Psych · Consultation-liaison psychiatry
Wilson disease and metabolic neuropsychiatry
Also known as Wilson's disease psychiatry · Hepatolenticular degeneration psychiatry · ATP7B copper neuropsychiatry · Wilson disease psychosis · Wilson disease depression · Metabolic neuropsychiatry Wilson · Chelation penicillamine psychiatry · Kayser-Fleischer psychiatric presentation
Exam-exhaustive fellowship topic on Wilson disease and metabolic neuropsychiatry: ATP7B copper toxicity, personality change and irritability, depression, psychosis, cognition, Leipzig score diagnostics, lifelong chelation and zinc, paradoxical neurologic worsening, psychotropic choice with basal ganglia and liver disease, family screening, transplant interface, and treatable metabolic differentials. FRANZCP-primary, globally tagged.
On this page & tools
Your progress
Saved locally on this device.
Target exams
Red flags
Psychiatrists fail this topic when they diagnose schizophrenia in a young adult with subtle tremor and abnormal LFTs, when they use high-potency typicals that freeze a copper-laden basal ganglia system, when they start penicillamine aggressively without warning about paradoxical neurologic worsening, or when they treat mood without securing lifelong chelation adherence.[1][2][11]
Overview and definition
Wilson disease (hepatolenticular degeneration) is caused by biallelic pathogenic variants in ATP7B, impairing biliary copper excretion and incorporation of copper into ceruloplasmin. Copper accumulates in liver, basal ganglia, cornea, and other tissues, producing hepatic, neurologic, and psychiatric phenotypes that may present in childhood, adolescence, or adulthood.[7][10][14]
In DSM-5-TR language, presentations map to mental disorder due to another medical condition, substance/medication-induced differentials, and sometimes major neurocognitive disorder due to another medical condition. ICD-11 frames WD within diseases of the nervous system and liver with associated behavioural syndromes. The CL task is early organic recognition, joint anticopper therapy, syndrome-specific psychotropics, and long-term adherence support.[1][6][10]
Metabolic neuropsychiatry here means treatable inborn errors and copper/metabolic toxicities that masquerade as primary psychiatric illness. WD is the high-yield prototype; brief differentials include acute intermittent porphyria, urea-cycle disorders, and Niemann–Pick type C when red flags demand a wider net.[1][10]
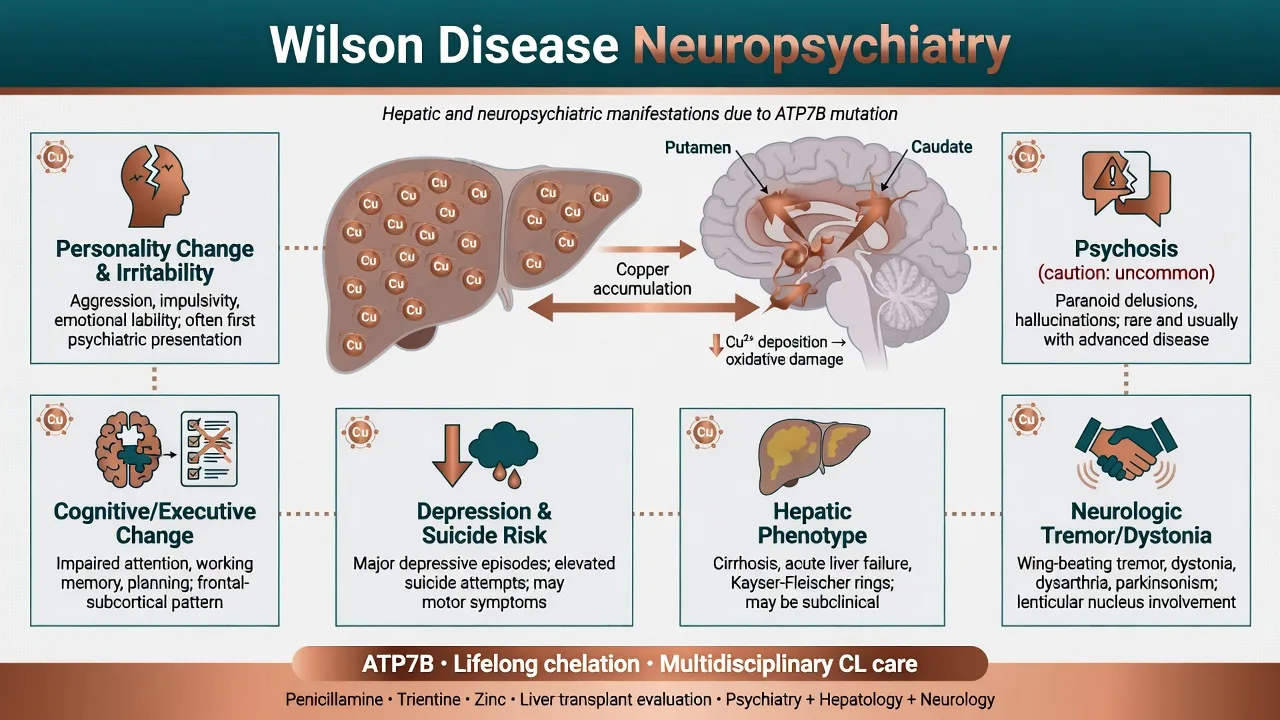
Epidemiology and burden
Population prevalence is often cited near 1 in 30,000, with higher rates in some founder or consanguineous populations; many pure psychiatric presentations are delayed or missed.[10][14] Reviews synthesise that about 30–40% of patients have psychiatric manifestations at diagnosis, and roughly one in five saw a psychiatrist before WD was recognised — an exam-critical delay statistic.[1]
Landmark psychiatric series show abnormal behaviour and personality change as the most common psychiatric features, with depression and cognitive impairment frequent; frank schizophrenia-like psychosis is less common than classic textbooks imply but remains a high-stakes mislabel risk.[3][4][5] Longitudinal work confirms incongruous behaviour, irritability, depression, and cognitive impairment as enduring themes across follow-up.[13] Psychiatric comorbidity worsens quality of life and psychosocial outcomes.[6]
Classification of key syndromes
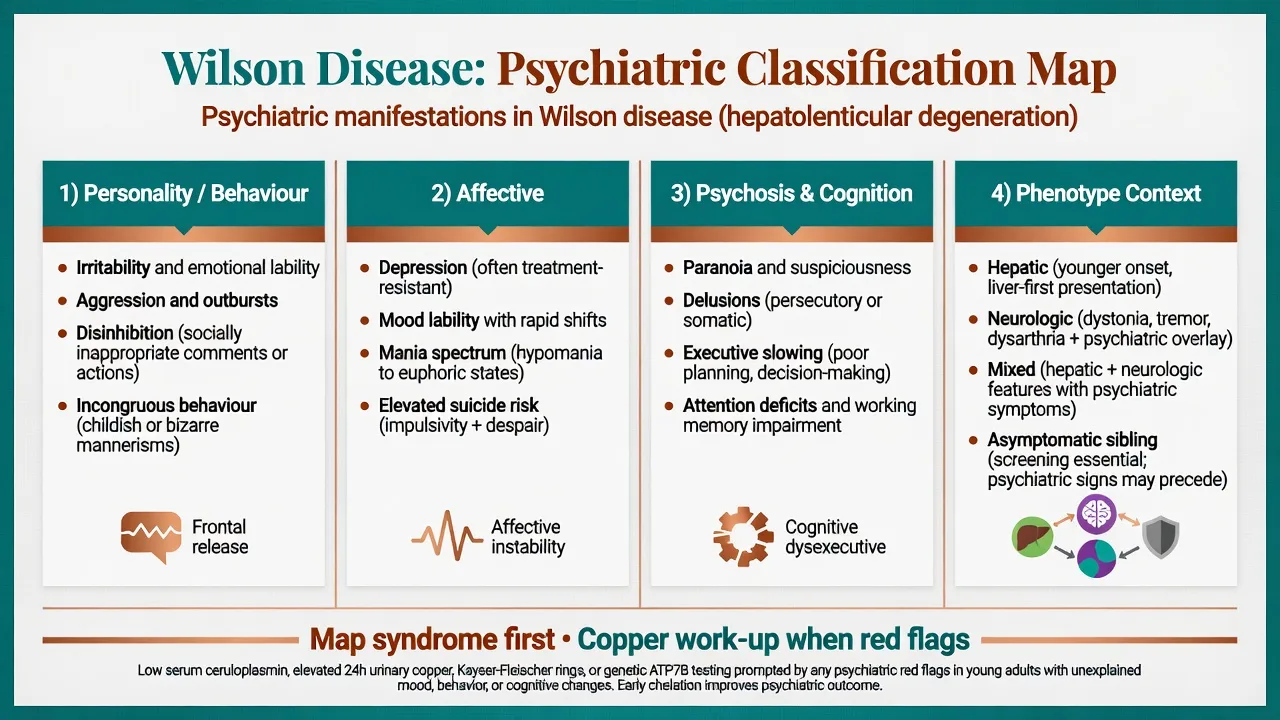
Personality and behavioural change
Irritability, aggression, disinhibition, and incongruous behaviour are classic. Carers describe short fuse, socially inappropriate comments, childish mannerisms, and relationship collapse — often mis-coded as primary personality or conduct pathology in adolescents and young adults.[3][4][5]
Affective syndromes
Major depression is common and must be treated actively; mood lability and bipolar-spectrum presentations occur. Suicide risk requires structured assessment, especially with despair about chronic neurologic disability or after diagnostic delay.[1][2][6]
Psychosis and cognition
Psychosis (paranoia, delusions, occasional hallucinations) can mimic primary schizophrenia; look for motor clues and liver clues rather than assuming pure idiopathic psychosis.[1][4] Cognitive change is typically subcortical-executive (attention, processing speed, planning) rather than a pure cortical aphasia/apraxia package.[1][10]
Phenotype context
- Hepatic-predominant: childhood/young adult liver disease, sometimes with subtler psychiatric load.
- Neurologic/neuropsychiatric: tremor, dystonia, dysarthria, parkinsonism plus behavioural/mood change.
- Mixed.
- Asymptomatic siblings found on cascade screening need preventive therapy planning.[7][10][12]
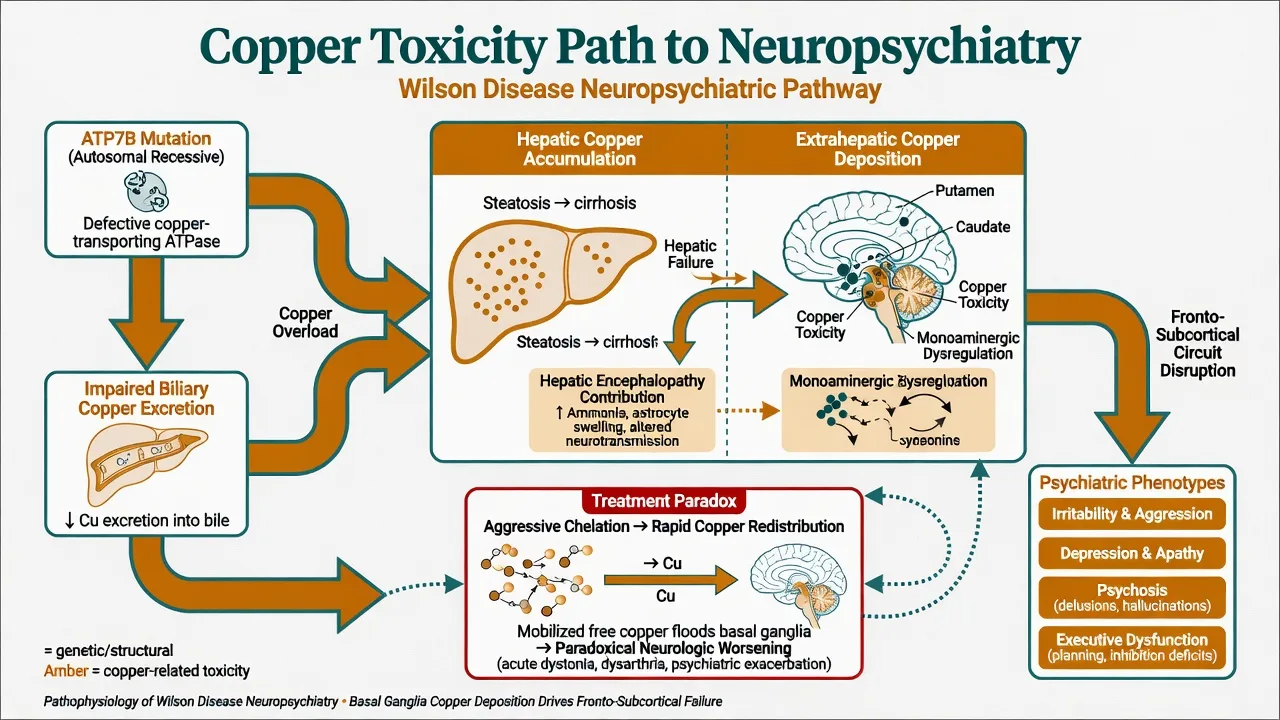
Pathophysiology (viva depth)
Loss of ATP7B function produces copper overload. Hepatic copper injury ranges from steatosis to cirrhosis and acute liver failure; free copper spills to the CNS with preferential basal ganglia (putamen, caudate, globus pallidus) and fronto-subcortical circuit toxicity.[10][14] This network failure links movement disorder with impulse control, affect regulation, and executive dysfunction. Hepatic encephalopathy may add a second, ammonia-related neuropsychiatric pathway when liver failure is advanced.[7][10]
Paradoxical neurologic worsening after aggressive chelation is attributed to rapid mobilisation and redistribution of copper — a mechanism every registrar must voice before starting D-penicillamine at full dose.[7][8][11]
Clinical assessment for CL
Structure the interview around tempo, motor clues, liver clues, full neuropsychiatric map, suicide risk, medications, capacity, and family history.[1][2][7]
- Onset and trajectory — school/work decline, personality change over months to years.
- Motor overlay — wing-beating tremor, dystonia, dysarthria, drooling, gait, parkinsonism.
- Hepatic clues — jaundice history, known cirrhosis, unexplained LFT abnormality, haemolysis.
- Psychiatric map — irritability, depression, mania, psychosis, cognition, sleep, substances.
- Suicide — ideation, plan, means, past attempts, hopelessness.
- Medications — prior high-potency antipsychotics, valproate, hepatotoxins, current chelators.
- Family — consanguinity, sibling liver/neurologic disease, prior genetic results.
- Capacity and adherence — understanding lifelong therapy; driving and safety.[1][7][10]
MSE language should capture both psychiatric content and soft neurologic signs without waiting for a fully developed dystonic face.[3][4]
Differential diagnosis
- Primary schizophrenia/bipolar/personality disorders without organic screen in a young patient with motor or liver red flags.
- Huntington disease, autoimmune encephalitis (including NMDAR), SLE cerebritis, neurosyphilis, HIV.
- Drug-induced EPS from antipsychotics — may coexist with or mask WD.
- Hepatic encephalopathy alone versus copper neurotoxicity (overlap possible in advanced hepatic WD).
- Other metabolic neuropsychiatry: acute intermittent porphyria (abdominal crises, dark urine, autonomic instability), urea-cycle disorders (hyperammonaemic encephalopathy), Niemann–Pick C (vertical supranuclear gaze palsy, cataplexy), severe B12/thyroid disease.[1][10][12]
Investigations
Diagnostic thinking is multi-parameter, not a single blood test.[7][8][12]
- Serum ceruloplasmin often low, but not diagnostic alone (acute-phase and analytic caveats).
- 24-hour urinary copper elevated in symptomatic disease.
- Slit-lamp examination for KF rings — nearly universal in neurologic WD; request ophthalmology when suspected.
- Leipzig (Ferenci) score integrates KF rings, neurologic signs, ceruloplasmin, urine copper, liver copper, Coombs-negative haemolysis, and genetics; score ≥4 makes diagnosis highly likely.
- ATP7B genetic testing and hepatic copper quantification when the score is intermediate or family screening is needed.
- Baseline LFTs, coagulation, FBC, renal function; ammonia if HE suspected; ECG and metabolic panel before psychotropics.
- Brain MRI may show basal ganglia signal change (educational correlation only — do not treat educational images as clinical scans).[7][8][12][14]
Acute management

- Safety — suicide and aggression risk; means restriction; hospital threshold when intent/plan/high lethality or carer safety fails.[1][2]
- Medical emergencies — acute liver failure, haemolytic crisis, severe HE → hepatology/ICU immediately.[7][9]
- Agitation — de-escalation; prefer lower-EPS strategies; avoid reflex high-dose haloperidol in a dystonic young adult.[2]
- Do not delay copper work-up while waiting for weeks of failed primary psychosis treatment if red flags are present.[1][12]
Definitive management
Anticopper therapy (disease-modifying core)
Lifelong therapy is mandatory. Oral chelators D-penicillamine and trientine promote copper excretion; zinc reduces intestinal copper absorption and is used for selected maintenance or asymptomatic patients. Dietary copper moderation is adjunctive, not sole therapy in symptomatic disease.[7][8][9][11]
Large comparative experience shows both major chelators control disease for most patients; D-penicillamine causes more treatment-limiting adverse events, while neurologic deterioration can occur with either agent if initiation is mishandled — start low and titrate, co-manage with hepatology/neurology, and warn patients about early neurologic worsening.[7][11] Example teaching starts (always individualise with specialist teams): D-penicillamine often begun at low divided oral doses (e.g. 250 mg oral daily class of start in many protocols, titrate slowly with monitoring) with pyridoxine co-prescription culture in some services; trientine similarly titrated from low oral divided doses; zinc acetate maintenance dosing is specialist-directed (commonly multi-times-daily separated from food/chelators). Monitor urine copper, LFTs, blood counts, and neurologic/psychiatric status.[7][8][11]
Non-adherence is a leading cause of late decompensation — psychiatry owns motivational and systems support as much as symptom control.[2][7]
Psychiatric pharmacotherapy by syndrome
Evidence is largely case series, cohort observation, and expert synthesis rather than large NPS RCTs — say this honestly in viva.[1][2]
Depression: treat with an SSRI while anticopper therapy proceeds. Pragmatic starts with hepatic caution: sertraline 25–50 mg oral daily or escitalopram 5–10 mg oral daily, titrate slowly, monitor hyponatraemia, activation, and suicidality on initiation.[2][6]
Irritability/behavioural dysregulation: structure, sleep, carer education; SSRI often first drug step; low-dose atypical antipsychotic if severe aggression or psychosis coexists.[2][5]
Psychosis: prefer lower-EPS atypical agents. Teaching examples: quetiapine 25–50 mg oral at night start, titrate cautiously; alternatives individualised (olanzapine 2.5–5 mg oral at night). Avoid high-potency typicals as first instinct. Clozapine appears in refractory case literature but adds agranulocytosis risk on top of penicillamine haematologic concerns — specialist only.[2]
Mania/mood instability with significant liver disease: lithium is attractive because it is not hepatically metabolised (monitor renal function, TSH, levels; start low, e.g. lithium carbonate 250–450 mg oral daily equivalent individualised, level-guided). Valproate is relatively hepatotoxic — use only with hepatology blessing if at all in active liver disease.[2]
ECT has case-level support for severe mood/psychosis when medical status allows and drugs fail or are unsafe.[2]
Special scenarios
First-episode psychosis clinic: any young patient with dystonia, tremor, dysarthria, KF suspicion, or LFT abnormality needs copper screening before years of depot antipsychotics.[1][4][12]
Paradoxical worsening on penicillamine: joint review — slow titration, consider switch to trientine/zinc strategies per specialist guidance, intensify rehab and psychiatric support.[7][11]
Transplant: fulminant hepatic WD or refractory liver failure may need liver transplant; CL assesses adherence capacity, mood, supports, and substance use.[7][9]
Family screening: siblings of index cases require evaluation even if asymptomatic; genetics counselling is standard.[7][14]
Pregnancy: coordinate hepatology; anticopper agent choice is specialist — psychiatry supports mood and adherence, does not freestyle chelator switches.[7][9]
Prognosis and disposition
Untreated WD progresses and can kill via liver failure or severe neurologic disability. With early consistent therapy, many patients stabilise; some neurologic and psychiatric features improve over months, though recovery may be incomplete if diagnosis was late.[7][10][11] Disposition defaults to a multidisciplinary WD pathway: hepatology, neurology, psychiatry/CL, ophthalmology, genetics, dietetics, and transplant services when needed. Advance planning for capacity, work, driving, and family cascade testing belongs in the CL note.[2][7][9]
Regional notes
ANZ (FRANZCP): Link early to tertiary hepatology/WD-experienced teams; access to trientine and specialist chelation monitoring varies by state — do not invent local formulary rules in exams, but always name multidisciplinary care and Mental Health Act principles without section-number invention. UK (MRCPsych): EASL/EASL-ERN guideline literacy and CASC communication about organic psychosis and lifelong therapy are high yield. US (ABPN): AASLD 2022 multidisciplinary guidance is the dominant North American reference; zinc and chelator pathways are well codified. South Asia (MD/DNB, NEET-SS): higher consanguinity in some communities, later presentation with dystonia/psychosis, and resource limits on genetics — prioritise KF rings, ceruloplasmin, urine copper, and pragmatic SSRI/low-EPS antipsychotic plans with available chelation.[7][8][9][10]
Exam pearls
Cu-RING
Summary
Fellowship-level WD psychiatry is metabolic neuropsychiatry done properly: recognise personality change, depression, psychosis, and executive decline as copper-related until proven otherwise when motor or hepatic clues exist; confirm with multi-parameter copper diagnostics and Leipzig scoring; start lifelong anticopper therapy low-and-slow with hepatology; choose hepatic- and EPS-aware psychotropics; and protect the family through cascade screening. A candidate who refuses a pure schizophrenia narrative in a tremulous young adult, names KF rings and paradoxical penicillamine worsening, and builds an adherence-centred multidisciplinary plan has mastered this CL atlas leaf.[1][2][7][10][11]
References
- [1]Zimbrean PC, Schilsky ML Psychiatric aspects of Wilson disease: a review Gen Hosp Psychiatry, 2014.PMID 24120023
- [2]Litwin T, Dusek P, Szafrański T, et al. Psychiatric manifestations in Wilson's disease: possibilities and difficulties for treatment Ther Adv Psychopharmacol, 2018.PMID 29977520
- [3]Dening TR, Berrios GE Wilson's disease. Psychiatric symptoms in 195 cases Arch Gen Psychiatry, 1989.PMID 2589927
- [4]Akil M, Schwartz JA, Dutchak D, et al. The psychiatric presentations of Wilson's disease J Neuropsychiatry Clin Neurosci, 1991.PMID 1821256
- [5]Akil M, Brewer GJ Psychiatric and behavioral abnormalities in Wilson's disease Adv Neurol, 1995.PMID 7872138
- [6]Mura G, Zimbrean PC, Demelia L, Carta MG Psychiatric comorbidity in Wilson's disease Int Rev Psychiatry, 2017.PMID 28681670
- [7]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: 2022 Practice Guidance on Wilson disease from the American Association for the Study of Liver Diseases Hepatology, 2025.PMID 36151586
- [8]European Association for Study of Liver EASL Clinical Practice Guidelines: Wilson's disease J Hepatol, 2012.PMID 22340672
- [9]European Association for the Study of the Liver EASL-ERN Clinical Practice Guidelines on Wilson's disease J Hepatol, 2025.PMID 40089450
- [10]Członkowska A, Litwin T, Dusek P, et al. Wilson disease Nat Rev Dis Primers, 2018.PMID 30190489
- [11]Weiss KH, Thurik F, Gotthardt DN, et al. Efficacy and safety of oral chelators in treatment of patients with Wilson disease Clin Gastroenterol Hepatol, 2013.PMID 23542331
- [12]Ferenci P Diagnosis of Wilson disease Handb Clin Neurol, 2017.PMID 28433100
- [13]Dening TR, Berrios GE Wilson's disease: a longitudinal study of psychiatric symptoms Biol Psychiatry, 1990.PMID 2378928
- [14]Merle U, Schaefer M, Ferenci P, Stremmel W Clinical presentation, diagnosis and long-term outcome of Wilson's disease: a cohort study Gut, 2007.PMID 16709660