Phys · rheumatological
IgG4-Related Disease
Also known as IgG4-related disease · IgG4-RD · IgG4-related sclerosing disease · type 1 autoimmune pancreatitis · lymphoplasmacytic sclerosing pancreatitis · IgG4-related sclerosing cholangitis · Mikulicz syndrome · IgG4-related sialadenitis · IgG4-related dacryoadenitis · Kuttner tumour · IgG4-related kidney disease · IgG4-related tubulointerstitial nephritis · retroperitoneal fibrosis · Ormond disease · IgG4-related aortitis · IgG4-related pachymeningitis · IgG4-related hypophysitis
Consultant-physician-depth guide to IgG4-related disease for FRACP DWE and DCE — the one-disease-many-organs fibroinflammatory concept linking type 1 autoimmune pancreatitis (painless obstructive jaundice, sausage-shaped pancreas, steroid-responsive), IgG4-related sclerosing cholangitis (distinguish from PSC and cholangiocarcinoma), Mikulicz syndrome (sialadenitis and dacryoadenitis, distinguish from Sjogren), IgG4-related kidney disease (tubulointerstitial nephritis), retroperitoneal fibrosis (Ormond disease, ureteric obstruction), aortitis, lung disease, pachymeningitis and hypophysitis; the diagnostic approach built on histopathology (dense lymphoplasmacytic infiltrate, storiform fibrosis, obliterative phlebitis, IgG4-positive plasma cells with IgG4 to IgG ratio above 40 per cent), the 2019 ACR and EULAR classification criteria and the 2020 revised comprehensive diagnostic criteria; the correct use and limits of serum IgG4 (elevated in 60 to 70 per cent, falsely positive in pancreatobiliary cancer and asthma, normal IgG4 does not exclude the disease); and the treatment ladder — glucocorticoids first-line, rituximab for relapsing or multi-organ disease, mycophenolate or azathioprine for maintenance, and the critical distinction of the steroid-responsive inflammatory phase from the steroid-resistant fibrotic phase.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
IgG4-Related Disease

The answer first
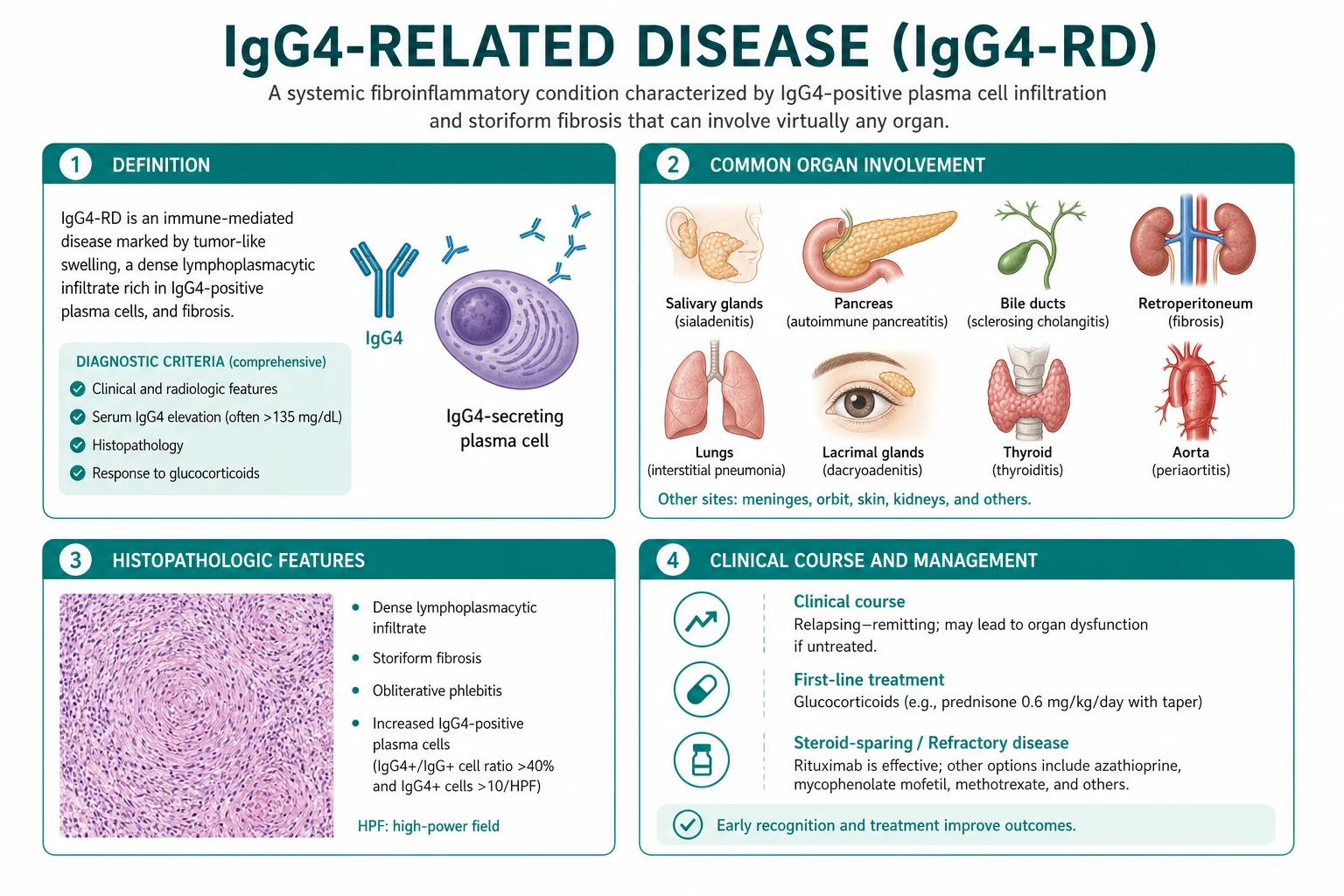
IgG4-related disease (IgG4-RD) is a systemic fibroinflammatory condition defined by a characteristic histopathology — a dense lymphoplasmacytic infiltrate rich in IgG4-positive plasma cells, storiform fibrosis and obliterative phlebitis — that can appear in almost any organ [1]. The central idea for the exam is one disease, many organs: type 1 autoimmune pancreatitis (AIP), IgG4-related sclerosing cholangitis, Mikulicz syndrome (sialadenitis and dacryoadenitis), IgG4-related kidney disease, retroperitoneal fibrosis, aortitis, lung disease, pachymeningitis and hypophysitis are all the same disease in different locations [1].
Three rules that change outcome [1]:
- Serum IgG4 is a biomarker, not a diagnosis. It is elevated in only 60 to 70 per cent of cases, it is falsely elevated in pancreatobiliary cancer, primary sclerosing cholangitis, asthma and atopy, and a normal IgG4 does not exclude the disease. The diagnosis rests on the clinicopathological pattern, with histopathology as the gold standard [1][2].
- IgG4-RD mimics cancer, and cancer can mimic IgG4-RD. A pancreatic head mass with painless jaundice is pancreatic cancer until proven otherwise. A modest IgG4 elevation occurs in 10 to 15 per cent of pancreatobiliary cancers. Never start steroids on serology alone when malignancy is plausible — obtain tissue and watch the steroid response with suspicion [1][3].
- Glucocorticoids work in the inflammatory phase and fail in the fibrotic phase. Prednisolone 30 to 40 mg daily produces a dramatic response within two weeks for active inflammatory disease, but burnt-out fibrotic lesions (a chronic biliary stricture, a mature retroperitoneal rind) are steroid-resistant — treat those with mechanical relief (stenting, drainage), not escalating immunosuppression [3].
The bedside reasoning moves in three steps. First, recognise the syndrome — is this a pancreatobiliary mass, a salivary or lacrimal gland enlargement, a renal lesion, a periaortic rind, or a dural or pituitary mass, and is there more than one organ involved? Second, confirm the histopathology and exclude the mimic — tissue biopsy for the triad of dense lymphoplasmacytic infiltrate, storiform fibrosis and obliterative phlebitis with IgG4-positive plasma cells and an IgG4 to IgG ratio above 40 per cent, while excluding malignancy, lymphoma and infection. Third, treat by phase and by organ — steroids for active inflammation, rituximab for relapsing or multi-organ disease, and mechanical relief for fibrotic obstruction [3][4][5].
The one disease, many organs concept

Before the unifying concept was recognised in 2003, the manifestations of IgG4-RD were scattered across specialties as unrelated organ-specific diseases [1]. The realisation that type 1 AIP, Mikulicz syndrome and idiopathic retroperitoneal fibrosis shared the same histopathology was the conceptual breakthrough. The examinable point is that a patient presenting with one organ should be actively screened for the others, because multi-organ involvement is the rule and changes the diagnosis, the treatment and the prognosis [1][3].
The organ map at a glance
| Organ | Lesion | Classic presentation | Key mimic |
|---|---|---|---|
| Pancreas | Type 1 AIP (lymphoplasmacytic sclerosing pancreatitis) | Painless obstructive jaundice, pancreatic mass, sausage-shaped pancreas | Pancreatic cancer |
| Biliary tree | IgG4-related sclerosing cholangitis | Intra- and extrahepatic strictures | PSC, cholangiocarcinoma |
| Salivary and lacrimal glands | Mikulicz syndrome, Kuttner tumour | Bilateral painless gland enlargement | Sjogren syndrome, lymphoma |
| Kidney | IgG4-related tubulointerstitial nephritis | Renal masses, AKI, low complement | Renal cell carcinoma, interstitial nephritis |
| Retroperitoneum | Retroperitoneal fibrosis (Ormond disease) | Ureteric obstruction, AKI, periaortic mass | Secondary RPF (malignancy, drugs) |
| Aorta | IgG4-related aortitis and periarteritis | Aneurysm, dissection, periaortic rind | Large-vessel vasculitis, infection |
| Lung | IgG4-related lung disease | Nodules, infiltrates, effusion | Lung cancer, sarcoidosis, infection |
| Meninges | IgG4-related pachymeningitis | Dural thickening, cranial nerve palsies, headache | Meningioma, infection, neurosarcoidosis |
| Pituitary | IgG4-related hypophysitis | Pituitary mass, hypopituitarism | Pituitary adenoma [1] |
The presence of more than one involved organ at baseline strongly supports IgG4-RD and lowers the chance that a single mass is malignant [1]. Conversely, a single mass with atypical features should always be biopsied rather than labelled on serology.
Organ-specific presentations in depth
Type 1 autoimmune pancreatitis
Type 1 AIP is the pancreatic manifestation of IgG4-RD and the most common way the disease comes to clinical attention. The patient is typically an older man who presents with painless obstructive jaundice, often with mild abdominal discomfort, weight loss (from biliary obstruction rather than cachexia) and occasionally new-onset diabetes [1]. The contrast CT shows a diffusely enlarged, sausage-shaped pancreas with delayed enhancement and a capsule-like rim — a pattern that differs from the focal atrophic or mass-forming pancreas of cancer [1][2].
The distinction from pancreatic cancer is the single most important decision. The discriminating features favouring type 1 AIP are a diffusely enlarged pancreas rather than a focal mass, a markedly elevated IgG4 (more than twice the upper limit), other-organ involvement (salivary glands, renal lesions, retroperitoneal rind), a low or normal CA19-9, and a dramatic radiological and biochemical response to steroids within two weeks [1][3]. The discriminating features favouring cancer are a focal low-attenuation mass with upstream duct dilatation, vascular encasement, a rising CA19-9, weight loss with cachexia, and the absence of other-organ involvement. When the picture is ambiguous, tissue is mandatory — endoscopic ultrasound-guided core biopsy or, where available, endoscopic forceps biopsy of the ampulla or bile duct [1].
The second thing to know is that type 2 AIP is a different disease. Idiopathic duct-centric pancreatitis affects younger patients, has an equal sex distribution, is associated with inflammatory bowel disease, has a normal serum IgG4 and no IgG4-positive cells on histology, and is not part of the IgG4-RD spectrum. The exam trap is to conflate the two [1].
IgG4-related sclerosing cholangitis
IgG4-related sclerosing cholangitis produces intra- and extrahepatic biliary strictures that are radiologically indistinguishable from primary sclerosing cholangitis (PSC) or cholangiocarcinoma [1]. The discriminating features are an elevated serum IgG4, the presence of type 1 AIP, the rapid response to steroids, and the absence of the strong inflammatory bowel disease association and the multifocal beaded strictures characteristic of PSC. Cholangiocarcinoma can elevate IgG4 mildly and can be fatal to miss — tissue diagnosis with intraductal sampling (spyglass, biopsy or brushings) is essential before steroids [1].
Mikulicz syndrome (sialadenitis and dacryoadenitis)
Bilateral, painless, persistent enlargement of the lacrimal and major salivary glands (submandibular more than parotid) in an older man is IgG4-RD until proven otherwise [1]. The distinction from Sjogren syndrome is a high-yield discriminator: IgG4-RD is male-predominant and older in onset, presents with prominent gland enlargement rather than pure sicca, has an elevated serum IgG4, and is anti-Ro and anti-La negative [1]. Sjogren is female-predominant, younger, sicca-dominant, and seropositive. Submandibular gland enlargement as a firm mass is also called Kuttner tumour and is a classic IgG4-RD presentation. Always consider lymphoma (marginal zone or MALT) when the glands are asymmetric or growing rapidly — flow cytometry on the biopsy resolves this.
IgG4-related kidney disease
The renal lesion is a tubulointerstitial nephritis with IgG4-positive plasma cells in the interstitium, often with immune-complex deposition and low complement. The contrast CT shows multiple, bilateral, wedge-shaped or round low-attenuation renal cortical lesions, which can be mistaken for renal cell carcinoma [1]. Patients present with renal impairment, sometimes with nephrotic-range proteinuria when there is membranous nephropathy overlap. The response to steroids is excellent when the lesion is inflammatory, but a missed diagnosis leads to irreversible renal scarring. Membranous nephropathy in IgG4-RD is linked to anti-laminin and to phospholipase A2 receptor antibodies in a subset.
Retroperitoneal fibrosis (Ormond disease)
Retroperitoneal fibrosis presents with flank or back pain, hypertension, hydronephrosis and acute kidney injury from bilateral ureteric obstruction, with a periaortic and peri-iliac fibrotic mass that encases rather than displaces the great vessels [1]. The first move is always to decompress the obstruction with ureteric stents or nephrostomy, because the renal recovery depends on the duration of obstruction. Steroids then address the active inflammatory component. The critical step is to exclude secondary causes — malignancy (the desmoplastic reaction of an occult retroperitoneal, colonic or urothelial tumour), drugs (ergot-derived, dopaminergic agents, beta-blockers), infection and prior radiotherapy — because secondary retroperitoneal fibrosis needs a different treatment and carries a different prognosis [1].
Aortitis, lung disease, pachymeningitis and hypophysitis
IgG4-related aortitis involves the thoracic and abdominal aorta, producing inflammatory aneurysms with a risk of dissection or rupture; it may coexist with retroperitoneal fibrosis. IgG4-related lung disease causes pulmonary nodules, consolidation, interstitial infiltrates, mediastinal lymphadenopathy and pleural effusion — biopsy is essential because the radiology overlaps with lung cancer and sarcoidosis. IgG4-related pachymeningitis causes dural thickening with headache and cranial nerve palsies, mimicking meningioma or neurosarcoidosis. IgG4-related hypophysitis causes a pituitary mass with hypopituitarism and can present in adrenal crisis — check all pituitary axes and give stress-dose hydrocortisone before imaging or steroid therapy [1].
Pathophysiology: why the disease behaves the way it does
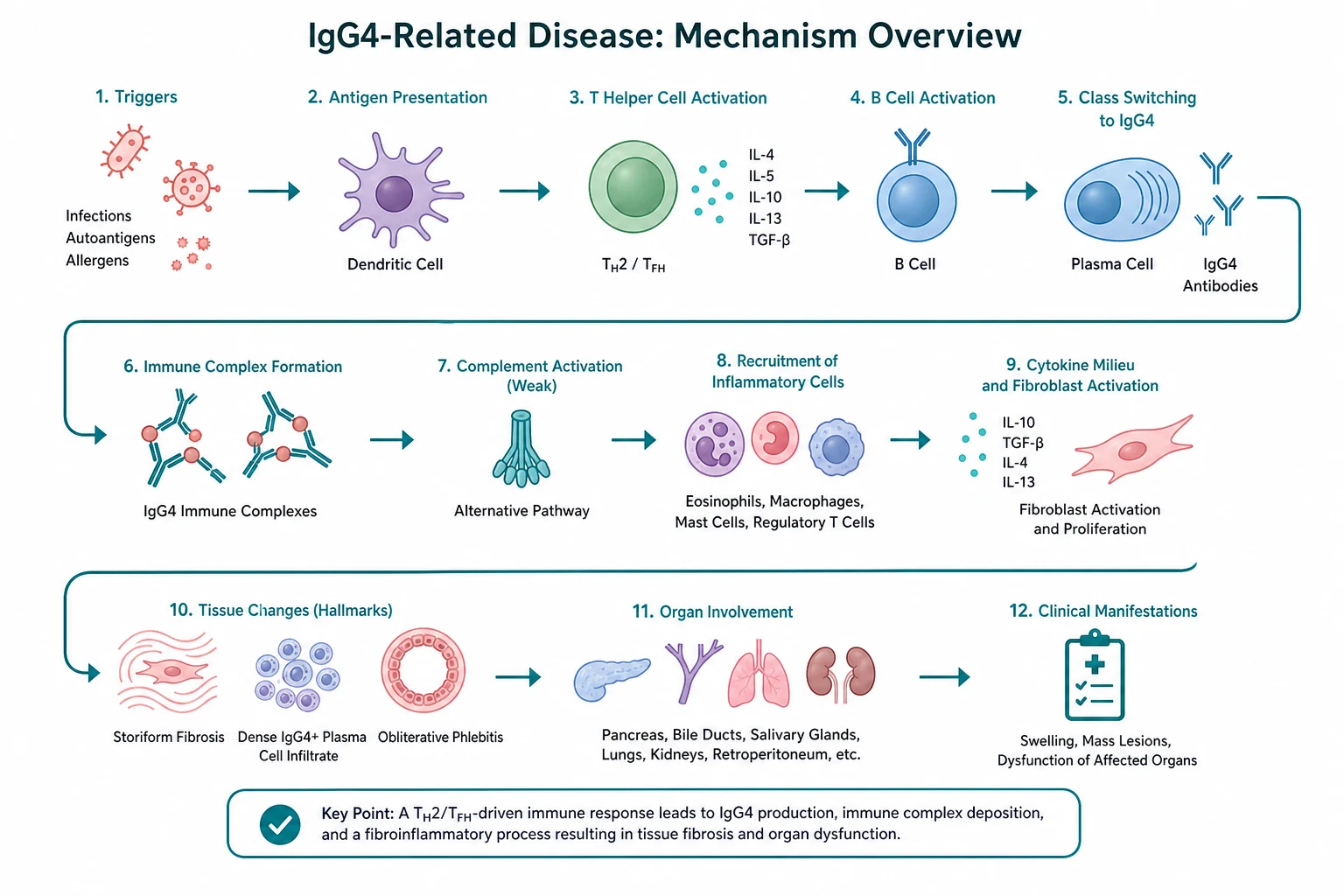

The histopathology is the defining feature and the gold standard for diagnosis [1]. Three findings together form the signature:
- A dense lymphoplasmacytic infiltrate rich in IgG4-positive plasma cells, often with lymphoid follicles and eosinophils.
- Storiform fibrosis — a swirling, cartwheel-like arrangement of spindle cells and collagen, so named because it resembles a woven mat.
- Obliterative phlebitis — veins that are destroyed or occluded by the inflammatory infiltrate, while arteries are typically spared [1].
Immunostaining shows an increased number of IgG4-positive plasma cells per high-power field and an IgG4 to total IgG plasma cell ratio above 40 per cent [1][6]. Two cautions. First, the IgG4 cell count threshold is organ-specific (for example, above 50 per high-power field for salivary gland, above 30 for pancreas), because IgG4-positive cells are present in normal and inflamed tissue. Second, the count is not diagnostic in isolation — lymphoma, multicentric Castleman disease, Rosai-Dorfman disease and even some cancers can have IgG4-positive cells; always interpret the count with the histological triad and the clinical context [1].
The mechanism is T-cell-driven, not antibody-driven. The effector cells are cytotoxic T cells (SLAMF7-positive CD4-positive cytotoxic T cells and CD8-positive cells) that release pro-fibrotic and pro-inflammatory cytokines (interferon-gamma, interleukin-1 beta, TGF-beta) [1]. IgG4 is the least abundant IgG subclass and has intrinsic anti-inflammatory properties (Fab-arm exchange, poor complement fixation), so the IgG4 elevation is now understood as a downstream marker of an ongoing immune response rather than the effector that damages tissue [1]. This T-cell-centric model is also why B-cell depletion with rituximab works without IgG4 being directly pathogenic — depleting the antigen-presenting and co-stimulatory B-cell compartment dampens the T-cell-driven inflammation [4][5].
The disease evolves in two phases that explain the treatment response. The early inflammatory phase is oedematous, IgG4-rich and highly steroid-responsive. The late fibrotic phase is collagen-dense, acellular and steroid-resistant. Fibrotic strictures, burnt-out retroperitoneal rinds and chronic biliary stenoses belong to the second phase and respond to mechanical relief rather than immunosuppression [1][3].
Diagnosis: the 2019 ACR and EULAR criteria, the 2020 revised criteria, and the role of tissue

Diagnosis integrates three streams: the clinical-radiological pattern, the serology and the histopathology. No single test is diagnostic. The two examinable frameworks are the 2019 ACR and EULAR classification criteria (an international weighted score for research and clinical classification) and the 2020 revised comprehensive diagnostic (RCD) criteria (a Japanese clinicopathological framework) [2][6].
The 2019 ACR and EULAR classification criteria
The criteria are a three-step process [2]:
- Entry criterion — characteristic clinical or radiological involvement of at least one of 11 typical organs (pancreas, biliary tree, salivary and lacrimal glands, kidney, lung, retroperitoneum, aorta, meninges, thyroid, breast, orbit). If the entry criterion is not met, the patient is not classified further.
- Exclusion criteria — a list of 32 clinical, serological, radiological and pathological items, any one of which excludes the diagnosis. The exclusion criteria are the safeguard against overdiagnosis: a rising CA19-9, clinical weight loss and cachexia, fever, a non-IgG4-elevated gammaglobulin, and atypical histology (granulomas, prominent neutrophils, necrosis, malignancy) all exclude IgG4-RD.
- Weighted inclusion criteria — points are scored across eight domains (clinical, serological, radiological and pathological) for a possible total well above 20. A total score of 20 or more classifies the case as IgG4-RD. The pathological domain carries the heaviest weight, reflecting that tissue is the gold standard [2].
The 2020 revised comprehensive diagnostic criteria
The Japanese criteria combine three domains [6]: (1) clinical-radiological (diffuse or localised organ swelling or a mass), (2) serological (serum IgG4 above 135 mg per decilitre), and (3) pathological (dense lymphoplasmacytic infiltrate and fibrosis, with IgG4-positive cells and an IgG4 to IgG ratio above 40 per cent). Definite IgG4-RD requires all three; probable requires clinical and pathological; possible requires clinical and serological. The criteria are designed to be used together with organ-specific criteria and emphasise that a normal IgG4 does not exclude the disease when the histology and the clinical pattern fit [6].
The correct use of serum IgG4
Serum IgG4 is supportive, not diagnostic [1][3]. It is elevated (above 135 mg per decilitre) in 60 to 70 per cent of patients with IgG4-RD. It is falsely elevated in 10 to 15 per cent of pancreatobiliary cancers and cholangiocarcinoma, and it is mildly elevated in primary sclerosing cholangitis, asthma, atopic dermatitis and other autoimmune diseases. It is normal in 20 to 30 per cent of biopsy-proven type 1 AIP. The practical use of the test is as follows: a markedly elevated IgG4 (more than twice the upper limit) with a normal CA19-9 favours IgG4-RD; a rising CA19-9 with a modestly elevated IgG4 favours malignancy; a normal IgG4 never excludes the disease [1].
Imaging and staging
Contrast CT and MRI (including MRCP) define the tumefactive lesions. FDG-PET-CT has two roles: it maps the disease extent at baseline (identifying metabolically active lesions suitable for biopsy and detecting occult aortitis, pachymeningitis or renal involvement) and it tracks the treatment response over time [1][3]. A baseline PET-CT is increasingly considered part of the workup of multi-organ IgG4-RD.
The investigations panel at a glance
At the bedside, order investigations in three tiers — supportive serology, definitive imaging and tissue, and the exclusion of mimics [1][2][3].
| Tier | Test | What it tells you | The pitfall |
|---|---|---|---|
| Serology | Serum IgG4 | Elevated above 135 mg per decilitre in 60 to 70 per cent | Falsely elevated in cancer, PSC, asthma; normal in 20 to 30 per cent of true disease |
| Serology | CA19-9, CEA | Exclusion of pancreatobiliary malignancy | Mildly elevated in cholangitis; rising trend favours cancer |
| Serology | Anti-Ro, anti-La, ANA, ANCA | Exclusion of Sjogren, lupus, vasculitis | IgG4-RD is typically anti-Ro and anti-La negative |
| Serology | Hepatitis B and C, HIV, IGRA | Pre-biologic and infection screen | Mandatory before rituximab |
| Imaging | Contrast CT, MRI and MRCP | Tumefactive lesions, sausage-shaped pancreas, biliary strictures, renal cortical nodules | A focal mass still needs tissue |
| Imaging | FDG-PET-CT | Disease extent and metabolically active biopsy targets; treatment response | Uptake is non-specific; it does not distinguish from malignancy |
| Tissue | Biopsy with IgG4 immunostaining | Dense lymphoplasmacytic infiltrate, storiform fibrosis, obliterative phlebitis | The count threshold is organ-specific and not diagnostic alone |
| Tissue | Flow cytometry, clonality | Exclusion of lymphoma | Required when a gland or mass is asymmetric or rapidly growing [1] |
The differential diagnosis: what to exclude, and in what order
The single most dangerous error is to treat cancer with steroids. The differentials fall into three groups [1].
Malignancy. Pancreatic cancer, cholangiocarcinoma, renal cell carcinoma, lung cancer, lymphoma (marginal zone, MALT, plasma cell) and metastatic disease can each produce a mass and can sometimes elevate IgG4. Exclude cancer with a combination of tumour markers (CA19-9), cross-sectional imaging, endoscopic sampling and tissue biopsy before committing to steroids. The diagnosis of IgG4-RD is never made on serology alone when a malignancy is plausible [1][3].
Mimic inflammatory disease. Sjogren syndrome (female, sicca-dominant, anti-Ro and anti-La, normal IgG4), primary sclerosing cholangitis (IBD association, beaded strictures, normal IgG4, poor steroid response), sarcoidosis (non-caseating granulomas, bilateral hilar lymphadenopathy), multicentric Castleman disease, Rosai-Dorfman disease and inflammatory myofibroblastic tumour all enter the differential and require histopathology to separate from IgG4-RD [1].
Infection and secondary causes. Tuberculosis, syphilis and fungal infection can cause bilateral salivary enlargement or mass lesions. Secondary retroperitoneal fibrosis (malignancy, drugs, radiation) must be excluded before labelling the periaortic rind IgG4-related [1].
Management: steroids, rituximab, and the phase of the disease
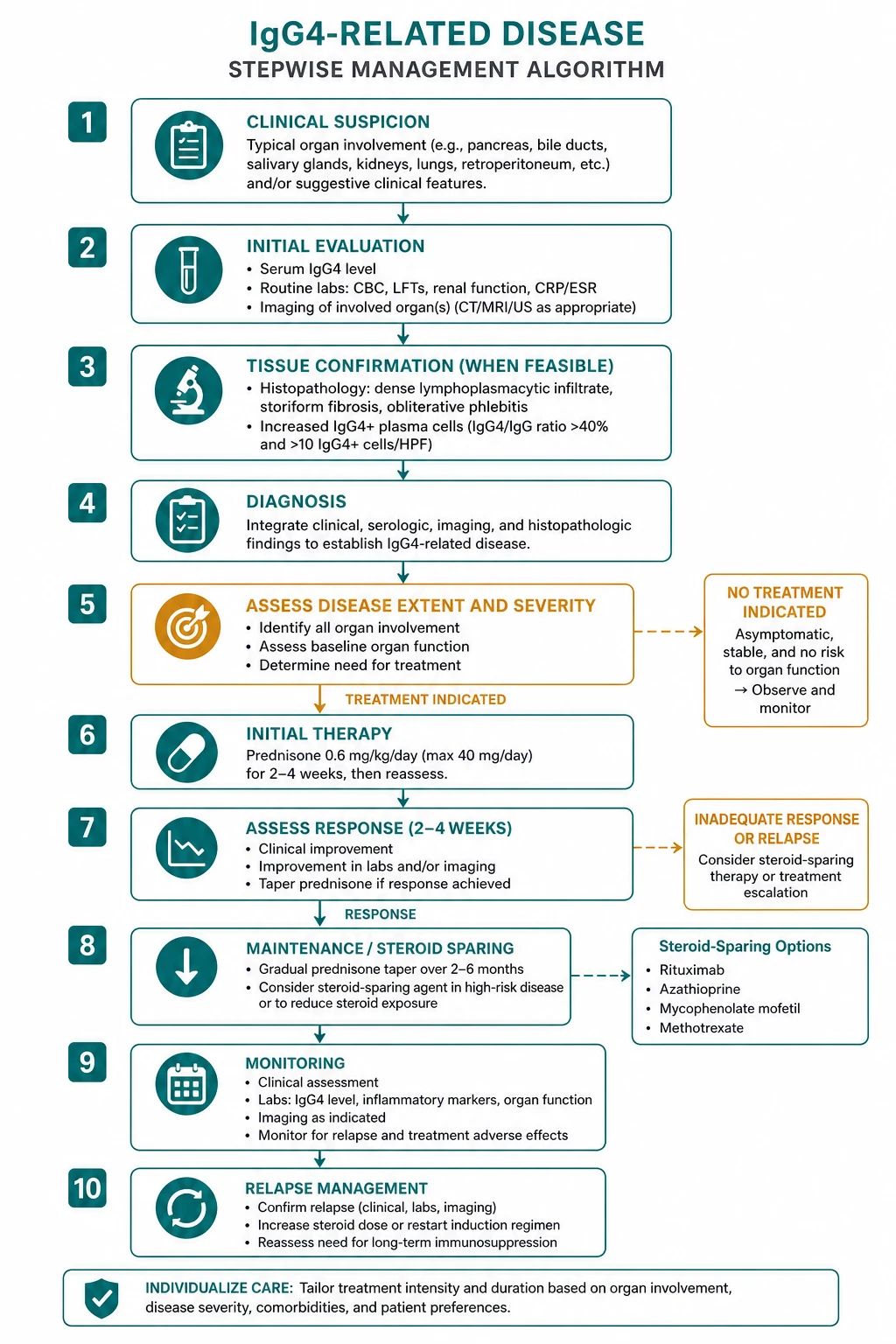
Glucocorticoids: first-line for active inflammatory disease
Prednisolone 30 to 40 mg daily for 2 to 4 weeks is the standard induction regimen, followed by a slow taper over 3 to 6 months [3]. The response in the inflammatory phase is dramatic — jaundice resolves, the pancreatic mass shrinks, the serum IgG4 falls and the PET-CT activity quenches within 2 weeks. A failure to respond within 2 weeks should prompt a search for malignancy or for a burnt-out fibrotic lesion [1][3]. Give bone protection (calcium, vitamin D, a bisphosphonate or denosumab where indicated) and consider pneumocystis prophylaxis with co-trimoxazole during high-dose steroids or combined immunosuppression.
Not every patient needs immediate steroids. Asymptomatic, mild, single-organ disease (for example, mild lacrimal gland enlargement) may be monitored, because the treatment burden is real and spontaneous remission, while rare, can occur [3].
Rituximab: the biologic of choice for relapsing or multi-organ disease
Rituximab (anti-CD20 B-cell depletion) is highly effective in IgG4-RD and is the biologic of choice for relapsing, multi-organ, steroid-dependent or steroid-refractory disease [4][5]. The regimen is 375 mg per square metre weekly for 4 doses, or 1 g on days 1 and 15. Before the first dose, screen for hepatitis B (HBsAg, anti-HBc and HBV DNA) and tuberculosis, because rituximab reactivates hepatitis B and can cause fatal fulminant hepatitis; give antiviral prophylaxis where indicated [4]. Premedicate with paracetamol, an antihistamine and an intravenous steroid, and monitor immunoglobulins for hypogammaglobulinaemia with repeat courses [5].
The mechanism is elegant. Rituximab depletes the B-cell compartment that provides antigen presentation and co-stimulation to the effector cytotoxic T cells; as the T-cell-driven inflammation subsides, the IgG4-positive plasma cells (which are CD20-negative and therefore not directly depleted) decline in parallel [4][5]. Relapse is common, and most patients relapse within a year, so rituximab is increasingly used as a planned maintenance strategy rather than only as rescue therapy [5].
Steroid-sparing and maintenance therapy
Conventional steroid-sparing agents — mycophenolate mofetil (2 g daily) and azathioprine (1 to 2 mg per kg daily) — are used for maintenance and in those who cannot have rituximab, but the evidence base is weaker than for rituximab [3]. Check thiopurine methyltransferase (TPMT) before azathioprine and monitor the full blood count for myelosuppression. Counsel on the teratogenicity of mycophenolate (pregnancy category D, effective contraception) [3].
The fibrotic phase: mechanical relief, not immunosuppression
Burnt-out fibrotic lesions are steroid-resistant [1][3]. For a chronic biliary stricture, place a stent. For bilateral ureteric obstruction from retroperitoneal fibrosis, stent or nephrostomise first and treat the inflammation second. For an aortic aneurysm, control blood pressure and involve vascular surgery. Escalating immunosuppression for a fibrotic lesion adds toxicity without benefit and is a classic exam trap.
Treating organ-threatening complications
Decompress first, immunosuppress second. Ureteric stents or nephrostomy for retroperitoneal fibrosis with obstruction; biliary drainage and antibiotics for an infected stricture with cholangitis; strict blood pressure control and vascular surgery for aortitis with aneurysm; stress-dose hydrocortisone for hypophysitis with adrenal insufficiency [1].
Emergencies and escalation
The IgG4-RD emergencies are mechanical, infective and endocrine, not merely inflammatory — address the threatened organ before the immunosuppression [1][3].
- Bilateral ureteric obstruction with acute kidney injury (retroperitoneal fibrosis) — decompress urgently with nephrostomy or ureteric stenting before imaging the mass in detail; renal recovery depends on the duration of obstruction. Start prednisolone 30 to 40 mg daily once the obstruction is relieved and the diagnosis is supported.
- Cholangitis or sepsis from an infected biliary stricture — broad-spectrum antibiotics and biliary drainage by ERCP take precedence over steroids; immunosuppression is added only once infection is controlled and the diagnosis is secure.
- Aortic dissection or rupture from IgG4-related aortitis — strict blood pressure control with an intravenous labetalol or esmolol infusion, urgent vascular surgery review, and cross-sectional imaging. Consider anticoagulation where thrombus coexists with aneurysm.
- Adrenal crisis from IgG4-related hypophysitis — check all pituitary axes and give parenteral hydrocortisone (100 mg intravenously, then 50 mg every 6 hours) with intravenous fluids before pituitary imaging; only then address the inflammatory mass with steroids.
- Acute visual change or progressive cranial nerve deficit from pachymeningitis — urgent MRI, high-dose methylprednisolone, and neurology or neurosurgery input to exclude meningioma, infection or neurosarcoidosis [1][3].
The general principle is that a patient who is deteriorating needs the organ threatened by the disease protected first, the diagnosis confirmed in parallel, and the immunosuppression calibrated to the phase of the lesion [3].
Relapse, surveillance and long-term outcomes
Relapse is the natural history of the disease. Roughly 40 to 50 per cent of patients relapse during or after the steroid taper, and most patients who respond to rituximab relapse within a year [3][5]. Plan a structured maintenance strategy from the outset rather than treating only flares — for multi-organ disease, this usually means a planned course of rituximab with monitoring of peripheral B-cell counts and immunoglobulins [5].
Surveillance combines symptom review with serial serum IgG4 (as a marker of activity, not a diagnostic test), renal and liver function, blood glucose, blood pressure, bone density, and periodic cross-sectional imaging for the involved organs. For aortitis, add periodic aortic imaging to detect aneurysm or dissection. For type 1 AIP, monitor for pancreatic exocrine and endocrine failure (steatorrhoea, diabetes) and for recurrent biliary obstruction [1][3].
The long-term outlook depends on the organ and the phase. Inflammatory disease caught early is highly treatable. Fibrotic disease, missed or late presentations, and the cumulative toxicity of repeated steroids all drive irreversible organ damage — pancreatic insufficiency, biliary cirrhosis, end-stage renal disease, and aortic dissection [1]. An unresolved question is whether IgG4-RD carries an increased risk of malignancy (pancreatic, biliary, lymphoma); cancer must always be excluded at diagnosis and reconsidered whenever the disease behaves atypically [1][3].
The FRACP DWE high-yield points
- The diagnosis is clinicopathological. Serum IgG4 is supportive; histopathology (dense lymphoplasmacytic infiltrate, storiform fibrosis, obliterative phlebitis, IgG4 to IgG ratio above 40 per cent) is the gold standard [1][6].
- A normal IgG4 does not exclude IgG4-RD (20 to 30 per cent of biopsy-proven type 1 AIP are seronegative). An elevated IgG4 does not confirm it (falsely elevated in cancer, PSC, asthma, atopy) [1].
- Type 1 AIP presents as painless jaundice with a sausage-shaped pancreas and responds to steroids; type 2 AIP is a separate IgG4-negative, IBD-associated disease [1].
- Mikulicz syndrome in an older man is IgG4-RD, not Sjogren — male sex, prominent gland enlargement, normal anti-Ro and anti-La, elevated IgG4 [1].
- Retroperitoneal fibrosis with ureteric obstruction — decompress first, then treat the inflammation; exclude secondary causes [1].
- Rituximab is the biologic of choice for relapsing, multi-organ or steroid-dependent disease; screen hepatitis B first [4][5].
- The fibrotic phase is steroid-resistant — treat the complication (stenting, drainage), not the burnt-out fibrosis [1][3].
- Always exclude cancer before a diagnostic steroid trial — a radiological response can occur with lymphoma and even transiently with cancer [1][3].
DCE short case: salivary and lacrimal gland examination for Mikulicz syndrome
A common short-case station is the older man with bilateral, painless gland enlargement. The instruction is to examine the head, neck and general systems [1].
The routine. Begin at the end of the bed: observe for the facial fullness of parotid or submandibular enlargement, the proptosis of lacrimal involvement, and any neck scars from previous biopsy. Examine the lacrimal glands first (evert the upper eyelid and palpate the superolateral orbit), then the parotid (inspect and palpate both lobes, note any ductal discharge), then the submandibular glands (bimanual palpation, feeling intra-orally), and then the cervical, supraclavicular and axillary lymph nodes [1].
The signs. Firm, non-tender, bilateral enlargement of the submandibular glands (Kuttner tumour) with lacrimal gland enlargement and firm, mobile cervical nodes is the classic IgG4-RD picture. Assess for sicca — dry eyes (Schirmer test if available), dry mouth, dental caries — and for the systemic features of multi-organ disease: examine the chest for pleural effusion, the abdomen for organomegaly, and the skin for excoriation from pruritus [1].
The discrimination. IgG4-RD (older man, prominent gland enlargement, normal anti-Ro and anti-La, elevated IgG4) versus Sjogren syndrome (younger woman, sicca-dominant, anti-Ro and anti-La positive, normal IgG4) versus lymphoma (asymmetric or rapidly growing, requires flow cytometry) versus sarcoidosis (non-caseating granulomas, bilateral hilar lymphadenopathy) [1].
The presentation. "I found firm, bilateral, non-tender enlargement of the submandibular and lacrimal glands in an older man with no sicca, no lymphadenopathy and no organomegaly. My leading diagnosis is IgG4-related disease causing Mikulicz syndrome. I would like to confirm the diagnosis with a serum IgG4 and IgG subclasses, autoimmune and infection serology, cross-sectional imaging and an FDG-PET-CT for extent, and a submandibular gland biopsy for histopathology and flow cytometry to exclude lymphoma." [1]
The discussion flows from the signs to the unifying diagnosis to the organ-specific differentials to the investigation to the management — lead with the answer and keep the structure tight [3].
Communication, consent and shared decision-making
Frame the diagnosis as a treatable but relapsing multisystem disease. Most patients respond to steroids, but relapse is common, so set the expectation for a maintenance strategy and long-term surveillance. Address the "is it cancer?" fear directly — explain that the disease mimics cancer and that cancer has been actively excluded by biopsy, imaging and the steroid response, because the question the patient is really asking is whether they have been misdiagnosed [3].
Discuss the steroid burden honestly — osteoporosis, diabetes, infection, cataracts and mood — and explain the rationale for steroid-sparing therapy (rituximab or mycophenolate) to reduce cumulative exposure. Coordinate multi-specialty care explicitly: rheumatology, gastroenterology or hepatology, nephrology, ophthalmology, surgery and endocrinology each own an organ; name the lead clinician so the patient knows who to call [3]. Address fertility and vaccination before rituximab — live vaccines are avoided during and after B-cell depletion, and hepatitis B screening precedes the first dose [4].
When the disease is burnt-out and organ damage is irreversible, the ethical shift is from immunosuppression to symptom control, rehabilitation and advance care planning. Early palliative care integration is appropriate for advanced fibrotic disease with end-organ failure [3].
References and sources
IgG4-related disease review (Kamisawa, Zen, Pillai, Stone, Lancet 2015) [1]; 2019 American College of Rheumatology and European League Against Rheumatism classification criteria for IgG4-related disease (Wallace, Naden, Chari, Stone, et al., Arthritis Rheumatol 2020) [2]; International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease (Khosroshahi, Wallace, Crowe, et al., Arthritis Rheumatol 2015) [3]; Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients (Khosroshahi, Carruthers, et al., Medicine 2012) [4]; Rituximab for IgG4-related disease: a prospective, open-label trial (Carruthers, Topazian, Khosroshahi, et al., Ann Rheum Dis 2015) [5]; 2020 Revised Comprehensive Diagnostic Criteria for IgG4-RD (Umehara, Okazaki, Kawa, et al., Modern Rheumatology 2021) [6]. All PMIDs verified live before use. Regional anchoring: Australian Rheumatology Association guidance on biological therapy access in ANZ; the 2019 ACR and EULAR criteria and the 2015 International Consensus Guidance Statement are the examinable international frameworks.
References
- [1]Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease Lancet, 2015.PMID 25481618
- [2]Wallace ZS, Naden RP, Chari ST, et al. The 2019 American College of Rheumatology/European League Against Rheumatism classification criteria for IgG4-related disease Ann Rheum Dis, 2020.PMID 31796497
- [3]Khosroshahi A, Wallace ZS, Crowe JL, et al. International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease Arthritis Rheumatol, 2015.PMID 25809420
- [4]Khosroshahi A, Carruthers MN, Deshpande V, et al. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients Medicine (Baltimore), 2012.PMID 22210556
- [5]Carruthers MN, Topazian MD, Khosroshahi A, et al. Rituximab for IgG4-related disease: a prospective, open-label trial Ann Rheum Dis, 2015.PMID 25667206
- [6]Umehara H, Okazaki K, Kawa S, et al. The 2020 revised comprehensive diagnostic (RCD) criteria for IgG4-RD Mod Rheumatol, 2021.PMID 33274670