Phys · rheumatological
The Systemic Vasculitides
Also known as systemic vasculitis · vasculitis · ANCA-associated vasculitis · giant cell arteritis · temporal arteritis · polymyalgia rheumatica · Takayasu arteritis · polyarteritis nodosa · granulomatosis with polyangiitis · Wegener granulomatosis · microscopic polyangiitis · eosinophilic granulomatosis with polyangiitis · Churg-Strauss syndrome · IgA vasculitis · Henoch-Schonlein purpura · cryoglobulinaemic vasculitis · Behcet disease
Consultant-physician-depth guide to the systemic vasculitides for FRACP DWE and DCE — the Chapel Hill 2012 vessel-size classification, large vessel disease (giant cell arteritis and polymyalgia rheumatica with urgent steroids and tocilizumab, Takayasu arteritis in young Asian women), medium vessel disease (polyarteritis nodosa with microaneurysms, HBV association and mononeuritis multiplex, Kawasaki disease), ANCA-associated small vessel vasculitis (granulomatosis with polyangiitis with c-ANCA/PR3 and ENT-lung-kidney disease, microscopic polyangiitis with p-ANCA/MPO and renal-pulmonary disease, eosinophilic granulomatosis with polyangiitis with asthma and eosinophilia), immune complex small vessel vasculitis (cryoglobulinaemic vasculitis with HCV and low complement, IgA vasculitis in children with palpable purpura and glomerulonephritis), variable vessel vasculitis (Behcet disease), the diagnostic approach using biopsy, angiography, ANCA pattern and complement, and the treatment framework of steroids first-line, cyclophosphamide or rituximab for organ-threatening disease, plasma exchange for anti-GBM overlap and severe RPGN, and maintenance therapy with azathioprine, mycophenolate or methotrexate.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
The Systemic Vasculitides
The answer first
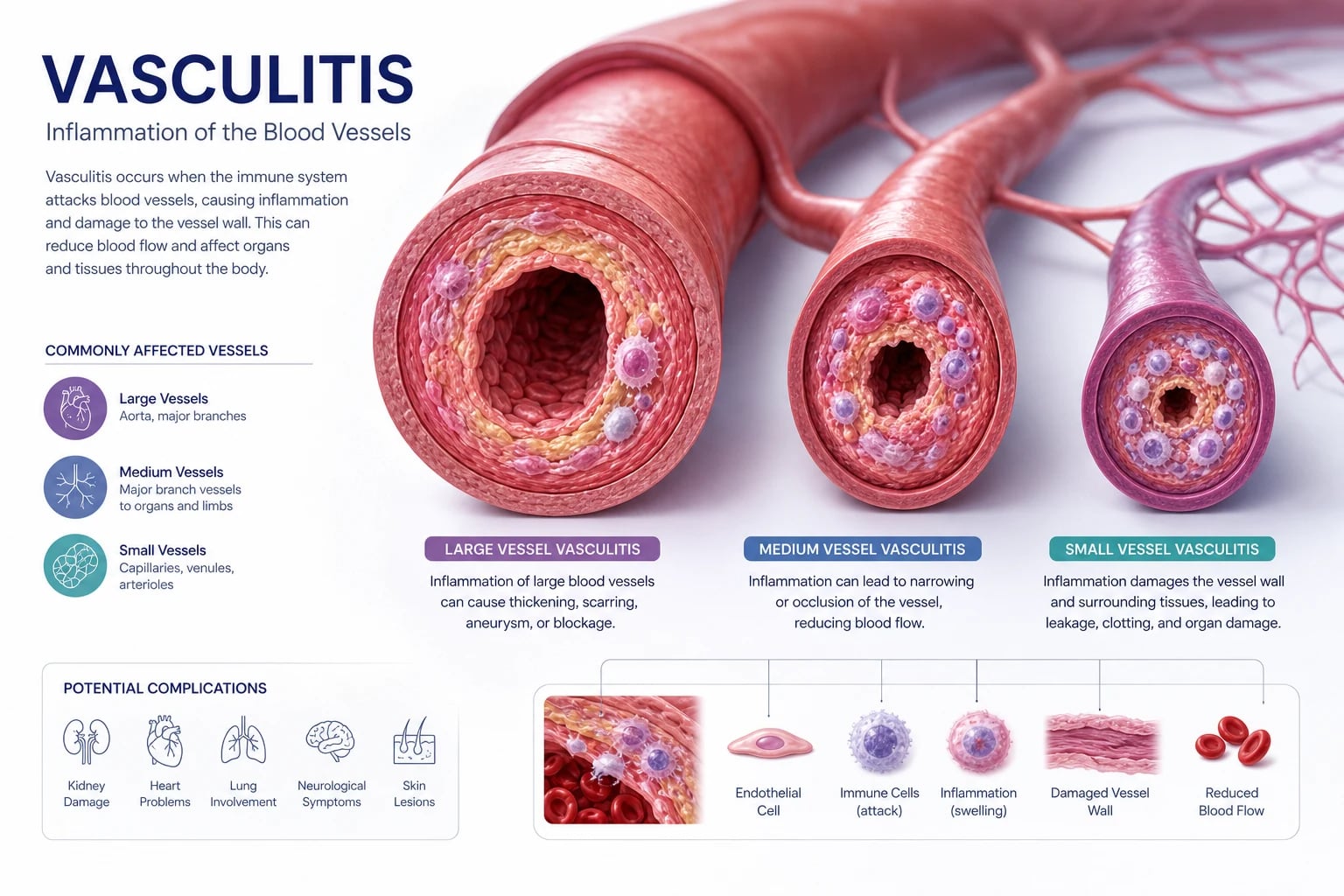
The systemic vasculitides are a group of inflammatory diseases of blood vessel walls classified by the predominant size of vessel involved. That single organising principle — large, medium or small — is how you must approach every vasculitis question, because vessel size predicts the clinical syndrome, the organ at risk, the investigation strategy and the treatment [1].
The 2012 revised International Chapel Hill Consensus Conference (CHCC) nomenclature is the examinable framework [1]. It renamed the eponyms: Wegener granulomatosis became granulomatosis with polyangiitis (GPA), Churg-Strauss syndrome became eosinophilic granulomatosis with polyangiitis (EGPA), and Henoch-Schonlein purpura became IgA vasculitis. The eponyms persist in clinical practice and exams, so learn both names.
Three rules that change outcome: [1]
- Giant cell arteritis with visual symptoms is an emergency. Start high-dose glucocorticoids immediately — do not wait for biopsy or a rheumatology review. Anterior ischaemic optic neuropathy causes irreversible blindness within hours. Tocilizumab is the evidence-based steroid-sparing agent for relapsing or refractory disease [2].
- A pulmonary-renal syndrome is a medical emergency. Haemoptysis with rapidly progressive glomerulonephritis is GPA or microscopic polyangiitis (MPA) until proven otherwise. Start high-dose glucocorticoids plus rituximab or cyclophosphamide within hours. Plasma exchange is reserved for anti-GBM overlap or dialysis-dependent disease [3][5][6].
- Rituximab has replaced cyclophosphamide as first-line induction for most ANCA-associated vasculitis. The RAVE and RITUXVAS trials established rituximab as non-inferior to cyclophosphamide for remission induction, with the advantage of being preferred in relapsing disease and in patients wishing to preserve fertility [3][4].
The clinical reasoning moves in three steps at the bedside. First, recognise the syndrome — is this a large-vessel problem (headache, jaw claudication, absent pulses), a medium-vessel problem (mononeuritis multiplex, mesenteric ischaemia, nodules), or a small-vessel problem (palpable purpura, glomerulonephritis, alveolar haemorrhage)? Second, confirm the vessel and the mechanism — biopsy the affected tissue, check ANCA pattern, complement levels, cryoglobulins and infection screen. Third, treat by severity — glucocorticoids for everything, but add rituximab or cyclophosphamide for organ- or life-threatening disease. [1]
Classification by vessel size
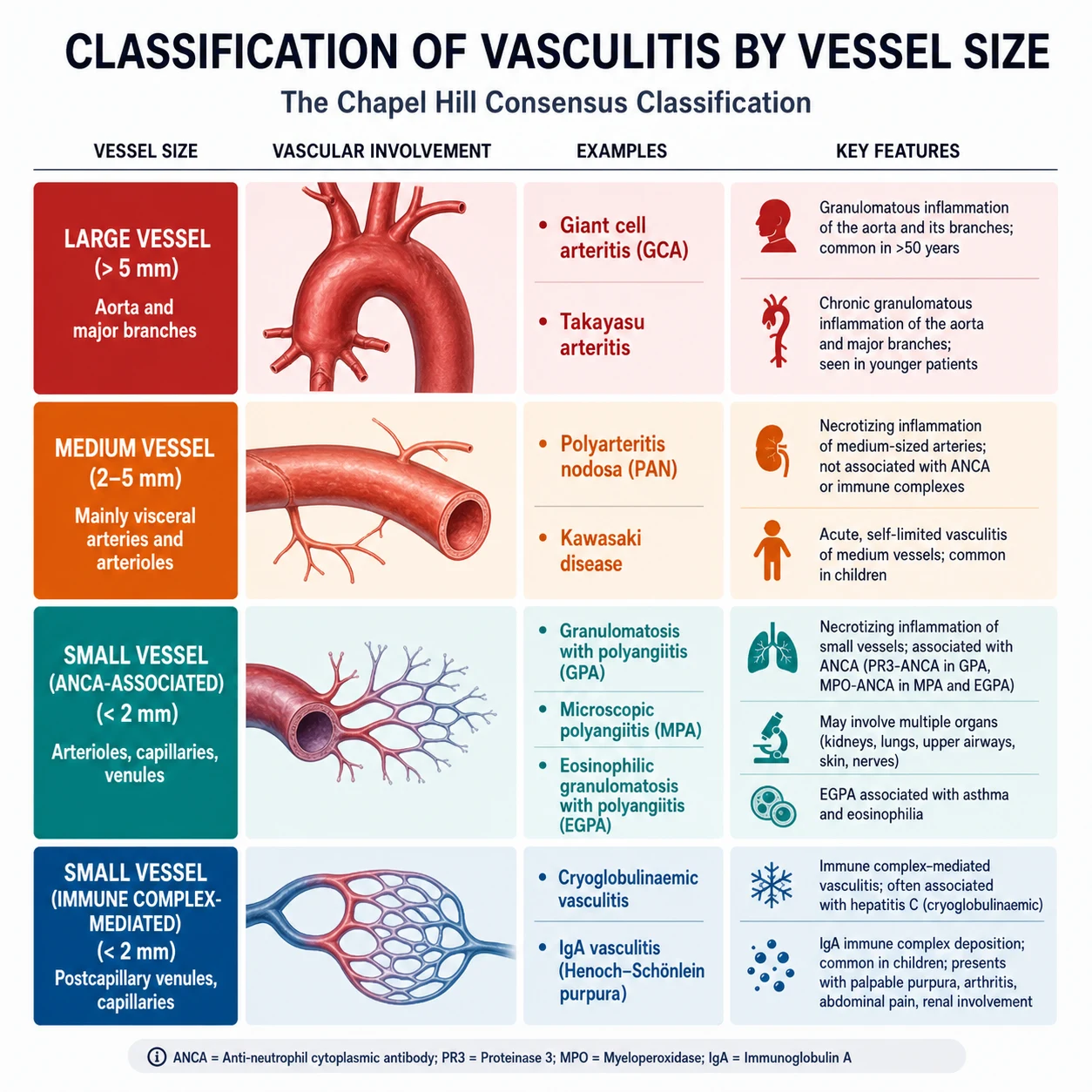
The CHCC 2012 nomenclature groups the vasculitides into seven categories [1]. The first three — large vessel, medium vessel, and small vessel — are the core examinable categories. The remaining four (variable vessel, single-organ, vasculitis secondary to another disease, and vasculitis of undetermined cause) capture the edges of the taxonomy.
The vessel-size framework at a glance
| Category | Predominant vessel | Key diseases | Hallmark clinical features |
|---|---|---|---|
| Large vessel | Aorta and major branches | Giant cell arteritis, Takayasu arteritis | Headache, jaw claudication, visual loss, absent pulses, bruits, limb claudication |
| Medium vessel | Main visceral arteries and their branches | Polyarteritis nodosa, Kawasaki disease | Microaneurysms, mesenteric ischaemia, mononeuritis multiplex, mucocutaneous lymph node syndrome |
| Small vessel — ANCA-associated | Arterioles, capillaries, venules (pauci-immune) | GPA, MPA, EGPA | Palpable purpura, rapidly progressive GN, alveolar haemorrhage, ENT disease |
| Small vessel — immune complex | Arterioles, capillaries, venules (immune deposits) | IgA vasculitis, cryoglobulinaemic, hypocomplementaemic urticarial | Palpable purpura, low complement, cryoglobulins, urticaria |
| Variable vessel | Any size, any location | Behcet disease, Cogan syndrome | Oral and genital ulcers, uveitis, pathergy, arterial aneurysms |
| Single-organ | Any size, one organ | Cutaneous leukocytoclastic angiitis, primary CNS angiitis | Isolated to one organ system |
DWE high-yield: The single most important discrimination at the bedside is the ANCA pattern and the complement level in suspected small-vessel vasculitis. A pauci-immune glomerulonephritis with c-ANCA/PR3 points to GPA; p-ANCA/MPO points to MPA or EGPA. Low complement points away from ANCA-associated disease and toward an immune-complex process (cryoglobulinaemic, IgA vasculitis, or lupus). [1]
Examiner trap: ANCA is not always positive in ANCA-associated vasculitis. Around 10 to 20 per cent of GPA and MPA cases are ANCA-negative, and EGPA is ANCA-positive in only about 40 per cent. A negative ANCA does not exclude a small-vessel vasculitis if the clinical and histological picture fits. Conversely, a positive ANCA does not prove vasculitis — it occurs in inflammatory bowel disease, infections, endocarditis and other conditions. [1]
Large vessel vasculitis
Large vessel vasculitis involves the aorta and its major branches. The two diseases are giant cell arteritis (in patients over 50, often with polymyalgia rheumatica) and Takayasu arteritis (in young patients, particularly Asian women). [1]
Giant cell arteritis and polymyalgia rheumatica
Giant cell arteritis (GCA, also called temporal arteritis) is a granulomatous vasculitis of the aorta and its branches, with a striking predilection for the extracranial branches of the carotid artery. It is the most common primary systemic vasculitis in adults. The cardinal clinical problem is threatened vision: anterior ischaemic optic neuropathy causes irreversible blindness that can be prevented only by immediate glucocorticoids. [1]
Clinical presentation — the features that must trigger action: [1]
- New-onset headache in a patient over 50 is the most common symptom. It is typically temporal, often severe, and different from previous headaches.
- Jaw claudication — pain in the masseter muscles on prolonged chewing that resolves with rest — is the single most specific symptom. Its presence should immediately raise GCA to the top of the differential.
- Visual symptoms — amaurosis fugax, diplopia, or sudden painless visual loss — are emergency features. Anterior ischaemic optic neuropathy is the mechanism, and the visual loss is often permanent within hours.
- Scalp tenderness — pain on combing hair or lying on a pillow — reflects temporal artery involvement.
- Constitutional features — fever, weight loss, fatigue, malaise — may be the only presentation, so GCA is a key cause of fever of unknown origin in an older patient.
- Polymyalgia rheumatica (PMR) coexists in about 40 to 50 per cent of patients with GCA. PMR causes severe morning stiffness and aching in the shoulder and pelvic girdles, without marked weakness. The ESR and CRP are almost always markedly elevated. [1]
Examination findings in GCA: [1]
| Site | Sign | Significance |
|---|---|---|
| Temporal artery | Thickened, tender, pulseless or nodular | Direct evidence of arteritis; palpate both temples |
| Upper limbs | Asymmetric blood pressure, bruits over subclavian or carotid | Large-vessel involvement in up to a third of patients |
| Eyes | Reduced acuity, relative afferent pupillary defect, disc swelling or pallor | Threatened or established anterior ischaemic optic neuropathy |
| Fundoscopy | Chalky white optic disc swelling (anterior ischaemic optic neuropathy) | Ophthalmic emergency |
Investigation — confirm but do not delay treatment: [1]
- Start glucocorticoids on suspicion, before biopsy. The 2022 ACR/EULAR classification criteria and all guidelines agree: do not wait for temporal artery biopsy to begin treatment. The biopsy should be done within two weeks of starting steroids, but remains useful for up to 2 to 4 weeks [8].
- Temporal artery biopsy is the gold standard. The finding is granulomatous inflammation with multinucleated giant cells and disruption of the internal elastic lamina. A unilateral biopsy has a sensitivity of about 77 per cent; a negative biopsy does not exclude GCA because of skip lesions.
- Temporal artery ultrasound showing the halo sign (hypoechogenic wall thickening from oedema) is an accepted diagnostic alternative; bilateral halos are highly specific [8].
- ESR and CRP are almost always elevated — typically ESR above 50 mm/hour. Remember that a normal ESR does not fully exclude GCA (about 4 to 15 per cent have a normal ESR, though CRP is usually still raised).
- Large-vessel imaging with CT angiography, MR angiography, or FDG-PET is increasingly used to detect extracranial large-vessel involvement (aortic inflammation, subclavian and axillary disease) that the temporal artery biopsy may miss.
The 2022 ACR/EULAR classification criteria for GCA use a scored system after first excluding mimics. Age 50 or older at diagnosis is mandatory, and a cumulative score of at least 5 classifies GCA [8]:
| Criterion | Points |
|---|---|
| Positive temporal artery biopsy OR temporal artery halo sign on ultrasound | +5 |
| ESR 50 mm/h or CRP 10 mg/L or higher; OR sudden visual loss | +3 |
| Jaw or tongue claudication | +2 |
| New temporal headache | +2 |
| Scalp tenderness | +2 |
| Temporal artery abnormality on vascular examination | +2 |
| Bilateral axillary involvement on imaging | +2 |
| FDG-PET activity throughout the aorta | +2 |
Management of GCA: [1]
- Immediate high-dose glucocorticoids. For GCA without visual symptoms: prednisolone 40 to 60 mg daily (not less than 40 mg, per EULAR guidance) [11]. The response is typically dramatic within 24 to 72 hours.
- For visual symptoms (threatened sight): give methylprednisolone 500 mg to 1 g IV daily for 3 days before transitioning to oral prednisolone. Refer to ophthalmology the same day.
- Taper slowly. The standard approach is to maintain 40 mg for 2 to 4 weeks, then taper by 10 per cent every 2 weeks or by 2.5 to 5 mg every 2 to 4 weeks, aiming for 10 to 15 mg by 3 months, then slower. Most patients need 1 to 2 years of treatment; relapse is common during taper.
- Tocilizumab (an IL-6 receptor blocker) is the evidence-based steroid-sparing agent. The GiACTA trial showed that subcutaneous tocilizumab (162 mg weekly or alternate-weekly, with a 26-week steroid taper) achieved sustained glucocorticoid-free remission in 53 to 56 per cent at 52 weeks versus 14 to 18 per cent with steroid taper alone [2]. Use it for relapsing disease, high steroid requirements, or difficulty tapering.
- Bone protection (calcium, vitamin D, bisphosphonate), gastric protection, and pneumocystis prophylaxis (co-trimoxazole) if on prolonged high-dose steroids.
- Aspirin is no longer routinely recommended for GCA by the 2018 EULAR update unless indicated for another reason [11].
Polymyalgia rheumatica is managed with lower-dose steroids than GCA: prednisolone 12.5 to 25 mg daily produces a dramatic response within days. The minimum effective dose is used and tapered over 1 to 2 years. If a patient with suspected PMR does not respond to 15 to 20 mg of prednisolone within a week, reconsider the diagnosis — late-onset rheumatoid arthritis, malignancy, infection and endocarditis are the key mimics. [1]
Takayasu arteritis
Takayasu arteritis (TAK) is a granulomatous large-vessel vasculitis of the aorta and its major branches. It affects young patients, particularly women of Asian descent, with a peak onset under 40 years — the mirror image of GCA in age and demographics. [1]
Clinical presentation: [1]
- Absent or unequal pulses (pulseless disease) and blood pressure discrepancy between arms are the hallmark. Always palpate pulses and measure blood pressure in both arms in a young patient with unexplained hypertension, claudication, or constitutional symptoms.
- Bruits over the subclavian, carotid, abdominal aorta or renal arteries.
- Limb claudication — arm claudication is particularly characteristic and unusual in atherosclerotic disease at a young age.
- Renovascular hypertension from renal artery stenosis.
- Neurological symptoms — dizziness, syncope, stroke, or transient ischaemic attacks from carotid or vertebral artery involvement.
- A prepulseless phase with fever, arthralgia, fatigue and weight loss may precede vascular signs by months. [1]
Diagnosis: [1]
- Vascular imaging is the cornerstone — digital subtraction angiography, CT angiography or MR angiography showing stenosis, occlusion or aneurysms of the aorta and its primary branches. The typical distribution is the aortic arch and its branches (type I), or the abdominal aorta and renal arteries (type III), or a combination (types II, IV, V).
- ESR and CRP are elevated but may be normal in chronic or burnt-out disease; they are unreliable markers of disease activity.
- FDG-PET can detect active vascular inflammation before structural changes appear on angiography. [1]
Management: [1]
- High-dose glucocorticoids (prednisolone 40 to 60 mg daily or 0.5 to 1 mg/kg) for induction. The response is generally good [11].
- Conventional immunosuppressants — methotrexate, azathioprine, mycophenolate or leflunomide — are used as steroid-sparing agents. TAK does not respond as predictably to tocilizumab as GCA does; evidence is emerging but weaker.
- Vascular intervention — angioplasty, stenting or bypass for critical stenoses, but only when disease is quiescent (to avoid restenosis), and ideally after inflammatory control.
- Revascularisation of renal artery stenosis for refractory renovascular hypertension.
DWE high-yield: The single most discriminating feature between GCA and Takayasu at the bedside is age. GCA is defined by onset at age 50 or older; Takayasu is overwhelmingly a disease of patients under 40. A young Asian woman with absent pulses, bruits and arm claudication is Takayasu until proven otherwise. [1]
Medium vessel vasculitis
Medium vessel vasculitis involves the main visceral arteries and their branches — the vessels between the arterioles and the aorta. The two diseases are polyarteritis nodosa and Kawasaki disease. [1]
Polyarteritis nodosa
Polyarteritis nodosa (PAN) is a necrotising vasculitis of medium-sized muscular arteries that causes microaneurysms, stenosis and thrombosis. It does not cause glomerulonephritis — this is a key distinguishing feature from small-vessel vasculitis. PAN does not involve the lung. [1]
Clinical presentation: [1]
- Constitutional symptoms — fever, weight loss, fatigue, myalgia — are near-universal and often precede organ-specific features by weeks.
- Mononeuritis multiplex is the classic neurological manifestation — simultaneous or sequential infarction of peripheral nerves producing foot drop, wrist drop, or sensory loss in distinct nerve territories. It reflects vasculitis of the vasa nervorum. This is a red-flag finding that demands urgent investigation.
- Abdominal pain from mesenteric vasculitis — post-prandial pain (intestinal angina), bowel ischaemia or perforation. This is a surgical emergency.
- Renal involvement causes renovascular hypertension and renal infarction, but not glomerulonephritis. (If there is glomerulonephritis, it is not PAN — reconsider MPA.)
- Skin — livedo reticularis, nodules, ulcers, digital gangrene.
- Testicular pain — testicular artery involvement is characteristic of PAN and a useful discriminating feature. [1]
The HBV association: [1]
A significant proportion of PAN cases are associated with hepatitis B virus infection. HBV-associated PAN is now much rarer in countries with universal HBV vaccination, but it remains an important exam point. The mechanism is immune-complex deposition in vessel walls. HBV-associated PAN is treated with antiviral therapy (entecavir or tenofovir) combined with plasmapheresis and a short course of steroids — a fundamentally different approach from idiopathic PAN, which requires prolonged immunosuppression. [1]
Investigation: [1]
- Mesenteric, renal or hepatic angiography showing microaneurysms and arterial beading is the characteristic finding and may be diagnostic. These represent focal areas of vessel wall weakening and dilatation.
- Biopsy of involved tissue (skin, nerve, muscle) showing transmural necrotising inflammation of medium-sized arteries confirms the diagnosis. In suspected vasculitic neuropathy, a sural nerve biopsy combined with a muscle biopsy has the highest yield.
- HBV, HCV, HIV serology — essential in all patients with PAN or suspected PAN.
- ANCA is negative in PAN. If ANCA is positive, reconsider small-vessel vasculitis.
- Complement levels are usually normal or elevated in PAN (it is not an immune-complex disease in the same way as cryoglobulinaemia). [1]
Management: [1]
- Idiopathic PAN — high-dose glucocorticoids (prednisolone 1 mg/kg daily) with cyclophosphamide for severe or multi-organ disease. Maintenance with azathioprine or methotrexate.
- HBV-associated PAN — antiviral therapy (entecavir or tenofovir), plasmapheresis (3 to 6 sessions over 2 weeks), and a short course of prednisolone tapered rapidly once antiviral therapy is established. The goal is viral clearance, which usually induces remission. [1]
Kawasaki disease
Kawasaki disease is a medium-vessel vasculitis of children (almost exclusively under 5 years). It causes mucocutaneous lymph node syndrome and, critically, coronary artery aneurysms in untreated cases (up to 25 per cent of untreated children). It is a leading cause of acquired heart disease in children in developed countries. [1]
Diagnostic criteria require fever for 5 or more days plus at least four of five principal features: bilateral non-purulent conjunctivitis; oral changes (strawberry tongue, cracked red lips, oropharyngeal erythema); polymorphous rash; peripheral extremity changes (acute erythema and oedema of palms and soles, later periungual desquamation); and cervical lymphadenopathy. [1]
Management is aspirin (high-dose then low-dose) and intravenous immunoglobulin (2 g/kg single infusion) within the first 10 days of fever to prevent coronary aneurysms. An echocardiogram is mandatory at diagnosis and follow-up. This is a paediatric topic but appears in physician exams because of the coronary artery sequelae and the principle that IVIG and aspirin prevent aneurysms. [1]
Examiner trap: PAN does not cause glomerulonephritis and does not involve the lung. If a patient has medium-vessel features with glomerulonephritis, think MPA. If they have medium-vessel features with pulmonary involvement, think GPA or MPA, not PAN. This single distinction — renal or lung involvement — rules PAN in or out. [1]
Small vessel vasculitis — ANCA-associated
The ANCA-associated vasculitides (AAV) are a group of necrotising small-vessel vasculitides characterised by pauci-immune (minimal immune complex deposition) glomerulonephritis and circulating anti-neutrophil cytoplasmic antibodies (ANCA). They are the most common cause of rapidly progressive (crescentic) glomerulonephritis and the pulmonary-renal syndrome in adults. [1]
The three diseases are granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA). [1]
Understanding ANCA
ANCA are autoantibodies against proteins in the cytoplasmic granules of neutrophils. Two patterns are detected by indirect immunofluorescence and confirmed by antigen-specific ELISA: [1]
| IIF pattern | Target antigen | Disease association |
|---|---|---|
| c-ANCA (cytoplasmic) | Proteinase 3 (PR3) | GPA (about 75 to 90 per cent PR3-ANCA positive) |
| p-ANCA (perinuclear) | Myeloperoxidase (MPO) | MPA (about 40 to 80 per cent MPO-ANCA positive), EGPA (about 40 per cent) |
The antibody specificity (PR3 versus MPO) is now considered more important than the IIF pattern, because it carries prognostic weight — PR3-ANCA disease relapses more frequently than MPO-ANCA disease. [1]
Always order ANCA by both methods — indirect immunofluorescence plus antigen-specific ELISA. The IIF pattern alone is not sufficient; p-ANCA can be positive in non-vasculitic conditions (inflammatory bowel disease, autoimmune hepatitis, infections, endocarditis), so a confirmatory anti-MPO or anti-PR3 ELISA is essential. [1]
Granulomatosis with polyangiitis (GPA, formerly Wegener)
GPA is a necrotising granulomatous vasculitis of the upper and lower respiratory tract with variable renal involvement. It is the most common AAV. [1]
Classic triad — ENT, lungs, kidneys: [1]
- ENT and upper respiratory tract — the most common and often first manifestation. Epistaxis, nasal crusting, bloody nasal discharge, saddle-nose deformity (collapse of the nasal bridge from cartilage destruction), subglottic stenosis (stridor), serous or purulent otitis media from Eustachian tube involvement, and conductive or sensorineural hearing loss. Oral ulcers are common.
- Lungs — cough, dyspnoea, haemoptysis. Imaging shows pulmonary nodules (often cavitating), infiltrates, and alveolar haemorrhage. Large-volume alveolar haemorrhage causes haemoptysis, a rising haemoglobin and bilateral infiltrates — a life-threatening manifestation.
- Kidneys — rapidly progressive (crescentic) glomerulonephritis with haematuria, proteinuria, red cell casts and a rising creatinine. Untreated, this progresses to end-stage renal disease within weeks. [1]
Other features: [1]
- Eye involvement — orbital pseudotumour (proptosis), scleritis, episcleritis, nasolacrimal duct obstruction.
- Skin — palpable purpura, ulcers, subcutaneous nodules.
- Nervous system — cranial neuropathies, mononeuritis multiplex, chronic meningitis.
- Constitutional — fever, weight loss, fatigue. [1]
Investigation: [1]
- c-ANCA with anti-PR3 positivity is present in about 75 to 90 per cent of GPA cases [9].
- Biopsy of involved tissue confirms the diagnosis. Lung or sinus biopsy shows necrotising granulomatous inflammation; renal biopsy shows pauci-immune necrotising and crescentic glomerulonephritis (little or no immune complex deposition on immunofluorescence).
- Chest CT to define the extent of pulmonary disease (nodules, cavitation, infiltrates).
- Urinalysis for blood and protein — always. Red cell casts suggest active glomerulonephritis.
The 2022 ACR/EULAR classification criteria for GPA use a points-based system (score at least 5), giving high weight to c-ANCA/PR3 positivity (5 points), bloody nasal discharge and crusting (3 points), and cartilaginous and pulmonary nodular features [9].
Microscopic polyangiitis (MPA)
MPA is a necrotising small-vessel vasculitis that causes renal and pulmonary disease without granulomatous inflammation of the upper respiratory tract. It is distinguished from GPA by the absence of granulomatous ENT disease and by the p-ANCA/MPO pattern. [1]
Key clinical features: [1]
- Rapidly progressive glomerulonephritis — the most common and often presenting feature. Renal involvement is more common at presentation in MPA than in GPA.
- Alveolar haemorrhage and pulmonary fibrosis — MPA is the AAV most associated with interstitial lung disease and pulmonary fibrosis, which can predate the vasculitic presentation by years.
- Skin — palpable purpura is the most common skin manifestation.
- Neurological — mononeuritis multiplex.
- Gastrointestinal — abdominal pain, bleeding.
- Constitutional — fever, weight loss, arthralgia. [1]
Investigation: [1]
- p-ANCA with anti-MPO positivity in 40 to 80 per cent [10].
- Renal biopsy shows pauci-immune necrotising crescentic GN — indistinguishable histologically from GPA.
- No granulomatous inflammation — this distinguishes MPA from GPA.
- The 2022 ACR/EULAR classification criteria for MPA (score at least 5) give high weight to p-ANCA/MPO positivity (6 points), pauci-immune GN (3 points) and lung fibrosis (3 points), with negative scores for features pointing to GPA [10].
Eosinophilic granulomatosis with polyangiitis (EGPA, formerly Churg-Strauss)
EGPA is the AAV with the most distinctive clinical profile. It combines late-onset asthma, peripheral and tissue eosinophilia, and systemic vasculitis. It is the rarest AAV. [1]
Three clinical phases (not always sequential): [1]
- Prodromal phase — allergic rhinitis, nasal polyposis and adult-onset asthma that may precede vasculitis by years.
- Eosinophilic phase — marked peripheral eosinophilia (often above 1.5 to 10 to the 9 per litre) and tissue eosinophilic infiltration, particularly of the lung (eosinophilic pneumonia) and gut (eosinophilic gastroenteritis).
- Vasculitic phase — systemic small-vessel vasculitis causing mononeuritis multiplex, palpable purpura, and (less commonly than in GPA and MPA) glomerulonephritis. [1]
Key discriminating features: [1]
- Asthma and eosinophilia are required for the diagnosis. If a patient with suspected AAV does not have asthma and eosinophilia, it is not EGPA.
- Cardiac involvement is the leading cause of death in EGPA — eosinophilic myocarditis, heart failure, arrhythmia, and pericarditis. An echocardiogram and troponin are essential at diagnosis.
- Mononeuritis multiplex is very common and often severe.
- ANCA is positive in only about 40 per cent of cases (usually p-ANCA/MPO). ANCA-positive EGPA tends to have more renal and neuropathic involvement; ANCA-negative EGPA tends to have more cardiac and eosinophilic disease. [1]
Management follows the AAV framework (below), but milder EGPA (no organ-threatening disease) may respond to glucocorticoids alone. Mepolizumab (an anti-IL-5 monoclonal antibody) is now an approved option for EGPA, reducing steroid exposure. [1]
The AAV clinical phenotypes compared
| Feature | GPA | MPA | EGPA |
|---|---|---|---|
| ENT and upper airway | Prominent (saddle nose, crusting, subglottic stenosis) | Absent or mild | Nasal polyposis, allergic rhinitis |
| Lung | Nodules, cavitation, alveolar haemorrhage | Alveolar haemorrhage, ILD, pulmonary fibrosis | Asthma, eosinophilic infiltrates |
| Kidney | Pauci-immune crescentic GN | Pauci-immune crescentic GN (often more severe) | Less common, usually milder |
| ANCA pattern | c-ANCA, PR3 (75 to 90 per cent) | p-ANCA, MPO (40 to 80 per cent) | p-ANCA, MPO (about 40 per cent) |
| Eosinophilia | Absent | Absent | Required for diagnosis (more than 1.5) |
| Cardiac | Rare | Rare | Common and life-threatening (myocarditis) |
| Key risk | ENT destruction, renal failure | Renal failure, pulmonary fibrosis | Cardiac death, severe neuropathy |
DWE high-yield: The single most important clinical discriminator between GPA and MPA is the presence or absence of granulomatous ENT disease. Nasal crusting, saddle-nose deformity, subglottic stenosis and recurrent epistaxis point to GPA. Their absence with p-ANCA/MPO and rapidly progressive GN points to MPA. EGPA is identified by the triad of asthma, eosinophilia and vasculitis. [1]
Small vessel vasculitis — immune complex
The immune complex small-vessel vasculitides are distinguished from the ANCA-associated group by prominent immune complex deposition on biopsy and, clinically, by low complement and cryoglobulins. The three key diseases are cryoglobulinaemic vasculitis, IgA vasculitis, and hypersensitivity (drug-induced) vasculitis. [1]
Cryoglobulinaemic vasculitis
Cryoglobulinaemic vasculitis is caused by circulating cryoglobulins — immunoglobulins that precipitate in the cold and dissolve on rewarming. The classic association is with hepatitis C virus (HCV) infection. About 80 to 90 per cent of mixed cryoglobulinaemia is associated with HCV. [1]
Types of cryoglobulinaemia: [1]
| Type | Composition | Association |
|---|---|---|
| Type I | Monoclonal Ig (usually IgM) | Haematological malignancy (multiple myeloma, Waldenstrom, CLL) |
| Type II (mixed) | Monoclonal IgM with rheumatoid factor activity + polyclonal IgG | HCV (most common), B-cell lymphoma |
| Type III (mixed) | Polyclonal IgM + polyclonal IgG | HCV, autoimmune disease (Sjogren, SLE) |
Types II and III (mixed cryoglobulinaemia) are the ones that cause vasculitis. Type I causes hyperviscosity and thrombosis, not vasculitis. [1]
The classic triad (Meltzer triad) of mixed cryoglobulinaemia: [1]
- Palpable purpura of the lower limbs
- Arthralgia (usually symmetrical, small joints)
- Weakness and generalised asthenia [1]
Other features: [1]
- Peripheral neuropathy — painful sensory or sensorimotor neuropathy, or mononeuritis multiplex.
- Renal involvement — membranoproliferative glomerulonephritis with proteinuria, haematuria and progressive renal impairment.
- Raynaud phenomenon and livedo reticularis.
- Sicca symptoms (dry eyes, dry mouth) — overlaps with Sjogren syndrome. [1]
Investigation: [1]
- Cryoglobulins — blood must be drawn warm (kept at 37 degrees Celsius) to prevent in-vitro precipitation. A common practical error is collecting the sample cold, giving a false-negative result.
- Low complement — particularly low C4 with normal or mildly low C3. This is a hallmark and a key discriminator from ANCA-associated vasculitis, where complement is typically normal.
- Rheumatoid factor positive with negative ANA (the IgM has rheumatoid factor activity).
- HCV serology (anti-HCV antibody and HCV RNA) — the most important aetiological screen.
- Serum and urine electrophoresis and B-cell lymphoma screen — to detect the underlying B-cell clonality. [1]
Management: [1]
- Treat the underlying cause. For HCV-associated cryoglobulinaemic vasculitis: direct-acting antiviral agents (DAAs) achieve HCV cure and usually remit the vasculitis. DAAs have transformed this condition.
- For severe or life-threatening cryoglobulinaemic vasculitis (rapidly progressive GN, severe neuropathy, ulcers, gut ischaemia): rituximab is the treatment of choice, combined with plasmapheresis for fulminant disease.
- Glucocorticoids for acute control, tapered as the underlying disease is treated. [1]
IgA vasculitis (formerly Henoch-Schonlein purpura)
IgA vasculitis is the most common systemic vasculitis of children (peak age 4 to 6 years), though it occurs in adults (more severely). It is an IgA immune-complex small-vessel vasculitis triggered by upper respiratory tract infections (particularly group A streptococcus). [1]
The classic tetrad — remember four P's and abdominal pain: [1]
- Palpable purpura — on the lower limbs and buttocks, typically in crops. This is present in virtually all cases and is usually the presenting feature.
- Abdominal pain — colicky, from IgA deposition in the gut wall. May be complicated by intussusception (particularly in children) or gastrointestinal bleeding.
- Arthritis or arthralgia — usually of the knees and ankles; periarticular swelling rather than true synovitis.
- Renal disease — IgA nephropathy (mesangial IgA deposition) ranging from microscopic haematuria and proteinuria to nephrotic syndrome or, rarely, rapidly progressive glomerulonephritis. [1]
Other features: [1]
- Scrotal swelling and pain in boys (mimics torsion).
- Rare central nervous system involvement. [1]
Investigation: [1]
- The diagnosis is clinical in children with the typical tetrad.
- Urinalysis and blood pressure at presentation and regular monitoring for at least 6 to 12 months, as renal disease can develop or worsen over weeks.
- Renal biopsy for significant renal involvement shows mesangial IgA deposition (indistinguishable from IgA nephropathy).
- Skin biopsy of a purpuric lesion shows leukocytoclastic vasculitis with IgA deposition on immunofluorescence — this is the defining pathological feature. [1]
Management: [1]
- Supportive care for most children — rest, hydration, analgesia, monitoring of renal function. The disease is usually self-limiting over 1 to 2 weeks.
- Glucocorticoids for severe abdominal pain, arthritis, or renal involvement — but steroids do not prevent the development of renal disease and are not given routinely.
- Severe renal disease (nephrotic syndrome, crescentic GN) is treated with the AAV immunosuppressive regimen (steroids plus cyclophosphamide or mycophenolate). [1]
Hypersensitivity vasculitis (drug-induced)
Hypersensitivity vasculitis is a small-vessel leukocytoclastic vasculitis caused by a drug or infection, presenting with palpable purpura of the lower limbs 7 to 10 days after exposure to the trigger. Common culprits are beta-lactam antibiotics, sulfonamides, thiazides, allopurinol, phenytoin and propylthiouracil. It is usually self-limiting with withdrawal of the trigger, and severe systemic involvement is uncommon. [1]
The key clinical step is always to take a complete drug history, including over-the-counter and herbal medications, and to correlate the onset of the purpura with drug exposure. Skin biopsy confirms leukocytoclastic vasculitis. Management is drug withdrawal and supportive care, with steroids for extensive skin involvement. [1]
Examiner trap: Low complement is the key discriminator between immune-complex and ANCA-associated small-vessel vasculitis. Low complement (especially low C4) points to cryoglobulinaemia, IgA vasculitis or lupus — not to ANCA-associated vasculitis. Always request C3 and C4 alongside ANCA when investigating suspected small-vessel vasculitis. [1]
Variable vessel vasculitis — Behcet disease
Behcet disease is unique among the vasculitides because it can involve vessels of any size and type (arterial and venous, in any organ). It is a vasculitis with a distinctive geographic distribution — the Silk Road countries (Turkey, the Mediterranean, the Middle East, Japan, Korea) — and is strongly associated with HLA-B51. [1]
The international criteria (ICBD) for Behcet disease require oral aphthous ulcers plus any two of: [1]
- Oral aphthous ulcers (mandatory) — recurrent, minor or major, multiple at a time.
- Genital aphthous ulcers — on the scrotum, vulva or penis; heal with scarring.
- Eye lesions — panuveitis or retinal vasculitis; sight-threatening; a leading cause of morbidity.
- Skin lesions — erythema nodosum, acneiform nodules, pseudofolliculitis.
- Positive pathergy test — a papule or pustule forming 24 to 48 hours after a skin prick with a sterile needle. This is a highly specific finding. [1]
Other important features: [1]
- Vascular — venous thrombosis (deep vein, cerebral sinus, portal) and arterial aneurysms (pulmonary artery aneurysms cause massive haemoptysis and are life-threatening). Thrombosis is thought to be vasculitic rather than procoagulant, so conventional anticoagulation is debated.
- Neurological — parenchymal brain lesions (brainstem, basal ganglia) and dural sinus thrombosis.
- Gastrointestinal — aphthous ulcers of the ileocaecal region mimicking Crohn disease.
- Articular — non-erosive arthritis. [1]
Management: [1]
- Colchicine for mucocutaneous disease and arthritis.
- Topical and systemic steroids for acute eye, vascular and neurological flares.
- Azathioprine, cyclosporin, interferon-alpha or anti-TNF agents (infliximab, adalimumab) for severe or refractory disease, particularly eye and neurological involvement.
- Apremilast (a PDE4 inhibitor) is approved for oral ulcers.
- For arterial aneurysms: high-dose steroids, cyclophosphamide, and TNF inhibitors; anticoagulation only if there is clear thrombosis without aneurysm. [1]
Behcet disease is an exam favourite because of the pathergy test, the HLA-B51 association, and the unique combination of oral and genital ulcers with uveitis. [1]
Single-organ vasculitis
Single-organ vasculitis is vasculitis confined to a single organ at diagnosis and for which no underlying systemic disease is identified after thorough evaluation. Examples include cutaneous leukocytoclastic angiitis (skin only), primary angiitis of the central nervous system (CNS only), and isolated aortitis. [1]
Primary angiitis of the central nervous system (PACNS) deserves specific mention because it is a challenging diagnosis. It presents with subacute encephalopathy, headache, cognitive decline, stroke, or myelopathy in a previously well patient. Brain MRI shows multiple ischaemic lesions, and conventional angiography shows beading of cerebral vessels. Brain and meningeal biopsy is the gold standard. Treatment is high-dose steroids and cyclophosphamide. It must be distinguished from the much more common and benign reversible cerebral vasoconstriction syndrome (RCVS), which is thunderclap-headache-associated and self-limiting. [1]
The diagnostic approach
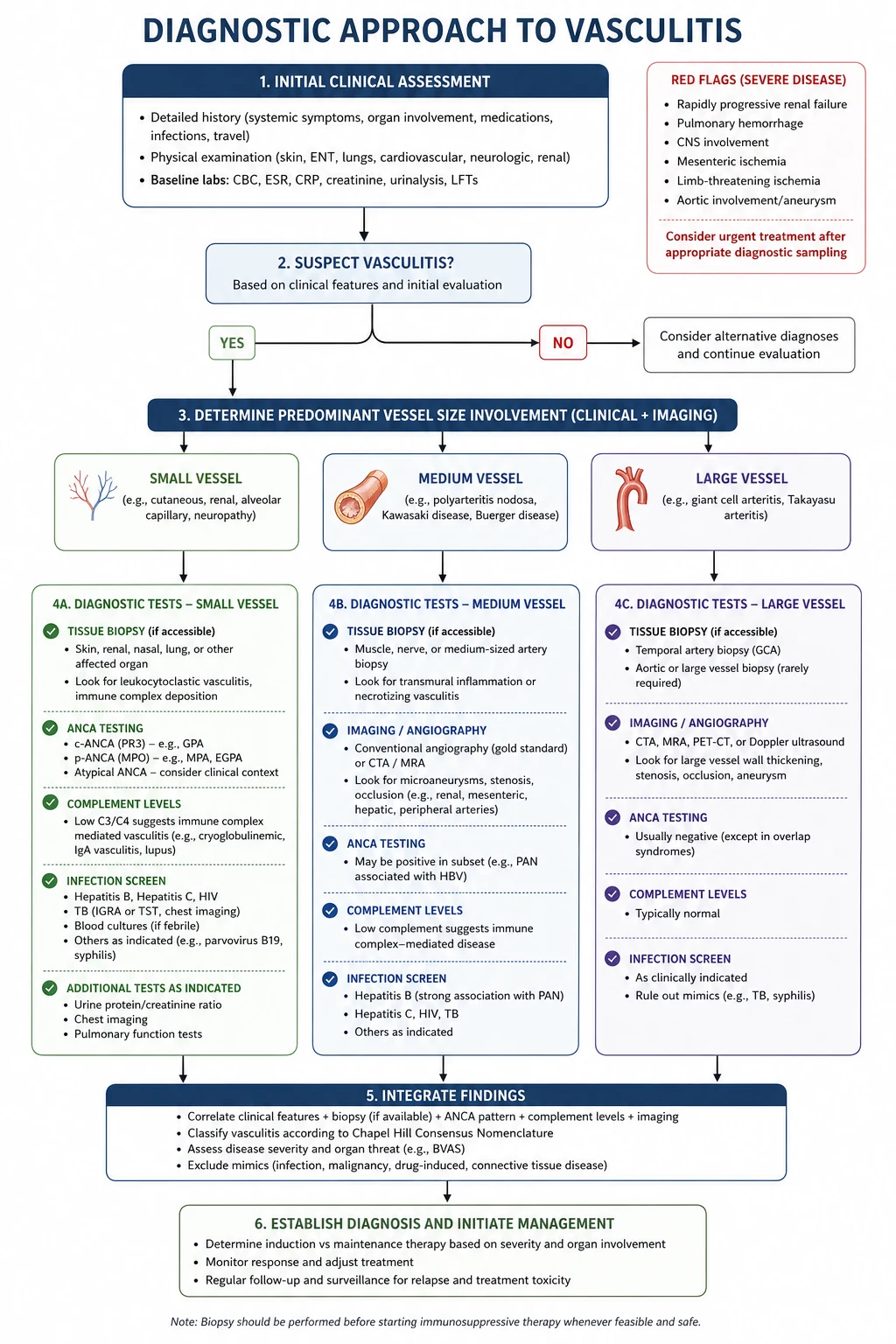
When you see a patient with suspected vasculitis, follow a structured approach. The goal is to answer three questions quickly: Is this vasculitis? What vessel size? What is the mechanism? [1]
Step 1 — Recognise the vasculitic syndrome
The presenting clinical pattern tells you the vessel size: [1]
| Clinical pattern | Vessel size | Diseases to consider |
|---|---|---|
| Headache, jaw claudication, visual loss, absent pulses | Large | GCA, Takayasu |
| Mononeuritis multiplex, mesenteric ischaemia, testicular pain, microaneurysms | Medium | PAN, Kawasaki |
| Palpable purpura, rapidly progressive GN, alveolar haemorrhage, ENT disease | Small | GPA, MPA, EGPA, cryoglobulinaemia, IgA vasculitis |
| Oral and genital ulcers with uveitis | Variable | Behcet |
Red-flag presentations that demand same-day action: [1]
- Pulmonary-renal syndrome (haemoptysis with rapidly progressive GN) — treat as GPA or MPA until proven otherwise.
- Sudden visual loss with headache in a patient over 50 — treat as GCA.
- Mononeuritis multiplex — this is vasculitis until proven otherwise.
- Abdominal pain with palpable purpura — consider mesenteric vasculitis or IgA vasculitis. [1]
Step 2 — Confirm the diagnosis
The laboratory and tissue panel: [1]
| Test | What it tells you |
|---|---|
| Full blood count, ESR, CRP | Inflammation; eosinophilia points to EGPA |
| ANCA (IIF plus anti-PR3 and anti-MPO ELISA) | GPA (c-ANCA/PR3), MPA and EGPA (p-ANCA/MPO) |
| Complement C3, C4 | Low in cryoglobulinaemia, IgA vasculitis and lupus; normal in AAV and PAN |
| Cryoglobulins (sample kept warm), RF | Cryoglobulinaemic vasculitis |
| ANA, anti-dsDNA, ENA | SLE and connective tissue disease overlap |
| Hepatitis B, C and HIV serology | PAN (HBV), cryoglobulinaemia (HCV), overlap with HIV |
| Urinalysis, urine microscopy, U&E, creatinine | Renal involvement — red cell casts indicate active GN |
| Tissue biopsy (skin, nerve, muscle, kidney, temporal artery, lung) | Definitive histology — the gold standard for most vasculitides |
| Angiography (mesenteric, renal, aortic) | Medium-vessel disease (PAN microaneurysms) and large-vessel disease (Takayasu) |
| FDG-PET and vascular ultrasound/MRI | Large-vessel inflammation (GCA extracranial disease, Takayasu) |
Biopsy principles: [1]
- Biopsy the most accessible involved tissue with the highest expected yield. In suspected cutaneous vasculitis, biopsy a fresh purpuric lesion (less than 24 to 48 hours old) for both light microscopy and immunofluorescence.
- In suspected vasculitic neuropathy, a combined sural nerve and muscle biopsy gives the highest yield.
- In suspected renal vasculitis, a renal biopsy is both diagnostic and prognostic (extent of crescents, tubulointerstitial damage, chronicity).
- In suspected GCA, a temporal artery biopsy should include a long segment (more than 1 to 2 cm) to minimise the effect of skip lesions. [1]
Step 3 — Exclude the mimics
This is a critical and often-examined step. Vasculitis mimics are common, and treating a mimic with high-dose immunosuppression is dangerous. The key mimics to exclude before committing to a vasculitis diagnosis: [1]
| Category | Mimics |
|---|---|
| Infection | Endocarditis (can be ANCA-positive), septic emboli, disseminated gonococcal infection, rickettsial infection, hepatitis |
| Malignancy | Atrial myxoma (systemic emboli), lymphoma, leukaemia, tumour emboli |
| Thrombotic | Antiphospholipid syndrome, thrombotic thrombocytopenic purpura, disseminated intravascular coagulation |
| Drug toxicity | Cocaine-induced pseudovasculitis (levamisole contamination), amphetamines |
| Other | Cholesterol emboli (post-catheter), fibromuscular dysplasia, reversible cerebral vasoconstriction syndrome |
Endocarditis is the most important mimic to exclude in any patient with suspected vasculitis. Blood cultures, echocardiography, and careful attention to risk factors are essential. Endocarditis can be ANCA-positive and can cause a pulmonary-renal syndrome indistinguishable from AAV at first presentation — treating it with rituximab and cyclophosphamide rather than antibiotics would be catastrophic. [1]
Atrial myxoma causes systemic emboli with constitutional features and can perfectly mimic vasculitis — an echocardiogram should be part of the workup of any embolic vasculitis-like syndrome. [1]
Cocaine and levamisole cause a devastating pseudovasculitis with purpura (particularly of the ears and nose), arthralgia and ANCA positivity (often with both PR3 and MPO positivity, which is unusual in true AAV). [1]
DWE high-yield: Always exclude infection before starting immunosuppression. Blood cultures, echocardiogram, hepatitis serology and a careful drug history are non-negotiable in any patient with suspected vasculitis, especially before cyclophosphamide or rituximab. Atrial myxoma is the single most dangerous structural cardiac mimic — an echocardiogram is part of the workup of embolic disease. [1]
The treatment framework
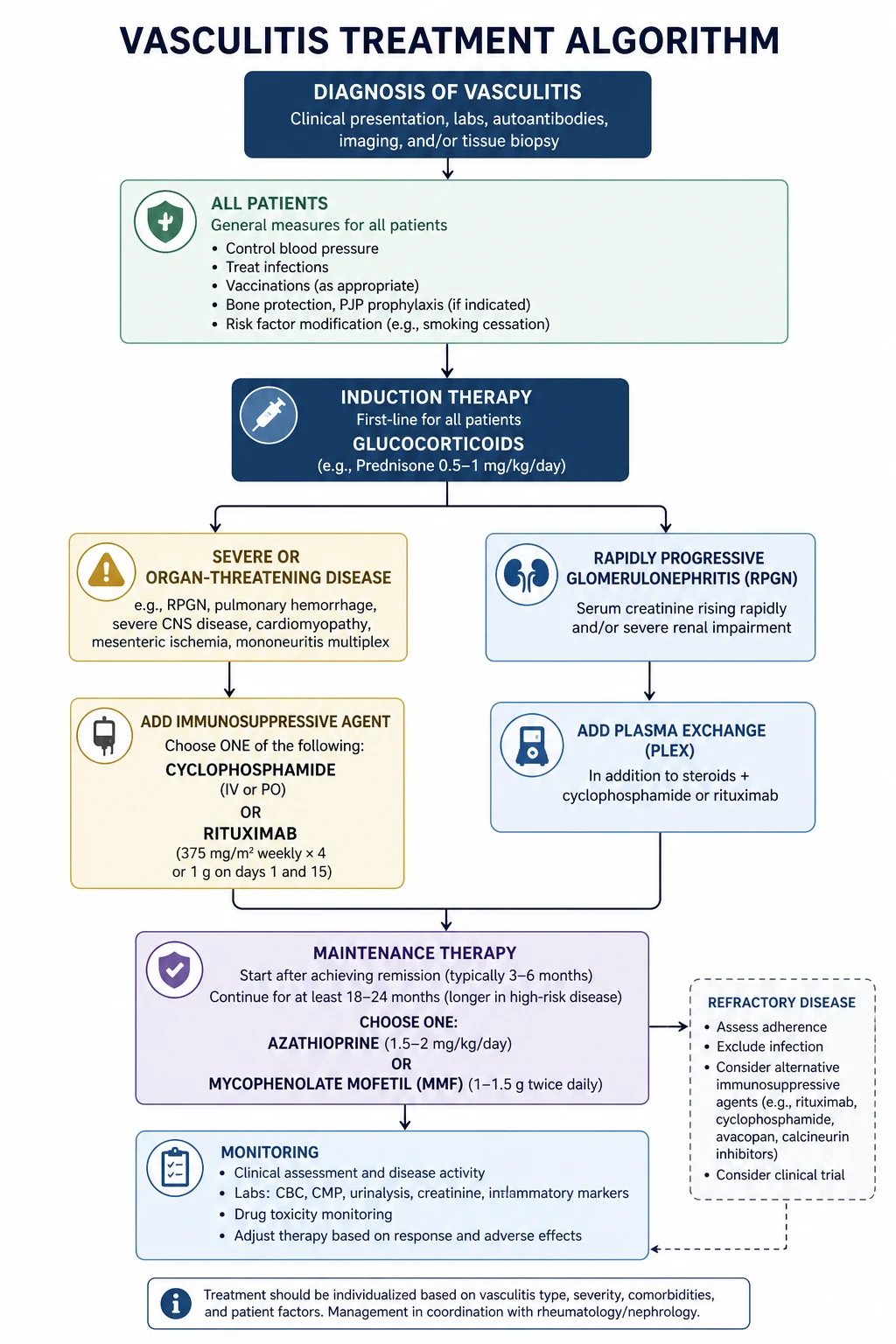
The treatment of vasculitis follows a risk-stratified framework that depends on the severity and extent of organ involvement, not on the specific diagnosis alone. The 2022 EULAR update on AAV management is the current evidence-based reference [7].
Principles of vasculitis treatment
Every vasculitis patient needs three questions answered: [1]
- Is this life-threatening or organ-threatening? If yes, the treatment is high-dose glucocorticoids plus an immunosuppressive agent (rituximab or cyclophosphamide) for induction.
- Is there an underlying trigger or cause? If yes, treat the cause — antivirals for HBV-PAN and HCV-cryoglobulinaemia, withdrawal for drug-induced, antibiotics for infection-triggered.
- What is the maintenance strategy after remission? Long-term immunosuppression to prevent relapse, with monitoring for treatment toxicity. [1]
Induction of remission for ANCA-associated vasculitis
For organ- or life-threatening AAV (rapidly progressive GN, alveolar haemorrhage, neuropathy, extensive disease), the induction regimen is: [1]
- Methylprednisolone 500 mg to 1 g IV daily for 3 days, followed by oral prednisolone 1 mg/kg daily (max 60 to 80 mg), tapered over the first 4 to 5 months to a target of 5 mg daily [7].
- PLUS rituximab 375 mg per square metre weekly for 4 weeks (RAVE regimen) or 1 g on days 1 and 15 (RITUXVAS regimen). Rituximab is now preferred over cyclophosphamide for most AAV, especially relapsing disease [3][4].
- OR cyclophosphamide 15 mg/kg IV every 2 weeks for 3 doses then every 3 weeks, or 2 mg/kg orally daily, for 3 to 6 months. Still used when rituximab is contraindicated, in pregnancy planning by men, or in some centres as first-line [7].
- Reduced-dose glucocorticoids (as shown by PEXIVAS) are non-inferior to standard high-dose regimens and reduce serious infections — a paradigm shift in AAV induction [6].
Plasma exchange is reserved for specific situations: [1]
- Anti-GBM overlap (dual-positive ANCA and anti-GBM) — always, to remove anti-GBM antibodies.
- Severe or dialysis-dependent renal vasculitis (creatinine above 500 micromol per litre or dialysis-dependent at presentation) — the MEPEX trial showed improved renal recovery at 3 months, though long-term survival benefit was not sustained [5]. The larger PEXIVAS trial did not show benefit in the composite of death or end-stage kidney disease [6], so plasma exchange is now more selectively used in expert hands [7].
- Severe diffuse alveolar haemorrhage with hypoxaemia — used in some centres, though evidence is limited.
Avacopan (a C5a receptor inhibitor) was approved for AAV based on the ADVOCATE trial, but this trial was retracted in June 2026 after data-integrity concerns were identified by the FDA and EMA. Avacopan is not currently recommended as standard therapy, and its regulatory status is under review. Do not cite avacopan as evidence-based in exams or clinical practice until the integrity issues are resolved. [1]
Maintenance of remission for AAV
After 3 to 6 months of successful induction, switch to maintenance therapy: [1]
- Rituximab 1 g every 4 months (or 500 mg every 6 months) for up to 24 to 36 months — now the preferred maintenance agent, particularly for PR3-ANCA disease, which has the highest relapse risk [7].
- Azathioprine 2 mg/kg daily — the traditional maintenance agent; used for 18 to 24 months after cyclophosphamide induction.
- Methotrexate 20 to 25 mg weekly — for non-renal or limited disease, or in patients who cannot tolerate azathioprine.
- Mycophenolate mofetil 2 g daily — an alternative, particularly in MPO-ANCA disease. [1]- Continue low-dose prednisolone (5 mg daily or less) for the first 4 to 6 months, then taper if possible.
The total duration of maintenance is typically 18 to 24 months after sustained remission, then cautious withdrawal with monitoring for relapse. [1]
Monitoring for relapse and treatment toxicity
- Relapse is suggested by a rising ANCA titre (particularly PR3-ANCA), rising inflammatory markers, recurrent haematuria, or new organ involvement. Treat a relapse with the induction regimen again (rituximab is effective in relapsing disease).
- Treatment toxicity — cyclophosphamide causes haemorrhagic cystitis, infertility, myelosuppression and malignancy; rituximab causes hypogammaglobulinaemia (monitor IgG levels) and opportunistic infection (PML is rare but real); prolonged steroids cause osteoporosis, diabetes, infection and cataracts.
- Co-trimoxazole prophylaxis against pneumocystis jirovecii is standard during induction.
- Bone protection, vaccination (before immunosuppression), cardiovascular risk management and psychosocial support are integral to care. [1]
Treatment of large and medium vessel vasculitis
| Disease | Induction | Maintenance |
|---|---|---|
| GCA | Prednisolone 40 to 60 mg daily; IV methylprednisolone if visual symptoms; tocilizumab for relapsing disease | Taper steroids over 12 to 24 months; tocilizumab for 12 months |
| PAN (idiopathic) | Prednisolone 1 mg/kg daily plus cyclophosphamide if severe | Azathioprine or methotrexate maintenance |
| HBV-PAN | Antiviral (entecavir or tenofovir) plus plasmapheresis plus short-course steroids | Monitor for viral clearance |
| Behcet | Colchicine for mucocutaneous; steroids for eye and vascular flares; anti-TNF for severe eye and neurological disease | Azathioprine, infliximab or apremilast for relapse prevention |
Long-term outcomes and surveillance
The systemic vasculitides are chronic relapsing diseases. Even after remission, patients require lifelong surveillance. [1]
AAV outcomes: [1]
- Five-year survival in AAV is now about 75 to 85 per cent with modern therapy, but mortality remains significantly higher than the general population, driven by cardiovascular disease, infection and malignancy.
- Renal outcome depends on the degree of renal damage at presentation — dialysis-dependent patients have a worse prognosis and a higher risk of end-stage kidney disease.
- Relapse is common, particularly in PR3-ANCA disease, GPA (versus MPA), and in patients who stop maintenance therapy early. PR3-ANCA disease relapses at roughly twice the rate of MPO-ANCA disease.
- Cardiovascular risk is significantly elevated in AAV and other vasculitides — aggressive management of traditional risk factors is essential. [1]
GCA outcomes: [1]
- Visual loss, once established, is usually permanent — prevention through early steroids is the only effective strategy.
- GCA increases the risk of aortic aneurysm (particularly thoracic) — screening with periodic imaging of the aorta is recommended for the first 5 years after diagnosis.
- PMR typically resolves over 2 to 5 years, but some patients need prolonged low-dose steroids. [1]
PAN outcomes: [1]
- Idiopathic PAN with modern therapy has a 5-year survival above 80 per cent.
- HBV-associated PAN with antiviral therapy has an excellent prognosis once the virus is cleared.
- PAN does not relapse after remission in most patients — a useful distinction from AAV, which relapses frequently. [1]
DCE exam preparation
Long case: suspected ANCA-associated vasculitis
A classic DCE long case is a patient with GPA or MPA presenting with a multisystem illness — constitutional symptoms, ENT complaints, haemoptysis, haematuria and a rising creatinine. The candidate must deliver a structured opening statement (SASPOP), a prioritised problem list, an integrated management plan, and show insight into patient perspective. [1]
Model opening statement (SASPOP format): [1]
"This is Mr David Chen, a 56-year-old engineer presenting with a 3-month history of progressive nasal crusting and epistaxis, followed by haemoptysis, rapidly progressive renal impairment with a creatinine of 340, and c-ANCA strongly positive with anti-PR3 specificity. His problem list is: (1) granulomatosis with polyangiitis with pulmonary-renal syndrome — an organ- and life-threatening presentation; (2) rapidly progressive glomerulonephritis with a creatinine of 340 and active urinary sediment; (3) pulmonary involvement with alveolar haemorrhage; (4) upper airway disease with nasal crusting; (5) treatment-related risks including immunosuppression and glucocorticoid toxicity; and (6) the psychosocial impact of a new diagnosis of a serious chronic illness on a working man with a family." [1]
Examiner probing questions and model answers: [1]
- What is your immediate management? — Admit, start IV methylprednisolone 1 g daily for 3 days, give rituximab 375 mg per square metre weekly for 4 doses, exclude infection with blood cultures and echocardiogram, and involve nephrology for renal biopsy. Consider plasma exchange given the severity and renal involvement.
- How will you confirm the diagnosis? — Renal biopsy for pauci-immune crescentic GN; chest CT for cavitating nodules; ANCA confirmed by ELISA.
- What are the key differential diagnoses to exclude? — Anti-GBM disease (check anti-GBM antibodies), lupus nephritis (ANA, anti-dsDNA, complement), endocarditis (blood cultures, echocardiogram), and malignancy.
- What is your maintenance plan? — After 6 months, switch to rituximab maintenance for at least 24 months, with bone protection, pneumocystis prophylaxis, cardiovascular risk management and vaccination.
- What is the prognostic discussion? — This is a chronic relapsing disease with significant treatment burden; sustained remission is achievable; renal outcome depends on response to induction; and cardiovascular and infection risk management are essential. [1]
Short case: palpable purpura on the lower limbs
A DCE short case may present a patient with palpable purpura and ask for a focused examination, differential and discussion. [1]
Examination routine: [1]
- Inspect the skin — describe the purpura (palpable, non-blanching, lower limbs, size, distribution, ulceration, livedo).
- Examine the hands and arms — splinter haemorrhages, nailfold changes, digital ulcers, nodules.
- Examine for systemic features — pulse, blood pressure (both arms), abdominal bruits, lymphadenopathy, joint swelling, oral and genital ulcers (Behcet), saddle-nose deformity (GPA).
- Neurological — test for mononeuritis multiplex (wrist drop, foot drop, sensory loss in distinct nerve territories). [1]
Presentation template: [1]
"This patient has palpable non-blanching purpura on the lower limbs consistent with a small-vessel vasculitis. The distribution and the presence of palpability point to leukocytoclastic vasculitis. Given the associated features (specify — ENT disease, asthma and eosinophilia, hepatitis C, recent drug exposure, joint pains and abdominal pain), the most likely diagnosis is (specify — GPA, EGPA, cryoglobulinaemia, IgA vasculitis, hypersensitivity vasculitis). I would confirm with a skin biopsy for light microscopy and immunofluorescence, and screen with ANCA, complement, cryoglobulins, hepatitis serology, urinalysis and a full drug history." [1]
Key references summary
2012 revised Chapel Hill Consensus Conference Nomenclature of Vasculitides (Jennette, Arthritis Rheum 2013) [1]; GiACTA trial of tocilizumab in giant cell arteritis (Stone, NEJM 2017) [2]; RAVE trial of rituximab versus cyclophosphamide for AAV (Stone, NEJM 2010) [3]; RITUXVAS trial of rituximab in renal AAV (Jones, NEJM 2010) [4]; MEPEX trial of plasma exchange in severe renal vasculitis (Jayne, JASN 2007) [5]; PEXIVAS trial of plasma exchange and reduced-dose glucocorticoids in severe AAV (Walsh, NEJM 2020) [6]; EULAR recommendations for the management of ANCA-associated vasculitis, 2022 update (Hellmich, Ann Rheum Dis 2024) [7]; 2022 ACR/EULAR classification criteria for giant cell arteritis (Buttgereit, Arthritis Rheumatol 2022) [8]; 2022 ACR/EULAR classification criteria for GPA (Robson, Ann Rheum Dis 2022) [9]; 2022 ACR/EULAR classification criteria for MPA (Suppiah, Ann Rheum Dis 2022) [10]; 2018 EULAR recommendations for management of large vessel vasculitis (Hellmich, Ann Rheum Dis 2020) [11]. Note: the ADVOCATE trial of avacopan (Jayne, NEJM 2021, PMID 33596356) was retracted in June 2026 after data-integrity concerns; avacopan is not currently recommended as standard therapy.
References
- [1]Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides Arthritis Rheum, 2013.PMID 23045170
- [2]Stone JH, Tuckwell K, Dimonaco S, et al. Trial of Tocilizumab in Giant-Cell Arteritis N Engl J Med, 2017.PMID 28745999
- [3]Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis N Engl J Med, 2010.PMID 20647199
- [4]Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis N Engl J Med, 2010.PMID 20647198
- [5]Jayne DR, Gaskin G, Rasmussen N, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis J Am Soc Nephrol, 2007.PMID 17582159
- [6]Walsh M, Merkel PA, Peh CA, et al. Plasma Exchange and Glucocorticoids in Severe ANCA-Associated Vasculitis N Engl J Med, 2020.PMID 32053298
- [7]Hellmich B, Sanchez-Alamo B, Schirmer JH, et al. EULAR recommendations for the management of ANCA-associated vasculitis: 2022 update Ann Rheum Dis, 2024.PMID 36927642
- [8]Buttgereit F, Matteson EL, Dejaco C, Dasgupta B. 2022 American College of Rheumatology/EULAR Classification Criteria for Giant Cell Arteritis Arthritis Rheumatol, 2022.PMID 36350123
- [9]Robson JC, Grayson PC, Ponte C, et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology Classification Criteria for Eosinophilic Granulomatosis with Polyangiitis Ann Rheum Dis, 2022.PMID 35110334
- [10]Suppiah R, Robson JC, Grayson PC, et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology classification criteria for microscopic polyangiitis Ann Rheum Dis, 2022.PMID 35110332
- [11]Hellmich B, Agueda A, Monti S, et al. 2018 Update of the EULAR recommendations for the management of large vessel vasculitis Ann Rheum Dis, 2020.PMID 31270110