Med, 2006.PMID 16790698
- [9]Collard HR, Ryerson CJ, Corte TJ, et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report Am J Respir Crit Care Med, 2016.PMID 27299520