Psych · foundations — psychiatric genetics and epigenetics
Psychiatric genetics and epigenetics
Also known as Psychiatric genomics · Heritability mental disorders · GWAS schizophrenia depression · Polygenic risk score psychiatry · Copy number variants psychosis · 22q11.2 deletion syndrome psychiatry · Epigenetics mental illness · Genetic counselling psychiatry · Gene environment interaction psychiatry
Exam-exhaustive psychiatric genetics and epigenetics for FRANZCP and MRCPsych: heritability and twin/adoption designs; liability-threshold and ACE models; GWAS architecture of schizophrenia and depression; CNVs including 22q11.2DS; polygenic risk scores and clinical limits; epigenetics as GxE mechanism; genetic counselling basics and ethics. FRANZCP-primary, globally tagged.
On this page & tools
Your progress
Saved locally on this device.
10 MCQs with explanations
Target exams
Red flags
1. Definitions examiners demand
Psychiatric genetics studies how DNA sequence variation (common SNPs, rare variants, structural variants) and de novo mutations contribute to risk for mental disorders and related traits. Psychiatric genomics emphasises genome-wide methods at scale (GWAS, sequencing, CNV arrays).[5][6]
Heritability (h2) is the proportion of phenotypic variance in a defined population and environment attributable to genetic differences. It is not the percentage of an individual's illness caused by genes, not a measure of immutability, and not fixed if environments change.[1][3]
| Term | Precise meaning | Classic trap |
|---|---|---|
| Narrow-sense h2 | Additive genetic variance / phenotypic variance | Equated with "inherited destiny" |
| Twin h2 | Estimated from MZ/DZ resemblance (ACE models) | Assumed equal environments always perfect |
| SNP-h2 | Variance tagged by common genotyped SNPs | Confused with twin h2 (usually lower) |
| GWAS | Association scan across common variants | "Found the gene" for a complex trait |
| CNV | Deletion/duplication of DNA segment | Treated as present in all cases |
| PRS/PGS | Weighted sum of risk alleles | Used as diagnostic test in clinic |
| Epigenetics | Regulation of expression without DNA sequence change | Confused with Lamarckian inheritance of acquired disease |
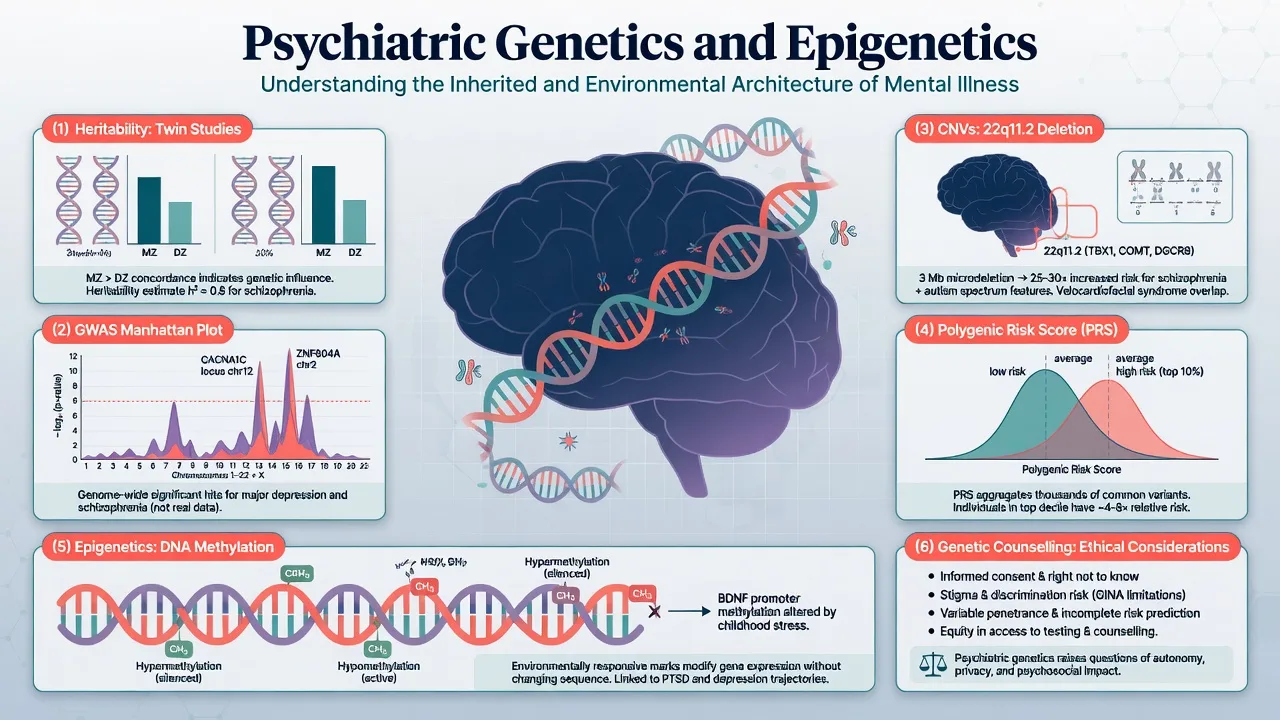
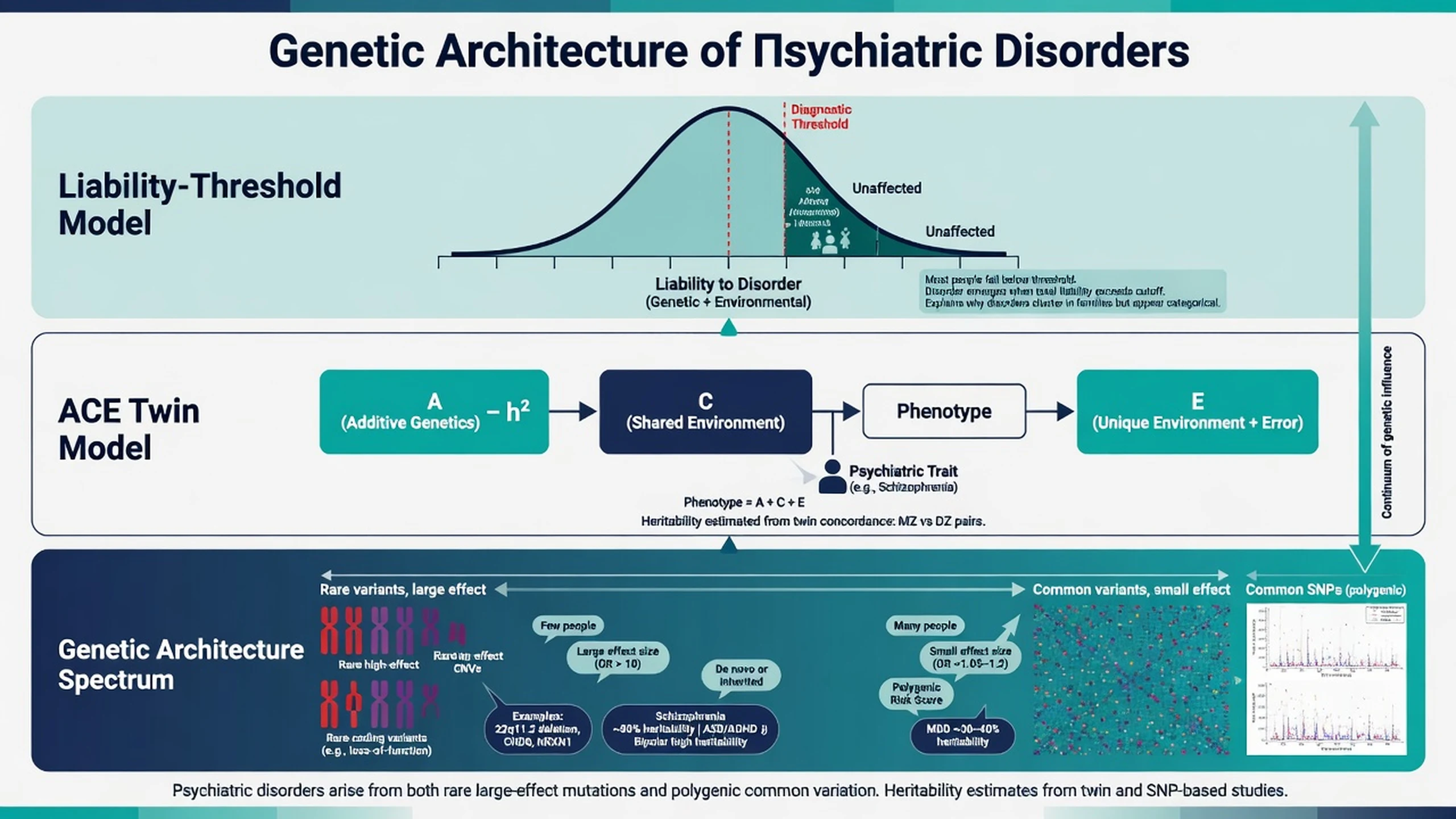
Liability-threshold (Falconer) model. Risk factors push a continuous liability distribution; diagnosis occurs when liability crosses a threshold. This reconciles polygenic inheritance with categorical DSM/ICD diagnoses and explains why relatives of severe cases have elevated risk — the same framework underlying twin analyses of schizophrenia and depression liability.[1][3]
2. Classical designs: family, twin, adoption
2.1 Family studies
First-degree relatives of people with schizophrenia have roughly an order-of-magnitude higher lifetime risk than the general population (~1%); risk falls with genetic distance and rises with multiple affected relatives. Similar familial aggregation holds for bipolar disorder, major depression, autism, and ADHD, with different absolute risks and heritabilities.[1][3][4]
2.2 Twin studies and ACE decomposition
Monozygotic (MZ) twins share essentially all segregating genes; dizygotic (DZ) twins share half on average like siblings. Greater MZ than DZ concordance implies genetic influence. The ACE model partitions variance into additive genetics (A), shared/common environment (C), and unique environment plus measurement error (E) — the decomposition used in the schizophrenia and depression twin meta-analyses.[1][2][3]
Sullivan and colleagues' meta-analysis of schizophrenia twin studies estimated heritability of liability about 81% (95% CI roughly 73–90%), with little shared environment under the fitted models — a staple exam number, always stated with the incomplete MZ concordance caveat.[1] Danish national twin data update heritability of schizophrenia to about 79%, with probandwise MZ concordance far below 100% (about one-third in that analysis), proving non-genetic contribution even for a highly heritable phenotype.[2]
For major depression, Sullivan and colleagues' meta-analysis estimated heritability of liability about 37% (roughly 31–42%), with substantial individual-specific environment and negligible shared environment in the summary twin models; recurrence is a key predictor of familial aggregation.[3] McGuffin and colleagues showed high heritability of bipolar disorder and substantial genetic correlation between mania and depression dimensions.[4]
2.3 Adoption studies
Adoption designs separate genetic relatedness from rearing environment. Elevated risk of schizophrenia in biological relatives of affected adoptees (versus adoptive relatives) supports genetic transmission beyond the shared household — examinable logic even when individual historic studies are not memorised cell-by-cell.[1]
3. Molecular architecture: GWAS and common variants
3.1 What GWAS does
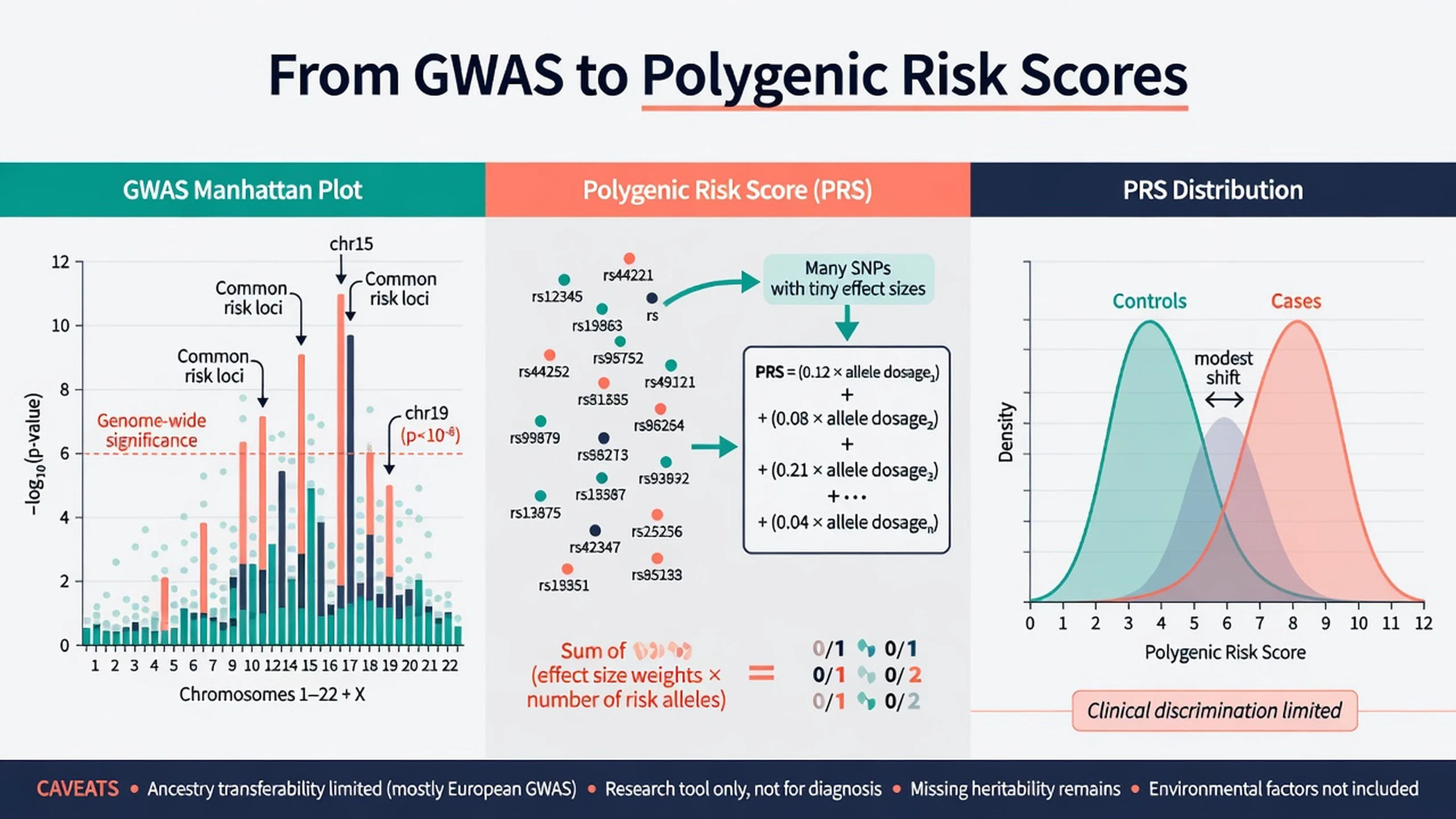
A genome-wide association study tests millions of common SNPs for case–control association. Multiple-testing control conventionally uses genome-wide significance near 5×10^-8. Sample size is destiny for polygenic traits: early underpowered studies failed; consortium meta-analyses succeeded.[5][6]
3.2 Schizophrenia
The PGC Schizophrenia Working Group identified 108 independent genome-wide significant loci, with biological signals including synaptic genes and immune/MHC regions among other pathways — a foundational 2014 result every registrar must name.[5] Trubetskoy and colleagues (2022) expanded the common-variant map substantially in multi-ancestry analyses and prioritised genes and synaptic biology at larger scale.[6]
3.3 Major depression
Wray and colleagues' PGC depression GWAS identified 44 risk variants and refined architecture: highly polygenic risk with correlations to educational attainment, body mass, and schizophrenia genetic risk — examiners want "polygenic, large-N dependent," not a single depression gene.[7]
3.4 Cross-disorder pleiotropy
The Cross-Disorder Group of the PGC showed shared genetic effects across five major psychiatric disorders and later mapped genomic relationships and pleiotropy across eight disorders, helping explain clinical comorbidity at the genomic level.[9][10]
3.5 Missing heritability
Twin h2 typically exceeds SNP-h2. Candidate explanations: rare variants and CNVs not tagged by common SNPs; imperfect LD tagging; structural variation; gene–gene and gene–environment interactions; phenotype heterogeneity and measurement error. Do not invent a single preferred fraction in viva — list mechanisms.[5][11][15]
4. Polygenic risk scores (PRS/PGS)
The International Schizophrenia Consortium showed that common polygenic variation contributes to schizophrenia risk and overlaps bipolar disorder — the intellectual origin of modern PRS applications in psychiatry.[8]
Construction (exam-level): sum of risk alleles weighted by GWAS effect sizes from an independent discovery sample, applied to a target individual of compatible ancestry. More alleles of small effect can yield scores that differ between case and control groups at population scale.[8][17]
Clinical utility today: Lewis and Vassos review PRS as promising research and future risk-stratification tools whose current discriminative ability is low in the general population for most psychiatric outcomes; they are not substitutes for clinical diagnosis, and ancestry mismatch degrades performance.[17] Exam stance: research tool; not routine diagnostic test; never sole basis for treatment choice or reproductive decisions.
5. Rare variants and CNVs (including 22q11.2)
5.1 CNV revolution
Malhotra and Sebat synthesised evidence that rare copy-number variants play causal roles especially in autism spectrum disorders, schizophrenia, and developmental delay — a "rare variant revolution" complementary to common-variant GWAS.[11] Marshall and colleagues quantified CNV contribution to schizophrenia in over 40,000 subjects, confirming enrichment of large rare CNVs and nominating loci of clinical relevance.[12]
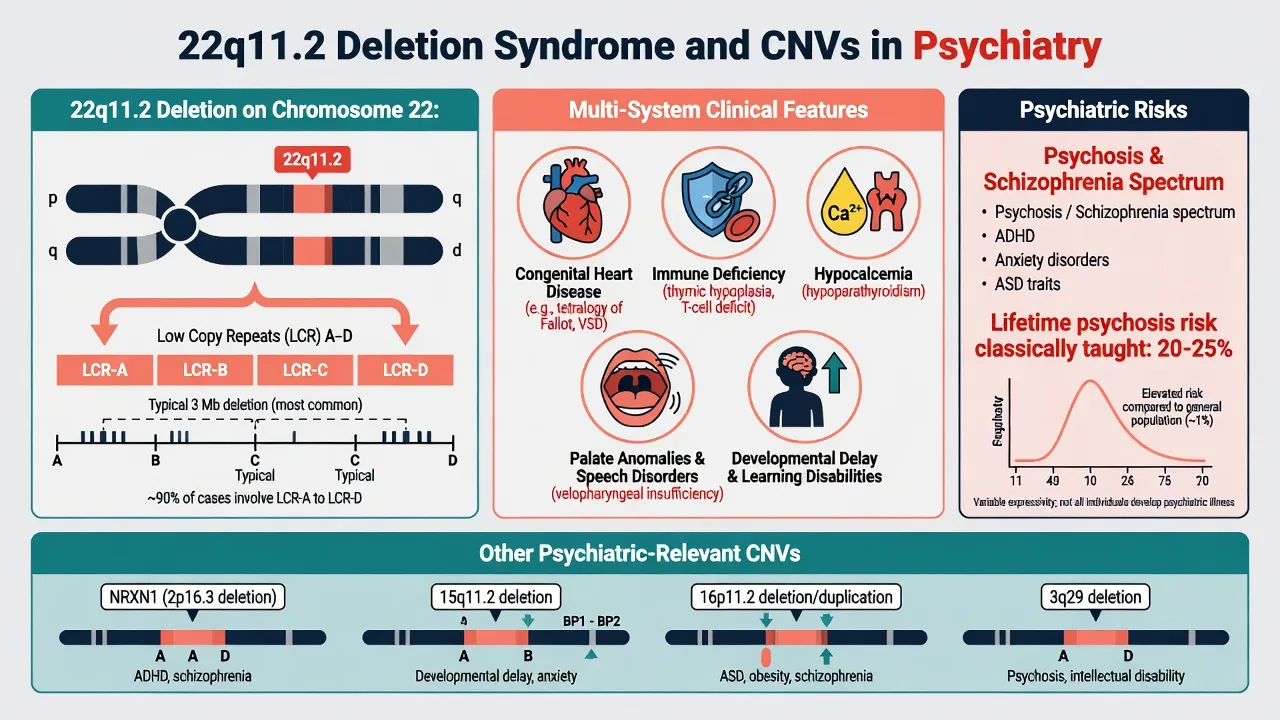
High-yield CNV loci (name recognition): 22q11.2 deletion, NRXN1 disruptions, 15q11.2, 16p11.2, 3q29, 1q21.1, and others — often with neurodevelopmental comorbidity (ID, epilepsy, congenital anomalies).[11][12]
5.2 Rare coding variants
SCHEMA consortium sequencing work showed rare coding variants in a set of genes confer substantial schizophrenia risk — rare alleles of larger effect sitting on the same liability continuum as polygenic common variation.[15]
5.3 22q11.2 deletion syndrome (22q11.2DS)
22q11.2DS is the most common chromosomal microdeletion syndrome (classically ~1 in 2000–4000 live births depending on ascertainment), usually de novo, with multisystem features: conotruncal cardiac defects, palatal anomalies, immune deficiency, hypocalcaemia, characteristic facial features, developmental delay, and high psychiatric morbidity.[13][14]
Psychiatry yield: elevated rates of psychosis/schizophrenia-spectrum disorders (classically taught lifetime risk on the order of 20–25% in many teaching sources), ADHD, anxiety disorders, and autism-spectrum features; adult neuropsychiatric care is essential as paediatric survival improves.[13][14] Bassett and the International 22q11.2 Deletion Syndrome Consortium published practical paediatric management guidelines; McDonald-McGinn and colleagues provide the disease primer synthesising medical and developmental complexity.[13][14]
Clinical pearl: new-onset psychosis plus subtle dysmorphology, hypocalcaemia, recurrent infection history, or repaired congenital heart disease should trigger consideration of 22q11 testing via clinical genetics pathways — not every first-episode psychosis needs microarray, but syndromic clues change the pretest probability.[13][14]
6. Epigenetics and gene–environment interplay
Epigenetics (exam definition): stable regulation of gene expression through DNA methylation, histone modifications, and non-coding RNAs without changing the DNA sequence. Mitotic heritability of marks is not the same as transgenerational inheritance of psychiatric disease in humans — avoid overclaiming.[16]
Weaver, Meaney, Szyf and colleagues showed that maternal care behaviours in rats alter offspring hippocampal glucocorticoid receptor promoter methylation and stress reactivity — the canonical molecular illustration of early environment programming gene expression.[16] Use this as a mechanism paradigm for GxE thinking, not as a clinical biomarker assay you order on the ward.
GxE vocabulary (conceptual scaffolding for interpreting twin residual environment and molecular embedding work such as the maternal-care methylation paradigm): Additive (genetic and environmental risks sum); Interaction (genetic effect differs by environment); Gene–environment correlation (rGE) (genotype influences exposure probability). Epigenetic processes are candidate mediators of lasting environmental effects; human psychiatric epigenetic epidemiology remains methodologically challenging (tissue specificity, reverse causation, cell-type heterogeneity). Clinical stance: no routine epigenetic diagnostic panel for MDD or schizophrenia in standard practice.[16][3][5]
7. Assessment: pedigree, red flags, investigations
7.1 Bedside genetics for psychiatrists
- Three-generation pedigree — mental illness, suicide, substance use, intellectual disability, epilepsy, congenital anomalies, infertility/recurrent miscarriage, consanguinity, adoption.[1][3]
- Age at onset and course — early onset and recurrence increase familial loading signals for mood disorders.[3]
- Syndromic screen — dysmorphology, short stature, cleft/palate history, hypocalcaemia, immune issues, cardiac surgery scars → consider 22q11/CNV pathway.[13][14]
- Capacity and family privacy — testing one person can reveal information about relatives; plan counselling before molecular tests when feasible.[13][17]
7.2 When to involve clinical genetics
Involve clinical genetics for known or suspected chromosomal syndrome, multiple congenital anomalies, or unexplained developmental delay; for formal recurrence-risk or reproductive counselling after a molecular diagnosis; and for cascade testing when a pathogenic CNV is already identified in a relative.[11][12][13][14]
Not indicated routinely: genome-wide testing for typical late-adolescent MDD without developmental red flags; commercial multi-gene "psych panels" sold as diagnostic certainty.[17]
7.3 Pharmacogenetics is adjacent, not identical
CYP2D6/CYP2C19 variation can inform drug metabolism for some antidepressants/antipsychotics in selected pathways — distinct from disease-risk genetics and from polygenic disorder scores. Do not conflate a pharmacogenetic report with a schizophrenia diagnosis or a clinical PRS.[5][17]
8. Management implications (what genomics changes — and does not)
Does not replace standard acute care for psychosis, depression, mania, or suicidality. First-episode pathways, metabolic monitoring, clozapine algorithms, and psychological therapies remain phenotype-driven.[5][6]
Does change: formulation (predisposing genetic loading with precipitating stress and perpetuating social factors); medical comorbidity surveillance in 22q11.2DS and other CNV syndromes; family psychoeducation with accurate risk language; research enrolment only with proper consent, kept distinct from clinical care.[1][2][3][13][14][17]
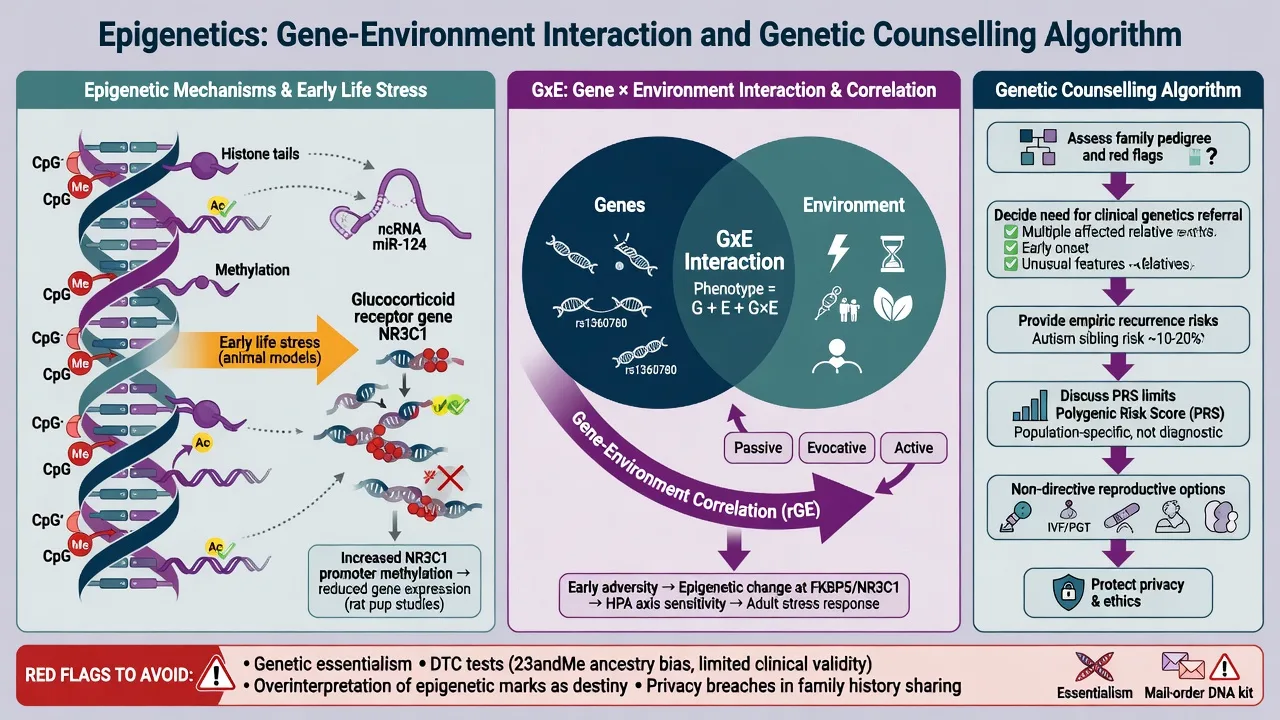
8.1 Genetic counselling basics (registrar-level)
Elicit agenda (blame, reproductive plans, testing requests); explain the multifactorial model with absolute and relative risks; give empiric recurrence risks by relationship when no pathogenic variant is known, or molecular risks when one is known; remain non-directive on reproductive options with clinical genetics involvement for formal sessions; document and offer a written summary; provide psychosocial support because results can relieve or intensify guilt.[1][8][13][14][17]
[1] [13] [14]9. Ethics, stigma, and professional practice
- Autonomy and non-directiveness in reproductive genetics.
- Privacy and duty to relatives — carefully balance confidentiality with serious preventable risks; follow local legal/professional guidance (jurisdiction-specific; do not invent section numbers).
- Justice and ancestry equity — Eurocentric GWAS limit PRS accuracy for many populations; acknowledge and avoid false reassurance/alarm.[17]
- Anti-stigma: genes contribute to risk for many people; they do not define moral worth or inevitability of violence.
- Forensic limit: genetic findings do not establish criminal responsibility; resist genetic essentialism in court reports.
- Commercial DTC testing: counsel on analytic validity, clinical validity, clinical utility gaps, and secondary findings.
10. Special populations
- Children and youth: higher yield of CNV diagnostics in developmental delay/ASD clinics; family counselling must match developmental stage.[11][14]
- Perinatal: separate medication teratogenicity discussions from inheritance risk; offer genetics referral when a parental pathogenic CNV is known.[13]
- Intellectual disability: dual diagnosis; lower threshold for chromosomal microarray in unexplained ID with behavioural phenotypes.[11]
- Older adults: late-onset presentations less often explained by highly penetrant CNVs; do not force a genetic narrative.
- Diverse ancestries: interpret PRS and some GWAS-derived claims cautiously.[17]
11. Evidence map (what to cite in essays)
| Claim | Anchor |
|---|---|
| SCZ twin heritability ~80% | Sullivan 2003; Hilker 2018 Danish registry |
| MDD twin heritability ~37% | Sullivan 2000 meta-analysis |
| Bipolar highly heritable; links to unipolar | McGuffin 2003 |
| 108 SCZ GWAS loci | PGC Nature 2014 |
| Expanded SCZ common-variant map / synaptic biology | Trubetskoy 2022 |
| MDD multi-locus GWAS | Wray 2018 |
| Polygenic schizophrenia–bipolar overlap | ISC Nature 2009 |
| Cross-disorder shared genetics | PGC Cross-Disorder 2013, 2019 |
| CNV role in psychiatry | Malhotra and Sebat 2012; Marshall 2017 |
| Rare coding SCZ genes | SCHEMA 2022 |
| 22q11 clinical care | Bassett 2011; McDonald-McGinn 2015 |
| Epigenetic programming paradigm | Weaver/Meaney 2004 |
| PRS clinical limits | Lewis and Vassos 2020 |
12. Exam pearls (rapid-fire)
- Heritability = population statistic, not personal percent genetic.[1][3]
- Incomplete MZ concordance = environment/stochasticity matter.[2]
- GWAS threshold ~5×10^-8; sample size unlocks polygenic signals.[5]
- PRS aggregates small effects; not a clinic diagnostic test today.[8][17]
- 22q11.2 deletion: multisystem + high psychosis relative risk; use guidelines and multidisciplinary care.[13][14]
- Cross-disorder pleiotropy helps explain comorbidity.[9][10]
- Epigenetics: mechanism story from animal models; not routine biomarkers.[16]
- Counselling is non-directive; protect privacy and autonomy.[13][17]
- Pharmacogenetics ≠ disease genetics.[17]
- Never invent "the schizophrenia gene."[5][8]
GENE-PRISM (psychiatric genetics stack)
13. Related topics
Link to schizophrenia spectrum, first-episode psychosis, bipolar, MDD, ASD/ADHD for phenotype management; formulation for integrating genetic predisposition; ethics for consent/privacy; critical appraisal for interpreting GWAS odds ratios and PRS AUC claims.[5][10][17]
References
- [1]Sullivan PF, Kendler KS, Neale MC Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies Arch Gen Psychiatry, 2003.PMID 14662550
- [2]Hilker R, Helenius D, Fagerlund B, et al. Heritability of Schizophrenia and Schizophrenia Spectrum Based on the Nationwide Danish Twin Register Biol Psychiatry, 2018.PMID 28987712
- [3]Sullivan PF, Neale MC, Kendler KS Genetic epidemiology of major depression: review and meta-analysis Am J Psychiatry, 2000.PMID 11007705
- [4]McGuffin P, Rijsdijk F, Andrew M, et al. The heritability of bipolar affective disorder and the genetic relationship to unipolar depression Arch Gen Psychiatry, 2003.PMID 12742871
- [5]Schizophrenia Working Group of the Psychiatric Genomics Consortium Biological insights from 108 schizophrenia-associated genetic loci Nature, 2014.PMID 25056061
- [6]Trubetskoy V, Pardiñas AF, Qi T, et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia Nature, 2022.PMID 35396580
- [7]Wray NR, Ripke S, Mattheisen M, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression Nat Genet, 2018.PMID 29700475
- [8]International Schizophrenia Consortium; Purcell SM, Wray NR, Stone JL, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder Nature, 2009.PMID 19571811
- [9]Cross-Disorder Group of the Psychiatric Genomics Consortium Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis Lancet, 2013.PMID 23453885
- [10]Cross-Disorder Group of the Psychiatric Genomics Consortium Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders Cell, 2019.PMID 31835028
- [11]Malhotra D, Sebat J CNVs: harbingers of a rare variant revolution in psychiatric genetics Cell, 2012.PMID 22424231
- [12]Marshall CR, Howrigan DP, Merico D, et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects Nat Genet, 2017.PMID 27869829
- [13]Bassett AS, McDonald-McGinn DM, Devriendt K, et al. Practical guidelines for managing patients with 22q11.2 deletion syndrome J Pediatr, 2011.PMID 21570089
- [14]McDonald-McGinn DM, Sullivan KE, Marino B, et al. 22q11.2 deletion syndrome Nat Rev Dis Primers, 2015.PMID 27189754
- [15]Singh T, Poterba T, Curtis D, et al. Rare coding variants in ten genes confer substantial risk for schizophrenia Nature, 2022.PMID 35396579
- [16]Weaver ICG, Cervoni N, Champagne FA, et al. Epigenetic programming by maternal behavior Nat Neurosci, 2004.PMID 15220929
- [17]Lewis CM, Vassos E Polygenic risk scores: from research tools to clinical instruments Genome Med, 2020.PMID 32423490