Phys · rheumatological
Systemic Sclerosis (Scleroderma)
Also known as scleroderma · systemic sclerosis · SSc · limited cutaneous systemic sclerosis · CREST syndrome · diffuse cutaneous systemic sclerosis · progressive systemic sclerosis
Consultant-physician-depth guide to systemic sclerosis for FRACP DWE and DCE — the three-hit pathophysiology, ACR/EULAR 2013 classification, limited versus diffuse cutaneous subtypes and their autoantibodies (anti-centromere, anti-Scl-70/topoisomerase I, anti-RNA polymerase III), multisystem organ involvement (Raynaud and nailfold capillaroscopy, oesophageal and small-bowel disease, NSIP-pattern interstitial lung disease, pulmonary arterial hypertension, scleroderma renal crisis), organ-based surveillance and management, the ACE-inhibitor first principle in renal crisis, mycophenolate and nintedanib for lung disease, and autologous stem cell transplant for severe early diffuse disease.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Systemic Sclerosis (Scleroderma)
The answer first
Systemic sclerosis (SSc, scleroderma) is a chronic multisystem autoimmune disease defined by three things happening together: microvascular injury, immune activation with autoantibody production, and fibroblast dysregulation causing excessive collagen deposition — the three-hit hypothesis. Those three processes explain every clinical feature, from Raynaud phenomenon and digital ulcers through to interstitial lung disease (ILD), pulmonary arterial hypertension (PAH), gut dysmotility and scleroderma renal crisis. [1]
The single most important classification at the bedside is limited versus diffuse cutaneous disease, because it predicts the timing and type of internal-organ involvement: [1]
- Limited cutaneous SSc (formerly CREST) — skin thickening confined to distal to the elbows and knees and the face; anti-centromere antibody; longstanding Raynaud; a later risk of pulmonary arterial hypertension, calcinosis and telangiectasia.
- Diffuse cutaneous SSc — skin thickening proximal to the elbows or knees or on the trunk; anti-Scl-70 (anti-topoisomerase I) or anti-RNA polymerase III antibody; earlier and more severe internal-organ disease including ILD, cardiac disease and scleroderma renal crisis [1].
Three rules that change outcome: [1]
- Scleroderma renal crisis is an emergency in which an ACE inhibitor saves lives and kidneys. New-onset hypertension with AKI (and often microangiopathic haemolytic anaemia) in early diffuse disease demands captopril immediately, up-titrated against blood pressure, even if the blood pressure is normal and even as the creatinine rises [4]. Prednisolone at 15 mg per day or more is a recognised trigger — avoid it [3].
- Dyspnoea in systemic sclerosis demands a same-visit lung and heart assessment. ILD and PAH are the leading disease-specific causes of death and are often clinically silent until advanced. Screen with PFTs and HRCT for ILD, and with annual transthoracic echo (and confirm by right heart catheterisation) for PAH.
- There is no disease-modifying cure. Management is organ-based: immunosuppression (mycophenolate, cyclophosphamide) and antifibrotic therapy (nintedanib) for ILD [5][6]; vasodilator therapy for Raynaud and digital ulcers; PAH-specific therapy for pulmonary hypertension; and ACE inhibitors for renal crisis. Autologous haematopoietic stem cell transplant is considered for selected patients with severe early diffuse disease [8][9].
Survival has improved markedly over the past three decades, driven by ACE inhibitors for renal crisis, modern PAH therapy, ILD screening and immunosuppression. Five-year survival is now around 80 to 85 per cent overall, but it varies widely by subtype, antibody and organ involvement — which is why structured surveillance, not episodic care, defines good management. [1]
Classification — limited versus diffuse cutaneous disease

The 2013 ACR/EULAR classification criteria are the examinable standard [1]. They were developed because the older 1980 ACR criteria missed early and limited disease. Two rules:
- Skin thickening proximal to the metacarpophalangeal joints of both hands is, on its own, sufficient to classify systemic sclerosis. No further scoring is required.
- If that sufficient criterion is absent, use the weighted additive score. A total of at least 9 out of 10 classifies definite SSc. [1]
| Domain | Item | Points |
|---|---|---|
| Skin thickening | Proximal to MCP joints (both hands) | Sufficient alone (9) |
| Fingertip lesions | Digital tip ulcers (2) or pitting scars (3) | 2 or 3 |
| Finger skin thickening | Puffy fingers (2) or sclerodactyly of fingers (proximal to MCP but distal to PIP) (4) | 2 or 4 |
| Telangiectasia | Present | 2 |
| Abnormal nailfold capillaries | Dilated, giant loops or dropout | 2 |
| PAH and/or ILD | Present (maximum 2) | 2 |
| Raynaud phenomenon | Present | 3 |
| SSc-related autoantibody | Anti-centromere, anti-topoisomerase I (anti-Scl-70) or anti-RNA polymerase III | 3 |
Once SSc is classified, assign the cutaneous subtype, because it drives the surveillance strategy and prognostic conversation. [1]
| Feature | Limited cutaneous SSc | Diffuse cutaneous SSc |
|---|---|---|
| Skin distribution | Distal to elbows and knees; face | Proximal to elbows or knees, or trunk; face |
| Onset of skin disease | Slow, often years of Raynaud first | Rapid, often within a year of Raynaud |
| Raynaud | Precedes skin disease by years | Near-concurrent with skin disease |
| Typical autoantibody | Anti-centromere | Anti-Scl-70 (topoisomerase I) or anti-RNA polymerase III |
| Peak organ risk | Late pulmonary arterial hypertension, calcinosis, telangiectasia, biliary disease | Early ILD, scleroderma renal crisis, cardiac disease |
| Prognosis | Better overall, but PAH is the killer | Worse early mortality, then plateau as skin softens |
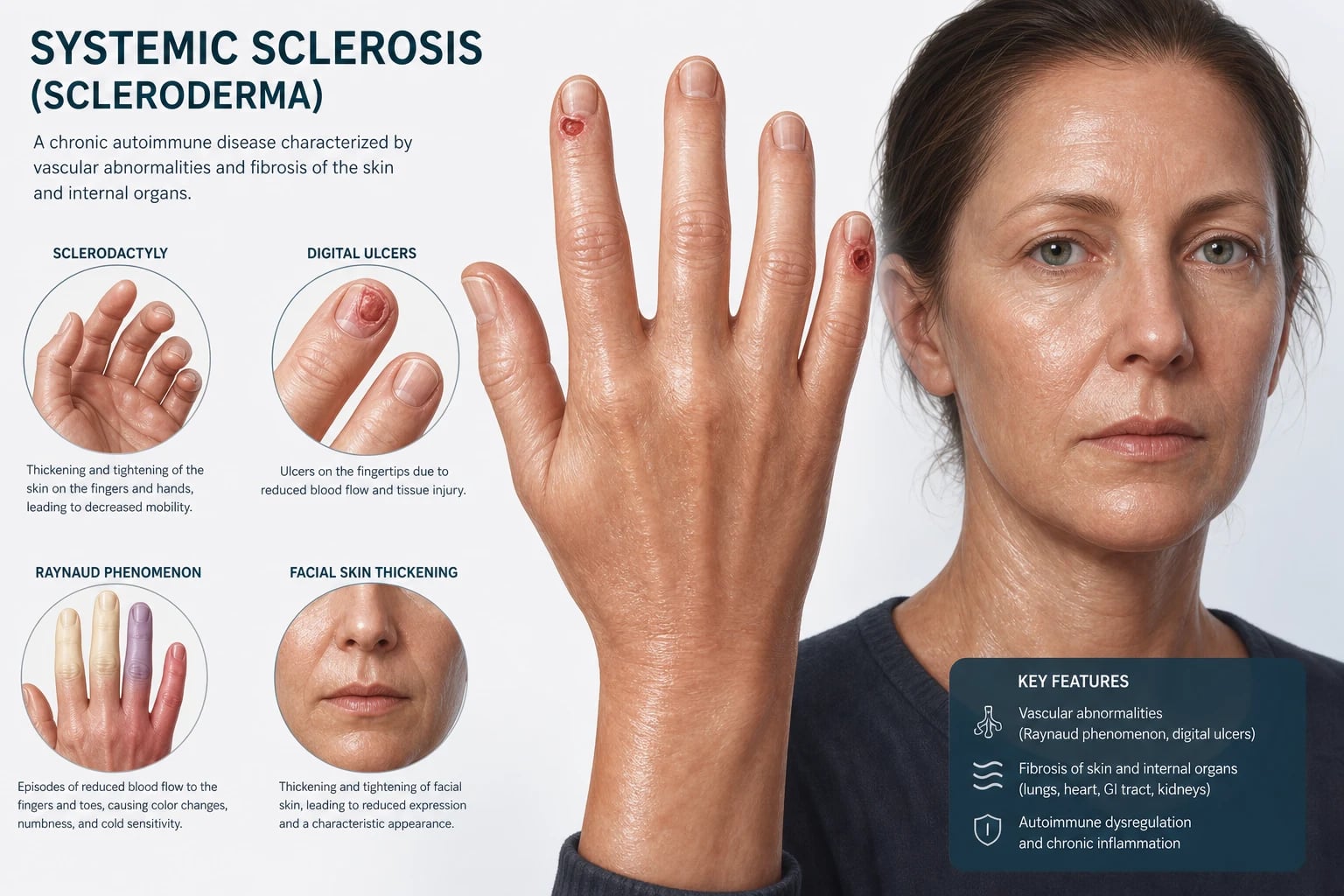
The CREST acronym (limited cutaneous SSc)
CREST is the mnemonic for the five features that cluster in limited cutaneous disease. It remains a useful teaching handle, but modern practice classifies by the skin extent and the antibody rather than by the acronym alone. [1]
- C — Calcinosis cutis — subcutaneous calcium deposits, typically over the fingertips, extensor surfaces and around joints; painful, can ulcerate and discharge.
- R — Raynaud phenomenon — virtually universal in limited disease; may precede other features by decades.
- E — Esophageal dysmotility — smooth-muscle atrophy of the lower two-thirds produces absent peristalsis, reflux, strictures and dysphagia.
- S — Sclerodactyly — skin thickening confined to the digits distal to the MCP joints.
- T — Telangiectasia — mat-like vascular lesions on the face, hands, lips and oral mucosa; a clue to the vasculopathic phenotype. [1]
DWE high-yield: Do not say "the patient has CREST therefore has limited disease." A patient with diffuse cutaneous skin involvement can still have calcinosis, Raynaud, oesophageal dysmotility and telangiectasia. The limited-versus-diffuse call is made on skin distribution, not on the presence of CREST features. [1]
Examiner trap: Anti-centromere and anti-Scl-70 are mutually exclusive in most patients. If both come back positive, suspect a lab error or an overlap syndrome. Anti-RNA polymerase III is the third major antibody and the one most candidates forget — it identifies a group with rapid diffuse skin disease, a high renal-crisis risk and a close temporal cancer association. [1]
Pathophysiology — the three-hit hypothesis
Systemic sclerosis is not primarily a skin disease. It is a vasculopathy with secondary fibrosis and immune dysregulation, and understanding the sequence unlocks every management decision. [1]
Hit 1 — microvascular injury
The earliest pathological event is repeated endothelial injury to small arteries and capillaries, the trigger for which is incompletely understood (genetic susceptibility, environmental exposures such as silica and organic solvents, viral triggers and immune complex deposition have all been implicated). Endothelial cell damage produces: [1]
- Vasospasm — the basis of Raynaud phenomenon, where cold or stress triggers an exaggerated digital vasoconstriction producing the classic white (ischaemic), blue (cyanotic) then red (reperfusion) colour sequence.
- Intimal proliferation and structural narrowing — chronic injury drives smooth-muscle proliferation and luminal narrowing, the basis of fixed (rather than reversible) digital ischaemia, PAH and renal crisis.
- Capillary loss — visible on nailfold capillaroscopy as enlarged, tortuous giant loops, microhaemorrhages and ultimately capillary dropout producing avascular areas. The capillaroscopy pattern is disease-defining in early SSc and is one of the 2013 criteria items. [1]
The vascular hypothesis matters clinically because it explains why vasodilator therapy (calcium channel blockers, PDE5 inhibitors, prostacyclin) is the backbone of Raynaud and digital ulcer management, and why endothelin receptor antagonists and prostacyclin analogues are foundational for PAH. [1]
Hit 2 — immune activation and autoantibodies
In a genetically susceptible host, the vascular injury triggers an autoimmune response characterised by CD4-positive T-cell activation, a type I interferon signature and the production of disease-specific antinuclear antibodies. The three major SSc-specific antibodies are mutually exclusive, define the clinical subtype and carry prognostic weight: [1]
| Antibody | Target antigen | Subtype | Clinical associations |
|---|---|---|---|
| Anti-centromere | Centromere proteins (CENP-A, CENP-B) | Limited cutaneous | Late PAH, calcinosis, telangiectasia; lower ILD and renal-crisis risk |
| Anti-Scl-70 (anti-topoisomerase I) | DNA topoisomerase I | Diffuse cutaneous | ILD (the strongest association), more severe skin and lung disease |
| Anti-RNA polymerase III | RNA polymerase III | Diffuse cutaneous | Rapid severe skin disease, high renal-crisis risk, and a close temporal association with cancer |
The anti-RNA polymerase III and cancer link is a high-yield exam point and a genuine clinical concern. In patients with this antibody, cancer is often diagnosed within 12 to 36 months of SSc onset, suggesting a paraneoplastic trigger in which the tumour drives the autoimmunity [10][11]. A new diagnosis of anti-RNA polymerase III-positive systemic sclerosis warrants age-appropriate cancer screening plus a low threshold for targeted investigation.
Hit 3 — fibroblast activation and collagen deposition
The third and final hit is the dysregulation of fibroblasts, which differentiate into myofibroblasts and overproduce type I and type III collagen, fibronectin and other extracellular matrix proteins. Transforming growth factor beta (TGF-beta) is the master signalling driver, amplified by connective tissue growth factor (CTGF), platelet-derived growth factor and endothelin-1. The result is progressive fibrosis of: [1]
- the dermis — the hardened, tethered skin and the digital contractures;
- the lung interstitium — the NSIP-pattern fibrosis;
- the gut wall — the dysmotility, bacterial overgrowth and malabsorption;
- the myocardium — patchy fibrosis, conduction disease and diastolic dysfunction;
- and the perivascular adventitia — which narrows the vessel lumen further and compounds the primary vasculopathy. [1]
The fibrotic hypothesis matters clinically because it explains why nintedanib (an antifibrotic that inhibits tyrosine kinases including the TGF-beta pathway) slows FVC decline in SSc-ILD [6], and why immunosuppression alone cannot reverse established fibrosis — only prevent its progression if started early.
Clinical features — the multisystem phenotype

Systemic sclerosis involves virtually every organ. The clinical skill is to anticipate the organ at risk based on subtype and antibody, screen for it proactively, and treat early. [1]
Skin and the vascular tree
- Raynaud phenomenon is virtually universal (>90 per cent) and often precedes other features by years. It may be the only feature of pre-SSc (Raynaud plus capillaroscopy change plus SSc-specific antibody, before skin or organ disease).
- Puffy fingers are the earliest skin sign, evolving over months into sclerodactyly (thickened, tethered digital skin). In diffuse disease this extends proximally — the modified Rodnan skin score (mRSS) at 17 body sites quantifies skin thickness and is a prognostic and trial endpoint.
- Digital pitting scars and fingertip ulcers reflect critical ischaemia; they cause severe pain, risk osteomyelitis and gangrene, and predict internal vascular disease.
- Calcinosis cutis is subcutaneous calcium deposition, typically over the fingertips and extensor surfaces; it ulcerates, discharges chalky material and is exquisitely painful.
- Telangiectasia are mat-like macular vascular lesions on the face, hands, lips and oral mucosa — a clue to the vasculopathic phenotype and the most visible cosmetic burden for many patients.
- Microstomia and a pinched (beak-like) nose reflect perioral and facial skin tightening. [1]
Gastrointestinal tract — involved in nearly every patient
GI involvement is present in most patients and is a major source of morbidity: [1]
- Oesophageal dysmotility — smooth-muscle atrophy of the lower two-thirds of the oesophagus produces absent peristalsis and a hypotensive lower oesophageal sphincter. The result is severe gastro-oesophageal reflux (with strictures, Barrett oesophagus and an adenocarcinoma risk), dysphagia and chest pain. A high-dose proton pump inhibitor is first-line; prokinetics and alginate are adjuncts.
- Gastroparesis and gastric antral vascular ectasia (GAVE, watermelon stomach) — delayed gastric emptying causes early satiety, nausea and bloating; GAVE causes occult iron-deficiency anaemia from chronic oozing and is treated endoscopically with argon plasma coagulation.
- Small bowel dysmotility and bacterial overgrowth — stasis drives small intestinal bacterial overgrowth (SIBO), producing bloating, diarrhoea, steatorrhoea and weight loss. Cyclic rotating antibiotics (for example rifaximin, metronidazole, ciprofloxacin) are the mainstay.
- Colonic dysmotility and pseudo-obstruction — constipation, megacolon and, rarely, colonic pseudo-obstruction; anorectal involvement causes faecal incontinence. [1]
Lung — the leading cause of death
Pulmonary involvement is now the leading overall cause of scleroderma-related death, ahead of renal crisis since the ACE inhibitor era. [1]
- Interstitial lung disease (SSc-ILD) occurs in about 40 per cent, more often in diffuse disease and with anti-Scl-70. The most common histological pattern is non-specific interstitial pneumonia (NSIP) (reticulation and ground-glass, often with traction bronchiectasis, basal and subpleural; the NSIP pattern is more fibrotic than cellular in advanced disease). Usual interstitial pneumonia (UIP) is less common. Presenting features are exertional dyspnoea and a dry cough; examination reveals fine basal crackles. Screen at baseline and then annually with PFTs (FVC, DLCO) and HRCT. An FVC below 70 per cent predicted, or a decline of 10 per cent or more, signals significant disease.
- Pulmonary arterial hypertension (SSc-PAH) occurs in 10 to 15 per cent, more often in limited disease and with anti-centromere antibody. It reflects isolated pulmonary vasculopathy, often with minimal or no ILD. Presenting features are exertional dyspnoea, fatigue, syncope or pre-syncope, and signs of right heart strain (loud P2, right ventricular heave, elevated JVP, peripheral oedema). Screen annually with transthoracic echo (estimating pulmonary artery systolic pressure and assessing right ventricular function); confirm by right heart catheterisation (mean pulmonary artery pressure at or above 25 mmHg, wedge pressure at or below 15 mmHg, pulmonary vascular resistance above 3 Wood units). The DLCO is disproportionately low for the FVC — a DLCO under about 55 per cent predicted with a preserved FVC is a clue to isolated vasculopathy. [1]
Cardiac involvement
- Myocardial fibrosis produces patchy fibrosis, diastolic dysfunction, systolic impairment (in advanced disease) and conduction disease (bundle branch block, heart block, ventricular arrhythmia). Cardiac MRI with late gadolinium enhancement is the most sensitive modality.
- Pericardial disease — pericardial effusion is common and may be haemodynamically significant; a large effusion in early diffuse disease is a red flag for renal crisis.
- Coronary vasospasm — microvascular disease can produce chest pain and troponin elevation with normal epicardial arteries. [1]
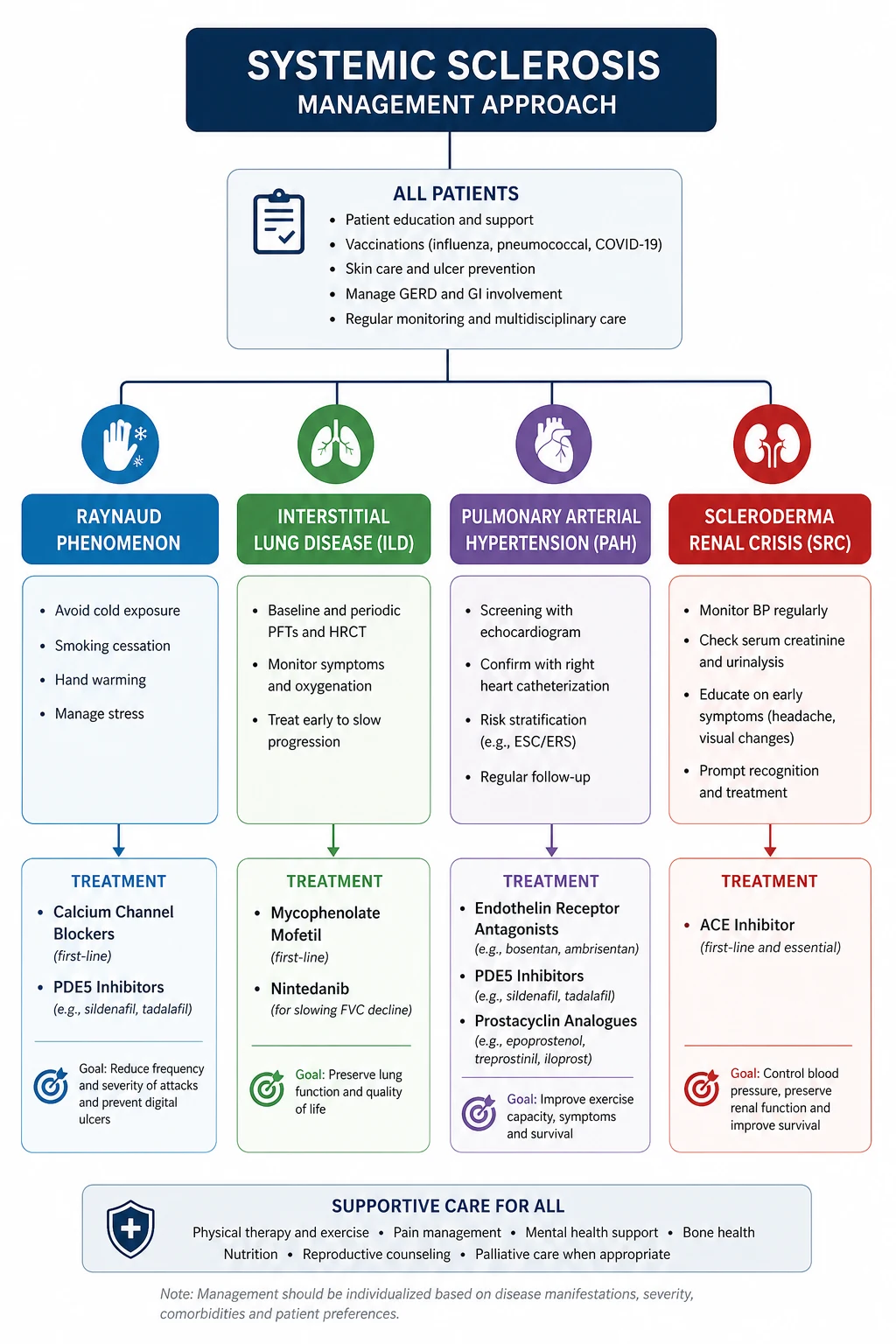
Renal — scleroderma renal crisis
Scleroderma renal crisis (SRC) is the classical emergency. It occurs in 5 to 10 per cent overall, almost exclusively in early diffuse cutaneous disease (within the first four years), and is strongly associated with anti-RNA polymerase III positivity and with prednisolone at or above 15 mg per day [3]. The pathophysiology is proliferative intimal arteriolopathy of the renal vasculature — the same vascular lesion as elsewhere, concentrated in the kidney — which drives renin-angiotensin-aldosterone system activation and a malignant-hypertension-like syndrome.
The clinical triad is: [1]
- New-onset or accelerating hypertension (though about 10 per cent are normotensive at presentation — a trap).
- Acute kidney injury with a rapidly rising creatinine.
- Microangiopathic haemolytic anaemia — schistocytes on the blood film, a low platelet count, elevated LDH and low haptoglobin; the picture mimics thrombotic thrombocytopenic purpura but the ADAMTS13 is normal. [1]
The non-negotiable rule: Start an ACE inhibitor immediately — captopril 6.25 to 25 mg orally, up-titrated every 6 to 12 hours against the blood pressure. Continue and increase the ACE inhibitor even if the creatinine rises and even if the patient becomes dialysis-dependent. The transient rise in creatinine reflects the necessary blood-pressure reduction and the recovery of the surviving nephrons; stopping the ACE inhibitor is the single most common avoidable error. ACE inhibitors transformed 1-year survival in SRC from under 20 per cent to about 76 per cent [4]. Up to half of patients need dialysis acutely, but a substantial proportion recover renal function and come off dialysis over weeks to months — so continue the ACE inhibitor through dialysis.
Less common but examinable features
- Sicca symptoms (dry eyes, dry mouth) from fibrosis of the minor salivary glands.
- Thyroid disease — autoimmune hypothyroidism and, less commonly, sclerosing thyroiditis.
- Joint contractures, tendon friction rubs (a specific sign of diffuse disease) and muscle weakness from disuse, myopathy or overlap myositis.
- Erectile dysfunction often precedes the diagnosis and reflects penile vascular disease.
- Peripheral and autonomic neuropathy.
- Osteoporosis — from malabsorption, immobility and glucocorticoid exposure. [1]
Investigations — confirm, subtype, screen
The workup has three goals: confirm the diagnosis with the 2013 ACR/EULAR criteria, subtype the disease with the antibody and skin extent, and define the organ burden with a structured screening bundle. [1]
Confirm the diagnosis
- Clinical examination — the skin distribution, the Raynaud history and the nailfold capillaroscopy.
- Nailfold capillaroscopy — a defining investigation. The early pattern shows dilated capillary loops; the active pattern adds giant loops and microhaemorrhages; the late pattern shows dropout and avascular areas. A normal capillaroscopy in a patient with apparent Raynaud argues strongly against SSc.
- 2013 ACR/EULAR scoring — as above [1].
Subtype the disease
- Full autoantibody panel — anti-centromere, anti-Scl-70 (topoisomerase I) and anti-RNA polymerase III. Also check anti-U1 RNP if an overlap syndrome is suspected, and routine ANA (a nucleolar or centromere pattern is a clue). The antibody result drives the surveillance strategy and prognostic discussion. [1]
Define the organ burden
| Organ | Investigation | Frequency |
|---|---|---|
| Skin | Modified Rodnan skin score (clinician) and skin photographs | Each visit |
| Lung (ILD) | Spirometry, lung volumes, DLCO; HRCT chest | Baseline, then annually (or sooner if symptomatic) |
| PAH | Transthoracic echo; right heart catheterisation if echo abnormal or symptoms | Baseline and annually thereafter |
| Renal | Blood pressure, serum creatinine, eGFR, urinalysis | Each visit; more frequently in the first 4 years of diffuse disease |
| Cardiac | ECG; cardiac MRI if symptoms or echo abnormal; troponin and NT-proBNP | Baseline and as clinically indicated |
| GI | Upper endoscopy, oesophageal manometry (selective), hydrogen or glucose breath test for SIBO, faecal calprotectin and elastase | Symptom-triggered |
| Haematology | Full blood count, blood film (schistocytes), LDH, haptoglobin if SRC suspected | Symptom-triggered |
| Malignancy | Age-appropriate screening plus targeted investigation if anti-RNA polymerase III positive | At diagnosis and per protocol |
The screening discipline: The biggest single error in SSc care is the failure to screen. ILD and PAH are silent until advanced; renal crisis develops over days; the only protection is a structured surveillance programme in a scleroderma clinic, with PFTs and echo at least annually for life, and renal monitoring monthly during the high-risk first four years of diffuse disease. [1]
Management — organ-based, lifelong
There is no disease-modifying cure for systemic sclerosis. The EULAR treatment framework is organ-based [2]: match the therapy to the active organ problem, monitor for response and toxicity, and treat early.
Scleroderma renal crisis — an emergency
- Start captopril 6.25 to 25 mg orally, up-titrating every 6 to 12 hours against blood pressure to a target of below 140/90 (lower if tolerated), as soon as SRC is suspected [4].
- Continue the ACE inhibitor even as the creatinine rises and even if dialysis is needed; up to half of patients who need dialysis recover renal function over weeks to months.
- Add renal replacement therapy (haemodialysis or peritoneal dialysis) for uraemia, fluid overload or hyperkalaemia; some centres use ACE-inhibitor continuation through dialysis to maximise renal recovery.
- Avoid intravenous or high-dose glucocorticoids (the trigger) and avoid ACE-inhibitor withdrawal.
- An angiotensin receptor blocker is a second-line substitute if an ACE inhibitor is not tolerated, but it is not equivalent in efficacy.
Raynaud phenomenon and digital ulcers
- First-line: a dihydropyridine calcium channel blocker — amlodipine 5 to 20 mg daily or nifedipine modified-release 30 to 60 mg daily. Titrate to the highest tolerated dose; warn about headache, flushing and ankle oedema. [1]- Second-line / combination: a phosphodiesterase type 5 inhibitor — sildenafil 25 to 50 mg three times daily or tadalafil. These improve both Raynaud and digital ulcer healing.
- Severe or critical digital ischaemia: intravenous iloprost (a prostacyclin analogue) over 3 to 5 days, in hospital. This is the therapy that saves threatened digits.
- Refractory digital ulcers: bosentan (an endothelin receptor antagonist) reduces the number of new digital ulcers (RAPIDS-2), though it does not heal existing ulcers [7]. Surgical digital sympathectomy or amputation for gangrene. Wound care, antibiotics for infection, and analgesia are essential adjuncts.
- Lifestyle: smoking cessation (non-negotiable), cold avoidance, hand warmers, and avoidance of beta-blockers (which worsen Raynaud) and sympathomimetic decongestants.
Gastrointestinal disease
- Reflux and oesophagitis: a high-dose proton pump inhibitor (for example pantoprazole 40 mg twice daily), continued long term; add a prokinetic (metoclopramide or domperidone) for delayed emptying; alginate and nocturnal dose timing; elevate the head of the bed. [1]- Dysphagia: exclude stricture and malignancy with endoscopy; consider dilation for strictures.
- SIBO and malabsorption: cyclic rotating antibiotics (rifaximin, metronidazole, ciprofloxacin), a lactose- and low-FODMAP-restricted trial, pancreatic enzyme supplementation if exocrine insufficiency is present, and nutritional support (oral supplements, enteral feeding via a PEG/J tube if needed).
- GAVE: endoscopic argon plasma coagulation; iron and transfusion support. [1]
Interstitial lung disease
- First-line immunosuppression: mycophenolate mofetil 2 to 3 g daily for at least two years. The Scleroderma Lung Study II established equivalence to oral cyclophosphamide with a substantially better safety profile, making mycophenolate the standard [5].
- Alternative / severe disease: intravenous or oral cyclophosphamide for rapidly progressive or severe inflammatory disease; rituximab in expert centres for refractory disease.
- Antifibrotic therapy: nintedanib 150 mg twice daily slows the annual rate of FVC decline in SSc-ILD (SENSCIS) [6]. It is added to (not replacing) immunosuppression for progressive fibrosing disease. Monitor LFTs (transaminitis) and for diarrhoea.
- Supportive: pneumococcal, influenza, COVID-19 and recombinant zoster vaccination before immunosuppression; oxygen for hypoxaemic respiratory failure; pulmonary rehabilitation; and lung transplant assessment for end-stage disease (SSc is a well-accepted transplant indication with outcomes comparable to idiopathic disease).
Pulmonary arterial hypertension
PAH in systemic sclerosis is managed in a specialist pulmonary hypertension centre following current ESC/ERS guidance. The strategy is initial combination therapy for most patients at diagnosis: [1]
- An endothelin receptor antagonist (bosentan, macitentan, ambrisentan) plus a phosphodiesterase type 5 inhibitor (sildenafil, tadalafil) as first-line combination.
- A soluble guanylate cyclase stimulator (riociguat — avoided if on a PDE5 inhibitor) is an alternative.
- A prostacyclin pathway agent (oral selexipag, inhaled or subcutaneous treprostinil, intravenous epoprostenol) is added for high-risk or escalating disease.
- Diuretics for right heart failure, oxygen for hypoxaemia, and anticoagulation only for selected patients. Pregnancy is contraindicated. [1]
Skin disease
- Methotrexate (10 to 25 mg weekly) for early inflammatory diffuse skin disease with evidence of activity (oedematous skin, tendon friction rubs). [1]- Immunosuppression with mycophenolate or cyclophosphamide also softens the skin as part of the ILD regimen.
- Intravenous immunoglobulin for overlap myositis and refractory skin disease.
- Physiotherapy and occupational therapy for contractures and hand function; surgical release for severe contractures. [1]
Autologous haematopoietic stem cell transplant
For carefully selected patients with severe early diffuse cutaneous disease (typically under 60, within 2 years of onset, with significant skin and/or lung involvement and no irreversible organ failure), autologous haematopoietic stem cell transplant is considered. Two landmark trials define the evidence: [1]
- ASTIS (van Laar, JAMA 2014) — HSCT versus monthly cyclophosphamide; long-term event-free and overall survival favoured transplant, at the cost of higher treatment-related mortality in the first year [8].
- SCOT (Sullivan, NEJM 2018) — myeloablative CD34-selected HSCT versus cyclophosphamide; superior global rank composite outcome and long-term survival favouring transplant, again with significant early toxicity [9].
The decision is a high-stakes shared one: transplant can halt or reverse the disease in responders, but carries a real treatment-related mortality (originally around 10 per cent, lower in modern non-myeloablative protocols and experienced centres). It is performed only in specialist centres after exhaustive counselling. [1]
Complications and prognosis
The leading causes of death in systemic sclerosis are pulmonary (ILD and PAH), followed by cardiac disease, GI complications and renal crisis. Cancer is an additional cause in anti-RNA polymerase III-positive patients. Overall five-year survival is around 80 to 85 per cent, but it varies by subtype, antibody and organ involvement: [1]
- Limited cutaneous SSc with anti-centromere — better long-term skin prognosis, but the late risk of PAH defines the trajectory; annual PAH screening is non-negotiable.
- Diffuse cutaneous SSc with anti-Scl-70 — the highest early ILD risk; PFT and HRCT surveillance drives outcome.
- Diffuse cutaneous SSc with anti-RNA polymerase III — the highest renal-crisis risk; close blood-pressure and creatinine monitoring in the first four years, with an immediate-response plan.
- Scleroderma renal crisis — with prompt ACE inhibitor therapy, 1-year survival is about 76 per cent; up to half need dialysis acutely but many recover renal function [4].
- Scleroderma with PAH — without PAH-specific therapy, median survival is under 3 years; with modern combination therapy, 3-year survival is now above 70 per cent.
Non-fatal morbidity is substantial and often under-recognised: chronic pain from calcinosis and ulcers, malnutrition from GI disease, body-image distress from the visible skin change, anxiety and depression, sexual dysfunction, and loss of employment. A multidisciplinary clinic — rheumatology, respiratory, cardiology, nephrology, gastroenterology, physiotherapy, occupational therapy, dietetics, psychology and specialist nursing — is the standard of care. [1]
How this is examined
DWE MCQ format
Expect clinical vignette stems testing: subtype identification from skin distribution and antibody; the ACE-inhibitor-first rule in renal crisis; the anti-RNA polymerase III and cancer association; the choice of mycophenolate (not cyclophosphamide) as first-line ILD therapy; the role of nintedanib; and the avoidance of high-dose prednisolone. The distractors are usually a plausible-but-wrong agent (an ARB or a calcium channel blocker for renal crisis), a plausible-but-wrong antibody pairing, or a plausible-but-wrong screening frequency. [1]
DCE long case format
A classic long case is a woman in her 50s or 60s with longstanding Raynaud, anti-centromere-positive limited cutaneous SSc, presenting with exertional dyspnoea from PAH (with or without co-existing ILD), reflux, and digital ulcers. The candidate must deliver the opening statement (SASPOP), the structured problem list, the integrated organ-based plan, and the probing-question defence (see the viva artifact). The examiner will probe the PAH pathway, the ILD screening, the vasodilator strategy, the renal-crisis contingency plan, and the pregnancy/contraception and psychosocial issues. [1]
DCE short case format
The hand is the single most common rheumatology short-case station. Examine the hands systematically — skin, digits, pulp, nailfold, joints, function — then the face (telangiectasia, microstomia), the chest (PAH, ILD), the abdomen and for telangiectasia elsewhere. Present the signs, offer systemic sclerosis as the unifying diagnosis, and request nailfold capillaroscopy, an autoantibody panel, and an organ-screening bundle. The discussion flows from signs to subtype, antibody, organ screening and management (see the case artifact). [1]
Common exam traps
- Confusing limited and diffuse subtypes on the skin distribution.
- Treating a rising creatinine in renal crisis by stopping the ACE inhibitor.
- Prescribing prednisolone above 15 mg per day in early diffuse disease. [1]- Missing pulmonary arterial hypertension because the patient "only" has dyspnoea.
- Calling the nailfold capillaroscopy non-specific.
- Forgetting the anti-RNA polymerase III cancer association.
- Using a beta-blocker for Raynaud (it worsens it).
- Pairing anti-centromere with anti-Scl-70 in one patient (they are mutually exclusive). [1]
Regional and guideline notes
- EULAR recommendations for the treatment of systemic sclerosis (Kowal-Bielecka, Ann Rheum Dis 2017, updated 2023) [2] — the primary European treatment framework, with organ-specific guidance on ILD, PAH, skin, GI, Raynaud, digital ulcers and renal crisis.
- ACR/EULAR 2013 classification criteria (van den Hoogen, Arthritis Rheumatol 2013) [1] — the examinable diagnostic standard.
- Australian Rheumatology Association and Scleroderma Australia — local guidance on screening, surveillance and access to subsidised therapies (including bosentan for digital ulcers and nintedanib for SSc-ILD under the PBS).
- British Society for Rheumatology and NHS England specialised commissioning for SSc-ILD and PAH pathways define the UK surveillance structure.
- ATS/ERS/JRS/ALAT progressive fibrosing ILD and IPF guidance inform nintedanib use; ESC/ERS pulmonary hypertension guidelines define PAH classification, screening and combination therapy.
- Drug doses verified against EULAR 2017 and the cited primary trials; where regional practice differs (for example captopril versus other ACE inhibitors in SRC), the ANZ preference for captopril (short half-life, rapid titration) is primary, with long-acting ACE inhibitors an acceptable alternative.
Key references
2013 ACR/EULAR classification criteria for systemic sclerosis (van den Hoogen, Arthritis Rheum 2013) [1]; EULAR recommendations for the treatment of systemic sclerosis, 2017 update (Kowal-Bielecka, Ann Rheum Dis 2017) [2]; prednisolone and scleroderma renal crisis risk (Steen and Medsger, Arthritis Rheum 1998) [3]; ACE inhibitors and outcome in scleroderma renal crisis (Steen, Ann Intern Med 1990) [4]; Scleroderma Lung Study II — mycophenolate versus cyclophosphamide (Tashkin, Lancet Respir Med 2016) [5]; SENSCIS — nintedanib for SSc-ILD (Distler, NEJM 2019) [6]; RAPIDS-2 — bosentan for prevention of new digital ulcers (Korn, Arthritis Rheum 2004) [7]; ASTIS — autologous HSCT versus cyclophosphamide (van Laar, JAMA 2014) [8]; SCOT — myeloablative HSCT for severe scleroderma (Sullivan, NEJM 2018) [9]; anti-RNA polymerase III and cancer in scleroderma (Shah, Arthritis Rheum 2010; Arthritis Rheumatol 2015) [10][11]. EULAR 2023 update of the treatment recommendations; Scleroderma Australia and Australian Rheumatology Association resources.
References
- [1]van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative Arthritis Rheum, 2013.PMID 24122180
- [2]Kowal-Bielecka O, Fransen J, Avouac J, et al. Update of EULAR recommendations for the treatment of systemic sclerosis Ann Rheum Dis, 2017.PMID 27941129
- [3]Steen VD, Medsger TA Jr. Long-term outcomes of scleroderma renal crisis Ann Intern Med, 2000.PMID 11033587
- [4]Steen VD, Costantino JP, Shapiro AP, Medsger TA Jr. Outcome of renal crisis in systemic sclerosis: relation to availability of angiotensin converting enzyme (ACE) inhibitors Ann Intern Med, 1990.PMID 2382917
- [5]Tashkin DP, Roth MD, Clements PJ, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial Lancet Respir Med, 2016.PMID 27469583
- [6]Distler O, Highland KB, Gahlemann M, et al. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease N Engl J Med, 2019.PMID 31112379
- [7]Korn JH, Mayes M, Matucci Cerinic M, et al. Digital ulcers in systemic sclerosis: prevention by treatment with bosentan, an oral endothelin receptor antagonist Arthritis Rheum, 2004.PMID 15593188
- [8]van Laar JM, Farge D, Sont JK, et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial JAMA, 2014.PMID 25058083
- [9]Sullivan KM, Goldmuntz EA, Keyes-Elstein L, et al. Myeloablative Autologous Stem-Cell Transplantation for Severe Scleroderma N Engl J Med, 2018.PMID 29298160
- [10]Shah AA, Rosen A, Hummers LK, Wigley F, Casciola-Rosen L. Close temporal relationship between onset of cancer and scleroderma in patients with RNA polymerase I/III antibodies Arthritis Rheum, 2010.PMID 20506513
- [11]Shah AA, Casciola-Rosen L, Rosen A. Cancer and systemic sclerosis: novel insights into pathogenesis and clinical implications Curr Opin Rheumatol, 2011.PMID 21825998