Phys · rheumatological
Systemic Autoinflammatory Syndromes
Also known as autoinflammatory disease · periodic fever syndrome · hereditary recurrent fever · familial Mediterranean fever · FMF · TNF receptor-associated periodic syndrome · TRAPS · hyper-IgD syndrome · HIDS · mevalonate kinase deficiency · MKD · cryopyrin-associated periodic syndrome · CAPS · familial cold autoinflammatory syndrome · FCAS · Muckle-Wells syndrome · NOMID · CINCA · adult-onset Still disease · AOSD · Schnitzler syndrome · Behcet disease
Consultant-physician-depth guide to the systemic autoinflammatory syndromes for FRACP DWE and DCE — the innate-versus-adaptive immune framework, the hereditary recurrent fevers (familial Mediterranean fever with MEFV and colchicine prophylaxis and AA amyloidosis, TNF receptor-associated periodic syndrome with TNFRSF1A and prolonged fever, hyper-IgD syndrome with mevalonate kinase deficiency), the cryopyrin-associated periodic syndromes (CAPS spectrum of FCAS, Muckle-Wells and NOMID), adult-onset Still disease with the Yamaguchi criteria and elevated ferritin, Schnitzler syndrome with chronic urticaria and monoclonal IgM gammopathy, Behcet disease cross-referenced with the vasculitis topic, the diagnostic approach of pattern recognition plus a targeted genetic panel, and the treatment principle that IL-1 blockade is transformative for NLRP3-driven disease, HIDS, colchicine-resistant FMF, TRAPS, AOSD and Schnitzler syndrome.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Systemic Autoinflammatory Syndromes
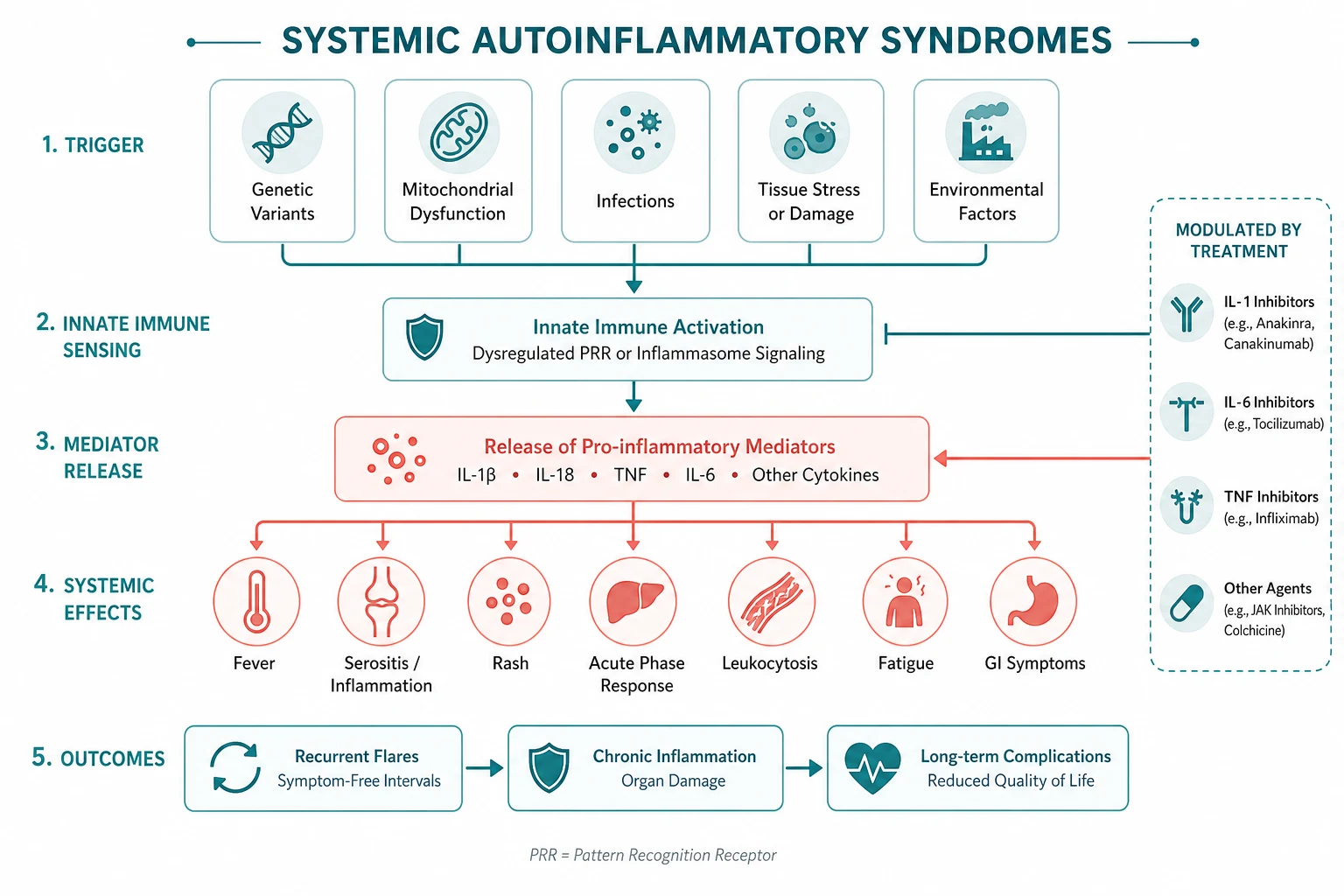

The answer first
Autoinflammatory diseases are defined by innate immune system dysregulation — recurrent, often unprovoked attacks of systemic inflammation driven by excess cytokine signalling (chiefly interleukin-1), without high-titre autoantibodies or antigen-specific lymphocyte responses. That single idea is the lens for the whole topic: where autoimmune disease (SLE, rheumatoid arthritis) is adaptive, antibody-driven and acquired, autoinflammatory disease is innate, cytokine-driven and often hereditary. [1]
Three rules that change outcome: [1]
- Interleukin-1 blockade is transformative for a defined group of these diseases. CAPS, HIDS, colchicine-resistant FMF, TRAPS, AOSD and Schnitzler syndrome respond dramatically to canakinumab or anakinra. Recognising an IL-1-responsive syndrome changes a patient's life.
- Colchicine prevents AA amyloidosis in familial Mediterranean fever. The goal of FMF treatment is complete attack prevention and amyloidosis prevention, not just fewer attacks. Adherence to lifelong colchicine is the single biggest determinant of long-term renal outcome.
- Attack duration is the best discriminator between the hereditary recurrent fevers. FMF attacks last 1 to 3 days; HIDS attacks last 3 to 7 days; TRAPS attacks last 1 to 3 weeks. When the genetic panel is pending or ambiguous, this single feature points to the diagnosis. [1]
The clinical reasoning moves in three steps at the bedside. First, exclude infection, malignancy and autoimmune disease — these are far more common than autoinflammatory disease and must never be missed. Second, characterise the attack pattern — duration, interval, family history, ancestry and accompanying organ features. Third, confirm with a targeted genetic panel and syndrome-specific tests, then treat by the dominant cytokine (colchicine for FMF; IL-1 for CAPS, HIDS, TRAPS, AOSD, Schnitzler; IL-6 for articular AOSD). [1]
Autoinflammatory versus autoimmune — the conceptual framework
The autoinflammatory-versus-autoimmune distinction is the conceptual foundation of this topic and is examinable. Autoimmune disease arises from loss of adaptive immune tolerance — autoreactive T cells and high-titre autoantibodies (anti-dsDNA, anti-CCP, ANCA). Autoinflammatory disease arises from excessive activation of innate immune pathways — inflammasomes, pattern-recognition receptors and the cytokines they release (interleukin-1 beta, interleukin-6, interleukin-18, TNF). [1]
The practical consequences matter at the bedside. In autoinflammatory disease, autoantibody panels are typically negative, the inflammation is episodic (in the periodic fever syndromes) or continuous (in the CAPS spectrum), and the treatment targets cytokines rather than lymphocytes. A patient with recurrent fever and a relentlessly negative autoimmune screen is the classic autoinflammatory presentation. [1]
Two diseases sit at the interface and are worth flagging. Adult-onset Still disease is autoinflammatory in mechanism (cytokine storm, IL-1 and IL-6 driven) but is acquired, not hereditary. Behcet disease is partly autoinflammatory (neutrophil hyperactivity) and partly vasculitic. In exams, the cleanest answer is that autoinflammatory disease is innate and cytokine-driven, while autoimmune disease is adaptive and antibody-driven. [1]
DWE high-yield: The single most testable distinction is innate (autoinflammatory) versus adaptive (autoimmune) immunity. Autoinflammatory diseases are driven by interleukin-1 in most cases, are often hereditary, and have negative autoantibody screens. AOSD and Behcet sit at the interface. [1]
Classification framework
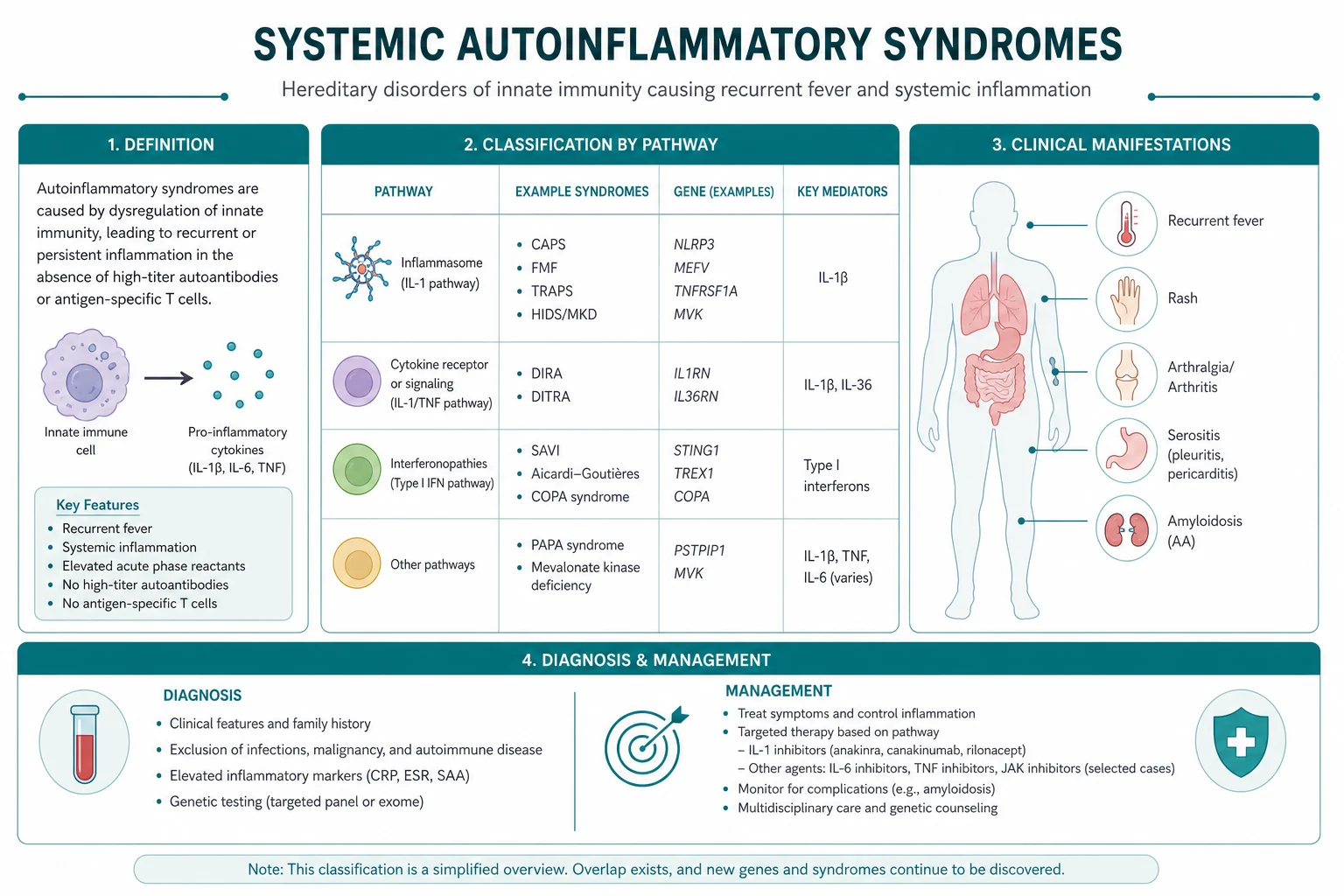

The autoinflammatory diseases divide into three clinically useful groups. The first is the hereditary recurrent fevers — FMF, TRAPS and HIDS (mevalonate kinase deficiency). The second is the cryopyrin-associated periodic syndromes (CAPS), a single NLRP3-driven spectrum running from mild cold-induced disease to devastating multisystem disease of infancy. The third is the polygenic or acquired autoinflammatory diseases — adult-onset Still disease and Schnitzler syndrome — which are not hereditary but share the innate, cytokine-driven mechanism and the IL-1 responsiveness. [1]
Behcet disease is best handled in its own vasculitis topic and cross-referenced here, because it straddles the autoinflammatory and vasculitic classifications. [1]
The comparison table is the high-yield revision aid. Memorise the gene, the inheritance, the attack duration and the first-line therapy for each hereditary recurrent fever; the rest follows from the cytokine biology. [1]
Familial Mediterranean fever
Familial Mediterranean fever (FMF) is the most common hereditary autoinflammatory disease and the archetype of the periodic fever syndromes. It is caused by autosomal recessive mutations in MEFV, which encodes pyrin, a protein that regulates the pyrin inflammasome. Mutant pyrin constitutively activates the inflammasome, driving interleukin-1 beta and interleukin-18 release and neutrophil-mediated serositis. The disease is concentrated in people of Mediterranean ancestry — Sephardic Jewish, Armenian, Turkish, Arabic and Greek populations — and carrier frequencies are high in these groups. [1]
Clinical presentation — the attack pattern that defines the disease: [1]
- Brief attacks lasting 1 to 3 days is the single most characteristic feature. The attacks are self-limiting and resolve completely between episodes, with the patient entirely well in the interval. A diary of attack duration is often diagnostic before any test.
- Fever is near-universal, typically reaching 38 to 40 degrees Celsius, and is the first feature of most attacks.
- Serositis is the hallmark. Peritonitis is most common and most clinically dramatic — severe diffuse abdominal pain with guarding and rigidity that mimics an acute surgical abdomen, driving a high rate of unnecessary laparotomy and appendicectomy before the diagnosis is made. Pleuritis (unilateral chest pain) and pericarditis also occur.
- Arthritis is typically a monoarticular arthritis of a large joint (knee, ankle, hip), distinct from rheumatoid arthritis.
- Erysipelas-like rash — a tender, warm, erythematous, non-blanching rash on the lower leg, typically below the knee, often mistaken for cellulitis — is the classic cutaneous sign. [1]
The Tel Hashomer criteria for FMF diagnosis are the examinable framework. Diagnosis is established by one major criterion or two minor criteria (the simple criteria set), with sensitivity above 95 per cent and specificity above 97 per cent [1]. The major criteria are typical attacks of peritonitis, pleuritis or pericarditis; monoarticular arthritis; fever alone; or incomplete abdominal attacks. A favourable response to colchicine and leg pain with exertion are supportive.
The feared complication — AA amyloidosis: [1]
AA amyloidosis is the principal cause of death in untreated FMF. Persistent inflammation drives overproduction of serum amyloid A, which deposits as AA amyloid in the kidney and presents as proteinuria progressing to nephrotic syndrome and renal failure. The risk is greatest in patients with the M694V homozygous genotype and in those with high attack frequency. Colchicine prevents AA amyloidosis, which is why the goal of FMF treatment is complete attack prevention, not merely attack reduction. [1]
Management: [1]
- Colchicine is first-line and lifelong. The dose is 1 to 2 mg daily (commonly 1.5 mg), titrated to the dose that completely prevents attacks. Gastrointestinal intolerance (diarrhoea) is the main adverse effect; reduce the dose in renal impairment and monitor full blood count for myelosuppression [2].
- Canakinumab for colchicine-resistant FMF. For patients who do not achieve complete attack control on the maximum tolerated colchicine dose, canakinumab (150 mg subcutaneously every 8 weeks) is the evidence-based IL-1 blocker, approved on the basis of the CLUSTER trial [3].
- Avoid laparotomy during an acute abdominal attack. A careful history of recurrent self-limiting attacks prevents unnecessary surgery. NSAIDs or colchicine manage the acute attack.
Examiner trap: A young patient of Mediterranean ancestry with recurrent abdominal pain, a previous normal appendicectomy and a scar is a classic FMF presentation — the prior surgery is a clue, not reassurance. A single urinalysis for proteinuria and a serum amyloid A screen for AA amyloidosis should be part of every suspected FMF assessment. [1]
TNF receptor-associated periodic syndrome
TNF receptor-associated periodic syndrome (TRAPS) is the second most common hereditary recurrent fever in many populations. It is caused by autosomal dominant mutations in TNFRSF1A, encoding the 55 kDa TNF receptor. The defective receptor fails to shed from the cell surface after TNF binding, trapping TNF signalling and driving mitochondrial reactive oxygen species and interleukin-1 beta production. TRAPS affects all ethnic groups, in contrast to the Mediterranean concentration of FMF. [1]
Clinical presentation — the prolonged attack that distinguishes TRAPS: [1]
- Prolonged attacks lasting 1 to 3 weeks is the cardinal discriminator from FMF. TRAPS attacks are far longer than FMF attacks, and this single feature often points to the diagnosis before genetic testing.
- Migratory centrifugal rash — an erythematous rash that begins on the extremity and migrates outward, often centripetally over the trunk, distinct from the FMF erysipelas-like rash.
- Periorbital oedema and conjunctivitis — periorbital swelling is a highly characteristic and nearly pathognomonic feature of TRAPS that should always be asked about in a recurrent fever history.
- Severe myalgia — migratory muscle pain, often localised to the affected limb, frequently prominent.
- Abdominal pain and serositis also occur, overlapping with FMF but without the brief 1 to 3 day cycle. [1]
Management: [1]
- Glucocorticoids as short courses are effective for acute attacks, but cumulative steroid toxicity is a major problem given the prolonged attack duration.
- Etanercept (a soluble TNF receptor fusion protein) provides partial benefit, exploiting the TNF-driven mechanism.
- Canakinumab is now the preferred long-term agent for TRAPS, targeting the interleukin-1 beta overproduction that underlies the inflammation. It is approved for TRAPS on the basis of the CLUSTER trial [3].
TRAPS also carries a risk of AA amyloidosis, though lower than in untreated FMF, so ongoing surveillance with urinalysis and serum amyloid A is appropriate. [1]
Hyper-IgD syndrome (mevalonate kinase deficiency)
Hyper-IgD syndrome (HIDS), now more accurately termed mevalonate kinase deficiency (MKD), is caused by autosomal recessive mutations in MVK, encoding mevalonate kinase. Deficient enzyme activity reduces isoprenoid synthesis, causing defective protein prenylation and interleukin-1 beta hypersecretion. The older name "hyper-IgD" reflects the variably elevated serum IgD, but IgD is an unreliable marker — the gene name MKD is preferred. [1]
Clinical presentation: [1]
- Attacks lasting 3 to 7 days of fever, with onset in early childhood.
- Cervical lymphadenopathy is a prominent and characteristic feature, distinguishing HIDS from FMF and TRAPS.
- Abdominal pain with vomiting and diarrhoea, often marked.
- Maculopapular rash, diffuse and non-specific.
- Attacks triggered by vaccination in childhood — a classic history that should prompt consideration of MKD.
- Elevated urinary mevalonic acid during attacks supports the diagnosis, as does a history of attacks after childhood vaccinations. [1]
Management: [1]
- Canakinumab is the evidence-based IL-1 blocker for HIDS, approved on the basis of the CLUSTER trial [3].
- Anakinra and etanercept have been used with variable response. NSAIDs and short courses of steroids manage individual attacks.
- HIDS is generally milder than FMF in terms of amyloidosis risk, but severe MKD variants cause neurological and developmental complications in children.
DWE high-yield: When the genetic panel is pending, attack duration is the discriminator: FMF 1 to 3 days, HIDS 3 to 7 days, TRAPS 1 to 3 weeks. Add the gene (MEFV, MVK, TNFRSF1A) and the cardinal extra feature (FMF serositis and erysipelas-like rash; HIDS cervical lymphadenopathy; TRAPS periorbital oedema and migratory rash) and the diagnosis is secure on clinical grounds. [1]
Cryopyrin-associated periodic syndromes (CAPS)
The cryopyrin-associated periodic syndromes (CAPS) are a single spectrum caused by autosomal dominant gain-of-function mutations in NLRP3, encoding cryopyrin (NLRP3), a component of the NLRP3 inflammasome. The mutant inflammasome is constitutively active, overproducing interleukin-1 beta. The IL-1 dependence of CAPS is the most dramatic in all of medicine — canakinumab can transform a child with NOMID into a normally developing child within weeks. [1]
CAPS runs along a spectrum of severity, from the mildest form to the most devastating multisystem disease of infancy. [1]
Familial cold autoinflammatory syndrome (FCAS)
FCAS is the mildest CAPS phenotype. It presents with cold-induced urticarial rash, fever and arthralgia within hours of cold exposure. Attacks are brief (less than 24 hours) and resolve on rewarming. Renal amyloidosis is rare. The cold trigger is the defining feature and should be sought in the history. [1]
Muckle-Wells syndrome
Muckle-Wells syndrome sits in the middle of the spectrum. It presents with: [1]
- Urticarial rash — continuous or recurrent, present from infancy, often mistaken for chronic urticaria.
- Progressive sensorineural hearing loss — the cardinal and feared feature. Hearing loss begins in childhood or adolescence and is irreversible once established. Early IL-1 blockade prevents further loss and is the single most important reason not to delay treatment.
- AA amyloidosis — a significant risk in untreated Muckle-Wells, presenting as proteinuria.
- Periodic fevers, arthralgia and conjunctivitis. [1]
The combination of chronic urticaria, hearing loss and amyloidosis in a family is Muckle-Wells until proven otherwise. [1]
NOMID/CINCA
NOMID (neonatal-onset multisystem inflammatory disease), also called CINCA (chronic infantile neurological cutaneous articular), is the most severe CAPS phenotype and one of the most severe autoinflammatory diseases. It presents in infancy with: [1]
- Chronic aseptic meningitis — continuous central nervous system inflammation causing headache, irritability, developmental delay and seizures; brain MRI shows leptomeningeal enhancement and hydrocephalus may develop.
- Characteristic arthropathy — central epiphyseal overgrowth of the knees and other large joints, causing bony swelling and contractures that are radiographically and clinically distinctive.
- Urticarial rash from birth.
- Papilloedema and optic atrophy from chronic raised intracranial pressure.
- Hearing loss, hepatosplenomegaly and chronic anaemia. [1]
NOMID/CINCA requires early and aggressive IL-1 blockade — canakinumab or anakinra — to prevent irreversible neurological and bony damage. Untreated, it carries a high mortality in childhood. [1]
The IL-1 dependence of CAPS

CAPS is the prototypical IL-1-driven disease. The demonstration that canakinumab produces rapid and sustained remission in CAPS, with normalisation of inflammatory markers and prevention of hearing loss progression, established IL-1 blockade as a transformative therapy [4]. Canakinumab 150 mg subcutaneously every 8 weeks (300 mg for severe CAPS, weight-adjusted in children) is the standard therapy. Anakinra and rilonacept are alternative IL-1 pathway agents.
Examiner trap: The hearing loss in Muckle-Wells is irreversible once established — IL-1 blockade halts further loss but does not restore hearing. This is the single best argument for early canakinumab. A child with chronic urticaria and progressive hearing loss must be referred urgently, not managed as ordinary urticaria. [1]
Adult-onset Still disease
Adult-onset Still disease (AOSD) is a polygenic, non-hereditary autoinflammatory disease that shares its mechanism and cytokine profile with systemic-onset juvenile idiopathic arthritis. It is driven by a cytokine storm of interleukin-1 beta (fever and systemic features), interleukin-6 (arthritis and the acute phase response) and interleukin-18 (macrophage activation). AOSD is explicitly a diagnosis of exclusion — infection, malignancy (especially lymphoma) and autoimmune disease must be systematically excluded first. [1]
Clinical presentation: [1]
- Spiking quotidian fever — typically one or two temperature spikes per day, often in the late afternoon or evening, reaching 39 degrees Celsius or higher, with a relatively normal or even low temperature between spikes.
- Evanescent salmon-pink rash — a macular or maculopapular rash on the trunk and proximal limbs, classically appearing with the fever spike and fading as the fever resolves. It is non-pruritic and leaves no residual pigmentation. The rash may be missed unless the patient is examined during the fever.
- Arthritis or arthralgia — typically a polyarthritis affecting wrists, knees, ankles and small joints; may evolve into a chronic destructive arthritis.
- Sore throat — a prominent and early feature that should be asked about.
- Lymphadenopathy, splenomegaly and abnormal liver function tests are common. [1]
The Yamaguchi criteria are the most widely used classification criteria for AOSD [5]. Diagnosis requires at least 5 criteria, of which at least 2 must be major criteria, after excluding infection, malignancy and other rheumatic disease.
The four major criteria are: fever of 39 degrees Celsius or higher lasting one week or more; arthralgia or arthritis lasting two weeks or more; a typical non-pruritic salmon-pink rash; and leukocytosis (10,000 per cubic millimetre or more with 80 per cent or more neutrophils). [1]
The four minor criteria are: sore throat; lymphadenopathy and/or splenomegaly; liver dysfunction; and negative tests for antinuclear antibody and rheumatoid factor. The negative autoantibody screen is a defining and frequently tested feature. [1]
Ferritin — the biomarker with a caveat: [1]
A markedly elevated ferritin, often above 1000 micrograms per litre and sometimes above 10,000, is characteristic of AOSD and reflects macrophage activation. The fraction of glycosylated ferritin is low (below 20 per cent), which increases specificity. The caveat — ferritin is also markedly elevated in infection, malignancy and haemophagocytic lymphohistiocytosis, so a high ferritin supports AOSD only after those are excluded. Ferritin above 10,000 with pancytopenia should trigger consideration of macrophage activation syndrome. [1]
Management: [1]
- NSAIDs and glucocorticoids for induction. Mild disease may respond to NSAIDs alone; most patients need prednisolone 0.5 to 1 mg/kg daily, tapered with a steroid-sparing agent.
- Interleukin-1 blockade (anakinra) early. Anakinra produces a rapid response in the systemic phenotype of AOSD and has a diagnostic-therapeutic overlap — a brisk response to anakinra supports the diagnosis. Early IL-1 blockade reduces cumulative steroid exposure.
- Interleukin-6 blockade (tocilizumab) for the articular or chronic phenotype, or when IL-1 blockade fails. Tocilizumab is particularly effective for the arthritis and for sustained remission.
- Macrophage activation syndrome is a feared complication (see below). [1]
Schnitzler syndrome
Schnitzler syndrome is an acquired autoinflammatory disease of older adults characterised by the triad of chronic urticarial rash, monoclonal IgM gammopathy and recurrent fever, driven by interleukin-1 beta overactivity. It is rare but increasingly recognised, and the diagnosis is rewarding because IL-1 blockade produces a dramatic response. [1]
The Strasbourg diagnostic criteria are the examinable framework [6]. The diagnosis requires both obligate criteria — chronic urticarial rash and a monoclonal IgM (or occasionally IgG) gammopathy — plus at least two minor criteria (for IgM) or three minor criteria (for IgG). The minor criteria are recurrent fever; objective findings of abnormal bone remodelling (bone pain or abnormal bone imaging); elevated CRP or leukocytosis; and a neutrophilic urticarial dermatosis on skin biopsy.
Clinical features: [1]
- Chronic urticarial rash — persistent or recurrent, present for years; the rash is a neutrophilic urticarial dermatosis on biopsy, distinguishing it from ordinary mast-cell-driven urticaria.
- Monoclonal IgM gammopathy on serum electrophoresis — the defining laboratory feature.
- Recurrent fever, bone pain and elevated inflammatory markers. [1]
Management: [1]
- Interleukin-1 blockade (anakinra or canakinumab) produces a dramatic and sustained response — the rash, fever and bone pain resolve rapidly. This IL-1 dependence is the therapeutic signature of Schnitzler syndrome [7].
- Haematology surveillance is mandatory. Schnitzler syndrome carries a long-term risk of lymphoproliferative malignancy — Waldenstrom macroglobulinaemia or other lymphoma — of approximately 15 to 20 per cent over years. The paraprotein requires annual haematology review, even though the autoinflammatory symptoms respond to IL-1 blockade.
Examiner trap: Do not treat Schnitzler syndrome with chemotherapy for the paraprotein. The rash and fever respond to anakinra; the paraprotein requires haematology surveillance, not immediate treatment, unless it progresses to overt lymphoma. [1]
Behcet disease (cross-reference the vasculitis topic)
Behcet disease straddles the autoinflammatory and vasculitic classifications and is covered in detail in the systemic vasculitides topic. The features relevant here are its autoinflammatory mechanism and its management framework. [1]
Behcet disease presents with recurrent oral aphthous ulceration (mandatory in the international criteria), genital ulceration, uveitis and pathergy. It has a striking geographic distribution along the ancient Silk Road (Turkey, the Mediterranean, the Middle East, Japan, Korea) and is strongly associated with HLA-B51. [1]
The management is severity-driven. Colchicine treats the mucocutaneous disease and arthritis. Azathioprine, ciclosporin or TNF inhibitors (infliximab, adalimumab) treat ocular and organ-threatening involvement. The 2018 EULAR update on Behcet syndrome is the current European framework [8]. The feared emergencies are pulmonary artery aneurysm rupture (massive haemoptysis) and cerebral venous sinus thrombosis.
The diagnostic approach

When you see a patient with recurrent unexplained fever or systemic inflammation, follow a structured approach. The goal is to answer four questions: Is this infection, malignancy or autoimmune disease? What is the attack pattern? What is the gene? What is the dominant cytokine? [1]
Step 1 — Exclude the common and dangerous mimics first
This is non-negotiable. Autoinflammatory disease is rare; infection, malignancy and autoimmune disease are common. [1]
| Mimic category | Examples | Key tests |
|---|---|---|
| Infection | Endocarditis, occult abscess, tuberculosis, malaria | Serial blood cultures, echocardiography, tuberculosis screening, travel history |
| Malignancy | Lymphoma, leukaemia | Lactate dehydrogenase, peripheral blood film, CT imaging, bone marrow biopsy if indicated |
| Autoimmune disease | SLE, systemic vasculitis, systemic-onset JIA | ANA, anti-dsDNA, ANCA, complement, cryoglobulins, biopsy |
| Haematological | Haemophagocytic lymphohistiocytosis | Ferritin, triglycerides, fibrinogen, soluble IL-2 receptor, bone marrow |
Step 2 — Characterise the attack pattern
The attack diary is the single most useful diagnostic tool. Ask the patient (or their parents) to record the date, duration, accompanying features and any trigger for every attack. From the pattern, identify the likely syndrome: [1]
| Pattern feature | Syndrome |
|---|---|
| Attacks 1 to 3 days, serositis, Mediterranean ancestry | FMF |
| Attacks 3 to 7 days, cervical lymphadenopathy, post-vaccination | HIDS/MKD |
| Attacks 1 to 3 weeks, periorbital oedema, migratory rash | TRAPS |
| Cold-induced urticaria and fever | FCAS (CAPS) |
| Chronic urticaria, hearing loss, amyloidosis | Muckle-Wells (CAPS) |
| Chronic meningitis, arthropathy, papilloedema in an infant | NOMID/CINCA (CAPS) |
| Spiking fever, salmon-pink rash, ferritin above 1000, negative ANA and RF | AOSD |
| Chronic urticaria, monoclonal IgM, fever in an older adult | Schnitzler |
Step 3 — Confirm with the targeted genetic panel
A targeted next-generation sequencing panel covering MEFV, TNFRSF1A, MVK, NLRP3 and the broader autoinflammatory genes is now the standard confirmatory test for the hereditary recurrent fevers and CAPS. Order it through clinical immunology or clinical genetics with appropriate counselling. [1]
Two cautions. First, a variant of uncertain significance is a common result and must be interpreted in the clinical context — treat the patient, not the genotype. Second, a negative panel does not exclude a hereditary recurrent fever, because novel genes, incomplete penetrance and somatic mosaicism (especially in NLRP3 in CAPS) occur. [1]
Step 4 — Syndrome-specific confirmatory tests
- Urinary mevalonic acid during an attack for HIDS/MKD.
- Serum protein electrophoresis and serum free light chains in any adult with chronic urticaria and fever (Schnitzler).
- Ferritin and glycosylated ferritin for AOSD.
- Audiometry and brain MRI for the CAPS spectrum.
- Skin biopsy of the urticarial rash for CAPS and Schnitzler — neutrophilic urticarial dermatosis.
- Serum amyloid A and urinalysis for proteinuria for all hereditary recurrent fevers at baseline and follow-up (AA amyloidosis surveillance). [1]
Treatment principles — interleukin-1 blockade and beyond

The unifying therapeutic principle of the autoinflammatory diseases is treat by the dominant cytokine. Five cytokine-targeting strategies cover the topic. [1]
1. Colchicine for FMF. Colchicine prevents attacks and prevents AA amyloidosis. The goal is complete attack prevention with lifelong therapy. Dose: 1 to 2 mg daily, titrated to tolerance and response [2].
2. Interleukin-1 blockade for the IL-1-driven syndromes. Three agents are available. Canakinumab (anti-IL-1 beta monoclonal antibody, 150 mg subcutaneously every 8 weeks) is approved for CAPS, colchicine-resistant FMF, HIDS, TRAPS and Schnitzler syndrome, with the CLUSTER trial underpinning approval in the hereditary recurrent fevers [3][4]. Anakinra (recombinant IL-1 receptor antagonist, 100 mg subcutaneously daily) is the preferred first IL-1 blocker in AOSD and Schnitzler and as a diagnostic-therapeutic probe. Rilonacept (an IL-1 trap) is an alternative for CAPS.
3. Interleukin-6 blockade for AOSD. Tocilizumab (8 mg/kg intravenously every 4 weeks, or 162 mg subcutaneously weekly) is effective in AOSD, particularly for the articular phenotype or when IL-1 blockade fails. [1]
4. TNF blockade. Etanercept for TRAPS; infliximab or adalimumab for refractory Behcet uveitis and severe organ involvement. [1]
5. Glucocorticoids. Used for AOSD induction (with NSAIDs), as short courses for TRAPS attacks, and for organ-threatening Behcet disease. They are not the backbone of hereditary recurrent fever management, where chronic steroid exposure causes cumulative harm. [1]
The practical decision at the bedside: for CAPS, HIDS, TRAPS, colchicine-resistant FMF, AOSD and Schnitzler, reach for IL-1 blockade early. For FMF, it is colchicine. For articular AOSD, consider IL-6. [1]
Complications and long-term outcomes
AA amyloidosis is the principal long-term complication of inadequately treated FMF (and of TRAPS and CAPS). It presents as proteinuria and progresses to nephrotic syndrome and renal failure. Colchicine prevents it; this is why complete attack prevention, not attack reduction, is the treatment goal. Screen with urinalysis and serum amyloid A at every follow-up. [1]
Macrophage activation syndrome (MAS) is a life-threatening complication of AOSD (and of systemic-onset JIA). It presents with persistent high fever, hepatosplenomegaly, pancytopenia, a markedly elevated ferritin (often above 10,000 micrograms per litre), hypertriglyceridaemia, hypofibrinogenaemia and elevated soluble interleukin-2 receptor. Treatment is high-dose glucocorticoids plus anakinra; etoposide is used in refractory HLH. MAS must be recognised early because it is rapidly fatal. [1]
Progressive sensorineural hearing loss in Muckle-Wells is irreversible once established — canakinumab halts further loss but does not restore hearing. This is the argument for early treatment. [1]
Schnitzler syndrome and lymphoproliferative malignancy — the risk of Waldenstrom macroglobulinaemia or lymphoma is approximately 15 to 20 per cent over years. Annual haematology surveillance with paraprotein monitoring is part of follow-up. [1]
Chronic steroid toxicity in AOSD and Behcet — osteoporosis, diabetes, infection, cataracts — drives the shift to early cytokine-directed therapy. [1]
Pulmonary artery aneurysm in Behcet causes massive haemoptysis and is life-threatening — high-dose steroids, cyclophosphamide, TNF inhibitors and embolisation. [1]
DCE exam preparation
Long case: undiagnosed familial Mediterranean fever with AA amyloidosis
A classic DCE long case is a young adult of Mediterranean ancestry with a 10-year history of recurrent abdominal pain and fever, a previous appendicectomy scar, now presenting with proteinuria and rising creatinine — undiagnosed FMF complicated by AA amyloidosis. The candidate must deliver a structured opening statement, a prioritised problem list, an integrated management plan, and show insight into the patient perspective. [1]
Model opening statement (SASPOP format): [1]
"This is Mr Yusuf Demir, a 28-year-old carpenter of Turkish ancestry presenting with a 10-year history of recurrent one-to-two-day attacks of fever and severe abdominal pain, a previous negative appendicectomy at age 20, and new-onset proteinuria on urinalysis with a serum creatinine of 130. His problem list is: (1) familial Mediterranean fever, almost certainly undiagnosed for a decade, now complicated by (2) AA amyloidosis with proteinuria and early chronic kidney disease; (3) a colchicine initiation and titration plan aimed at complete attack prevention and amyloidosis arrest; (4) genetic confirmation with MEFV testing and cascade family testing; (5) the psychosocial impact of a delayed diagnosis, a preventable complication, and the implications for his siblings and future children." [1]
Examiner probing questions and model answers: [1]
- What is your immediate management? — Confirm the diagnosis with MEFV genetic testing and a serum amyloid A level, start colchicine 1.5 mg daily, perform a renal biopsy to confirm AA amyloidosis, and screen siblings with a careful history, urinalysis and genetic counselling.
- How do you prevent further amyloidosis? — Complete attack prevention with the maximum tolerated colchicine dose, monitored by serum amyloid A and urinalysis. Canakinumab is the rescue therapy for colchicine-resistant disease.
- Why was the diagnosis missed? — FMF attacks mimic an acute abdomen; the episodic nature and full inter-attack wellness, plus low awareness, drive diagnostic delay. The average delay is several years. [1]
Short case: a chronic urticarial rash
A DCE short case may present a patient with a chronic urticarial rash and ask for a focused examination, differential and discussion. [1]
Examination routine: [1]
- Inspect the skin — describe the rash: chronic urticarial lesions, distribution (trunk and limbs), whether they leave bruising, whether they are cold-induced, and whether there is scarring.
- Examine for syndrome-specific signs — hearing aids or cochlear implants (Muckle-Wells), joint deformity (NOMID), hepatosplenomegaly (AOSD, MAS), lymphadenopathy (HIDS), periorbital oedema (TRAPS).
- Examine for complications — pitting oedema and hypertension (AA amyloid nephropathy), digital clubbing and papilloedema (NOMID).
- Take a targeted history — age of onset, family history, cold triggering, hearing loss, paraprotein on prior bloods. [1]
Presentation template: [1]
"This patient has a chronic urticarial rash present since childhood, with bilateral hearing aids indicating sensorineural hearing loss. The combination of chronic urticaria and progressive sensorineural hearing loss is characteristic of Muckle-Wells syndrome, a cryopyrin-associated periodic syndrome caused by a gain-of-function mutation in NLRP3. I would confirm with NLRP3 genetic testing, a skin biopsy to demonstrate neutrophilic urticarial dermatosis, and baseline urinalysis and serum amyloid A to screen for AA amyloidosis. The treatment is canakinumab 150 mg subcutaneously every 8 weeks, which halts further hearing loss and prevents amyloidosis." [1]
Key references summary
Livneh 1997 Tel Hashomer diagnostic criteria for familial Mediterranean fever (Arthritis Rheum, 9336425) [1]; Ozen 2016 EULAR recommendations for the management of familial Mediterranean fever (Ann Rheum Dis, 26802180) [2]; Ost 2020 CLUSTER trial of canakinumab in colchicine-resistant FMF, HIDS/MKD and TRAPS (Ann Rheum Dis, 32571870) [3]; Lachmann 2009 in vivo regulation of interleukin-1 beta in cryopyrin-associated periodic syndromes (J Exp Med, 19364880) [4]; Yamaguchi 1992 preliminary classification criteria for adult Still disease (J Rheumatol, 1578458) [5]; Simon 2013 Strasbourg diagnostic criteria for Schnitzler syndrome (Allergy, 23480774) [6]; de Koning 2007 Schnitzler syndrome review (Semin Arthritis Rheum, 17586002) [7]; Hatemi 2018 update of the EULAR recommendations for the management of Behcet syndrome (Ann Rheum Dis, 29625968) [8]. Local ANZ guidance: Australian Rheumatology Association and AHPA pathways for genetic testing and biological therapy access.
References
- [1]Livneh A, Langevitz P, Zemer D, et al. Criteria for the diagnosis of familial Mediterranean fever Arthritis Rheum, 1997.PMID 9336425
- [2]Ozen S, Demirkaya E, Erer B, et al. EULAR recommendations for the management of familial Mediterranean fever Ann Rheum Dis, 2016.PMID 26802180
- [3]Ost D, Laskari K, Simon A, et al. Long-term efficacy and safety of canakinumab in patients with colchicine-resistant familial Mediterranean fever: results from the randomised phase III CLUSTER trial Ann Rheum Dis, 2020.PMID 32571870
- [4]Lachmann HJ, Lowe P, Felix SD, et al. In vivo regulation of interleukin 1beta in patients with cryopyrin-associated periodic syndromes J Exp Med, 2009.PMID 19364880
- [5]Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still's disease J Rheumatol, 1992.PMID 1578458
- [6]Simon A, Asli B, Braun-Falco M, et al. Schnitzler's syndrome: diagnosis, treatment, and follow-up Allergy, 2013.PMID 23480774
- [7]de Koning HD, Bodar EJ, Simon A, et al. Schnitzler syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment Semin Arthritis Rheum, 2007.PMID 17586002
- [8]Hatemi G, Christensen R, Bang D, et al. 2018 update of the EULAR recommendations for the management of Behçet's syndrome Ann Rheum Dis, 2018.PMID 29625968