Psych · Foundations — psychoneuroendocrinology and psychoimmunology
Psychoneuroendocrinology and psychoimmunology
Also known as Psychoneuroimmunology · HPA axis psychiatry · Stress biology psychiatry · Cytokine hypothesis of depression · Sickness behaviour · Allostatic load mental health · Glucocorticoid receptor depression
Exam-exhaustive fellowship foundation on psychoneuroendocrinology and psychoimmunology: HPA/HPG/HPT axes, allostatic load, glucocorticoid receptor resistance, tuberoinfundibular prolactin pharmacology, cytokine–brain signalling, sickness behaviour, inflammatory depression and psychosis risk evidence, investigation limits, and clinical application without biomarker overclaim. FRANZCP-primary, globally tagged.
On this page & tools
Your progress
Saved locally on this device.
10 MCQs with explanations
Target exams
Red flags
Overview and definition
Fellowship exams (FRANZCP theory and clinical reasoning; MRCPsych Paper A/B; ABPN blueprint; MD/DNB viva) expect a fluent map from stress biology to clinical syndromes without collapsing psychiatry into a single chemical or a single cytokine. Psychoneuroendocrinology studies how hypothalamic–pituitary axes shape mood, cognition, arousal, reproduction, and drug adverse effects. Psychoimmunology (psychoneuroimmunology) studies how neural and immune systems regulate each other, including cytokine-driven changes in motivation and affect. [1][4][9]
| Domain | Core construct | Exam use |
|---|---|---|
| HPA axis | CRH → ACTH → cortisol; GR/MR feedback | Depression models, trauma, DST history |
| HPG / HPT | Sex steroids; thyroid hormones | Perinatal, lithium–thyroid, sexual function |
| Prolactin | Tuberoinfundibular dopamine inhibition | Antipsychotic adverse effects |
| Allostasis | Adaptive set-point change; load when chronic | Formulation language for chronic stress |
| Cytokine–brain | Sickness behaviour; inflammatory depression | Medical illness, IFN models, research adjuncts |
| Scaffold for multilevel viva answers. [1][2][4] |
What this topic is for. To answer mechanism questions, interpret physical-health labs, and avoid biomarker myths. What it is not. A substitute for endocrine psychiatry of primary Cushing/thyroid disease, autoimmune encephalitis work-up, descriptive psychopathology, or jurisdiction-specific mental health law. Cross-link those clinical topics when red flags appear. [2][16]
Classification of axes and frameworks
Clinical psychiatry still diagnoses with DSM-5-TR / ICD-11. Endocrine and immune findings are dimensional modifiers of risk, phenotype, and physical health — not free-standing diagnoses of "HPA disorder" as a psychiatric code. Research frameworks (e.g. RDoC arousal/regulatory systems) may organise constructs, but they do not replace clinical manuals. [2][6]
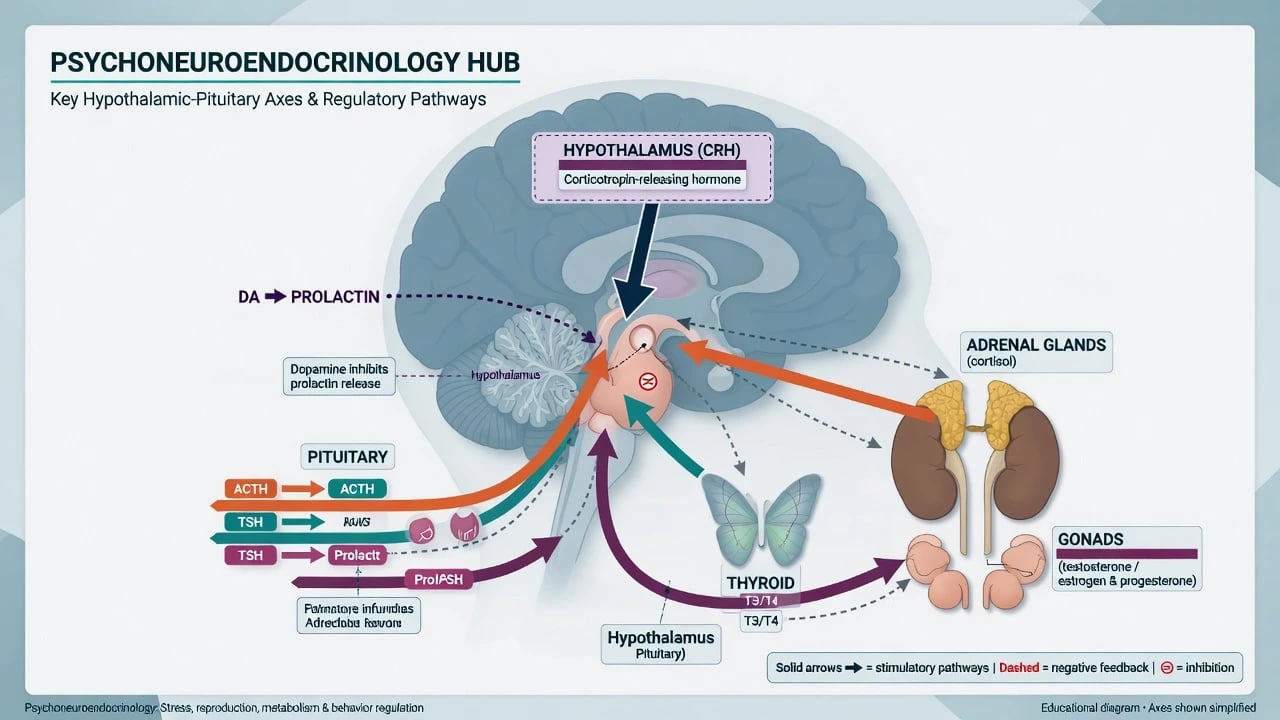
Major axes examiners expect
- HPA — limbic and hypothalamic CRH drive pituitary ACTH and adrenal cortisol; diurnal rhythm; negative feedback via mineralocorticoid (MR) and glucocorticoid (GR) receptors in hippocampus, hypothalamus, and pituitary. [2][13]
- HPT — TRH–TSH–T3/T4; clinically essential for differential of mood/psychosis and for lithium monitoring (detailed in endocrine psychiatry and lithium topics).
- HPG — GnRH–gonadotropins–sex steroids; links to libido, menstrual function, perinatal transitions, and hypogonadism secondary to hyperprolactinaemia. [15]
- Prolactin — tonically inhibited by tuberoinfundibular dopamine; D2 blockade removes the brake → hyperprolactinaemia. [15]
Pathophysiology: HPA, glucocorticoids, and allostatic load
Cascade and feedback
Stress and limbic inputs increase hypothalamic CRH, pituitary ACTH, and adrenal cortisol. Cortisol exerts permissive, stimulatory, suppressive, and preparative actions across systems — not a simple on/off stress switch. [13] Hippocampal and hypothalamic GR/MR signalling normally restrain the axis; impaired feedback perpetuates hypercortisolism. [2][3]
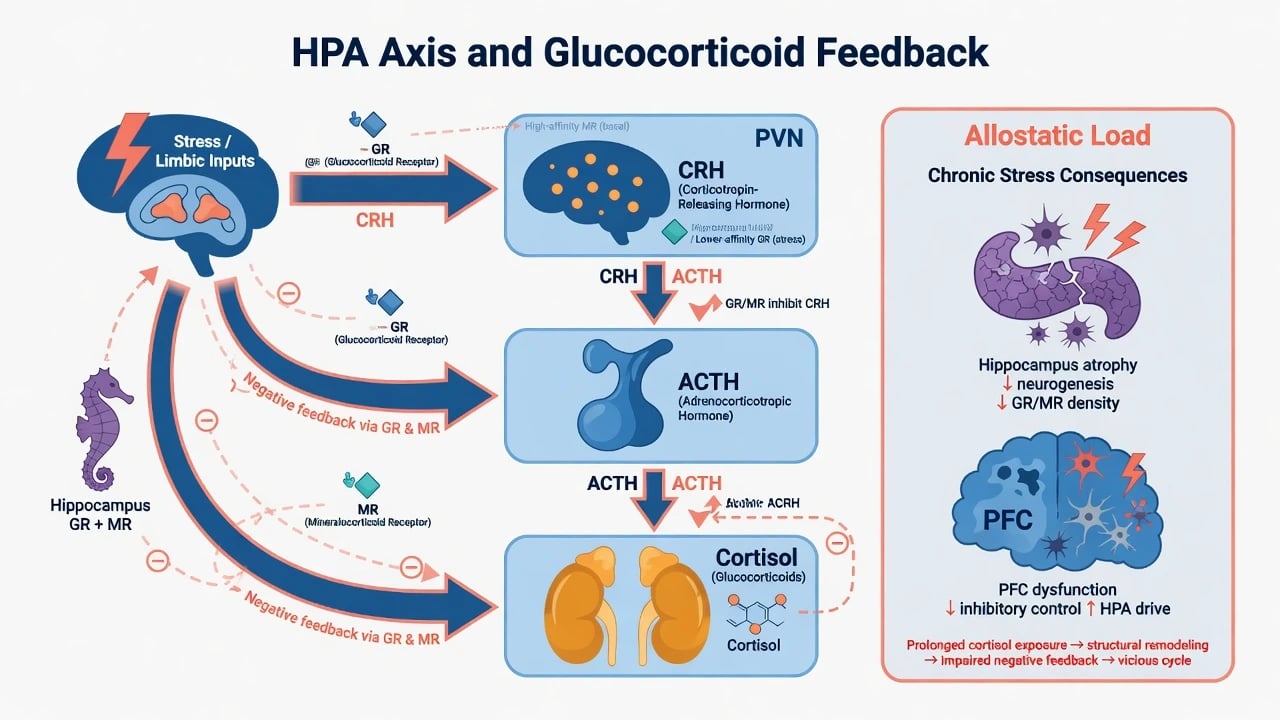
Depression models (viva depth)
Classical findings include basal hypercortisolism, flattened diurnal rhythm, and reduced feedback sensitivity in many depressed samples — especially severe/melancholic presentations. [2] Holsboer's corticosteroid receptor hypothesis emphasises impaired GR signalling as central to HPA dysregulation and as a target of antidepressant action over time. [3] Gold and Chrousos organised melancholic depression as a relatively high CRH/noradrenergic state versus atypical features as a relatively low CRH/noradrenergic organisation — a useful viva contrast, not a lab test. [11]
Childhood trauma can program enduring HPA and stress-reactivity changes relevant to later mood and anxiety disorders. [12] Glucocorticoids remodel neural circuits (hippocampus, amygdala, prefrontal cortex) and interact with neurotrophic stress models of mood illness. [1][13]
Psychoimmunology: cytokines, sickness behaviour, and brain signalling
Sickness behaviour
Acute infection or inflammation triggers an adaptive behavioural program — sickness behaviour — including fatigue, anhedonia, social withdrawal, anorexia, sleep change, and psychomotor slowing. When prolonged or contextually inappropriate, this phenotype overlaps major depression and offers a mechanistic model rather than a competing diagnosis. [4][5]
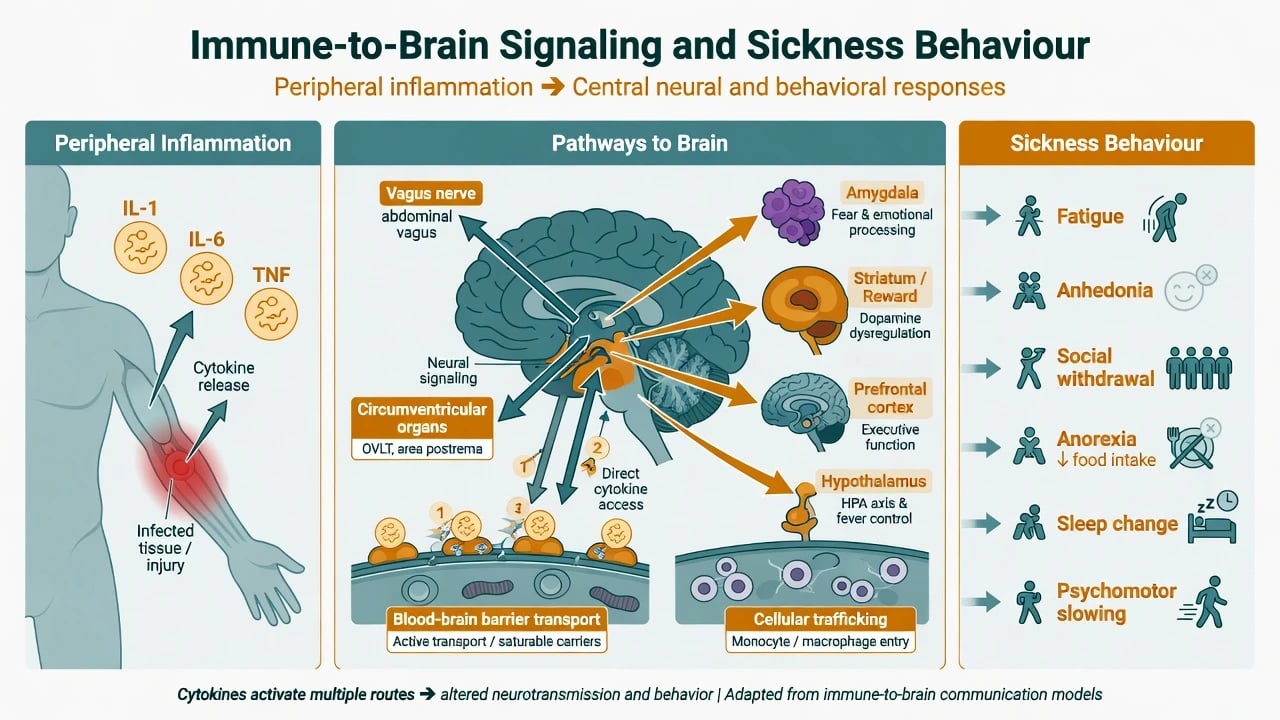
Routes and mechanisms
Immune-to-brain signalling uses neural pathways (including vagal afferents), humoral access at circumventricular organs and transporters, and cellular trafficking. Centrally, cytokines alter monoamine metabolism, glutamate signalling, and motivation/reward circuits; the kynurenine pathway is a frequently examined molecular bridge at overview depth. [7][10] Reciprocally, sympathetic and HPA outputs reprogram immune function — the neural and innate immune systems co-regulate rather than act in parallel isolation. [9]
IFN-alpha and related cytokine therapies produce a depression-like syndrome in a substantial minority of patients and remain a classic human model of cytokine-driven mood change. [5][7] Contemporary syntheses frame inflammation as an evolutionary trade-off that becomes a modern treatment research target in a subset of depressed patients — not the cause of all depression. [6]
Epidemiology and risk
| Risk domain | What evidence supports | Exam caution |
|---|---|---|
| Early adversity | Developmental HPA programming; later mood/anxiety risk | Biology does not erase social determinants [12] |
| Childhood inflammation | Higher childhood IL-6/CRP associated with later depression and psychosis risk in longitudinal cohorts | Association ≠ destiny or individual prediction [8] |
| Autoimmunity / severe infection | Elevated later schizophrenia-spectrum risk in large registers | Not proof that most psychosis is autoimmune encephalitis [16] |
| Iatrogenic endocrine | Antipsychotic hyperprolactinaemia common with strong D2 blockade | Agent-specific liability [15] |
| Medical inflammation | Higher depression rates in inflammatory diseases; cytokine therapies | Treat medical disease and depression together [5][6] |
Sex differences in HPA/HPG regulation and autoimmune prevalence appear in epidemiology; do not over-extend into unsupported biological essentialism in viva answers. [1][9]
Clinical presentation and MSE translation
Translate biology into MSE language, not lab printouts. Map HPA hyperdrive and high-arousal states to insomnia, anxiety, agitation, cognitive inefficiency, and melancholic features; map sickness or inflammatory phenotypes to fatigue, slowing, social withdrawal, reduced appetite, and low motivation; map hyperprolactinaemia to galactorrhoea, amenorrhoea or oligomenorrhoea, sexual dysfunction, hypogonadal symptoms, and longer-term bone risk; and treat primary endocrine red flags (cushingoid habitus, myxoedema features, adrenal crisis physiology) as medical pathways rather than pure functional stress. [2][4][11][15]
Differential diagnosis
| Presentation | Favours primary psychiatric syndrome | Favours primary medical/endocrine/immune disease |
|---|---|---|
| Depression + raised CRP | Idiopathic MDD with inflammatory comorbidity or lifestyle factors | Active infection, autoimmune flare, malignancy |
| Depression + hypercortisol signals | Stress-related HPA activation without endocrine disease | Cushing syndrome features, progressive cognition, hypokalaemia |
| Psychosis + mild inflammatory markers | Primary schizophrenia-spectrum illness | Autoimmune encephalitis pattern, delirium, systemic sepsis |
| Sexual dysfunction on antipsychotics | Drug-induced hyperprolactinaemia or other drug effects | Primary pituitary disease, pregnancy, hypothyroidism |
| Discriminators are time course, physical signs, investigations indicated by red flags, and syndrome structure — not a single cytokine number. [2][6][15][16] |
Assessment
History structure. Early trauma and chronic stress load; inflammatory diseases; cytokine therapies (IFN, IL-2); exogenous steroids; antipsychotic class and sexual/menstrual symptoms; thyroid symptoms; weight, sleep, substances. [12][15]
Risk. Suicide and violence assessment remains clinical. CRP, cortisol, and IL-6 do not triage risk. [6]
Communication. Accurate plain language: "Stress and inflammation can change brain systems that control energy, motivation and sleep; that helps explain symptoms in some people; we still diagnose by history and mental state, and we treat both the mental illness and physical health." Avoid chemical-imbalance cartoons and false certainty. [4][6]
Investigations
| Test | Role in this topic | Pitfall |
|---|---|---|
| Thyroid panel, glucose, lipids, electrolytes, FBC, LFT | Baseline physical health and psychotropic safety | Skipping metabolic monitoring |
| Prolactin | Symptomatic patients or high-risk agents | Ignoring amenorrhoea/galactorrhoea; forgetting pregnancy test when relevant [15] |
| Morning cortisol / DST | Historical research and endocrine diagnosis of Cushing when clinically indicated | Using Carroll-era DST as a modern diagnostic for MDD [14][2] |
| CRP / IL-6 | Research stratification interest; medical illness work-up | Treating as DSM diagnostic tests [6][8] |
| MRI / LP / EEG / autoimmune panels | When organic/autoimmune red flags present | Ordering to "prove stress axis dysfunction" |
The dexamethasone suppression test was standardised as a laboratory approach to melancholia in a landmark 1981 programme, but poor sensitivity/specificity and limited clinical utility mean it is not a routine diagnostic for depression today. [14][2]
Acute and emergency management
Medical emergencies at the interface include adrenal crisis, thyroid storm, severe corticosteroid-induced mania/psychosis, and behavioural presentations of delirium, NMS, or serotonin syndrome. Stabilise airway, circulation, temperature, and electrolytes; reverse endocrine crises with medical protocols; only then refine psychiatric formulation. Neuroscience framing must never delay safety or medical rescue. [13][16]
Definitive management and clinical application
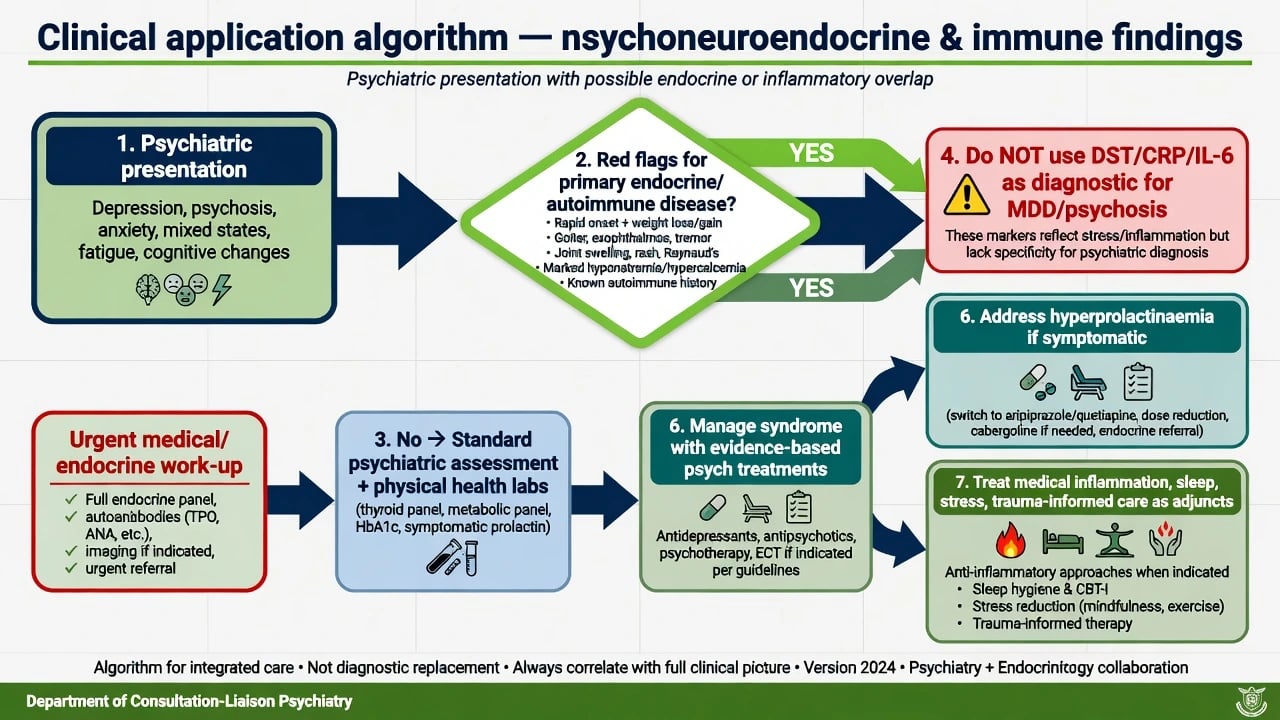
Principles
- Treat the psychiatric syndrome with evidence-based care (antidepressants, antipsychotics, mood stabilisers, psychological therapies, social interventions as indicated by the disorder). Endocrine/immune concepts refine formulation and physical health, they do not replace guidelines. [2][6]
- Hyperprolactinaemia management. Confirm symptoms and levels; exclude pregnancy where relevant; consider dose reduction if psychosis control allows, switch to lower-prolactin agents (e.g. aripiprazole, quetiapine), or other evidence-informed strategies; refer endocrinology for persistent symptomatic cases or uncertain aetiology. [15]
- Inflammatory depression research. Anti-inflammatory or anti-cytokine adjuncts remain investigational/selected-context for most candidates; do not claim them as universal first-line depression care in ANZ or NICE/APA-aligned practice without indication. [6][10]
- Biologically coherent adjuncts. Sleep restoration, exercise, treating active medical inflammation, and trauma-informed psychotherapies are compatible with HPA and immune models. [1][12]
RANZCP, NICE, and APA depression and psychosis guidance centre clinical diagnosis, risk, functional recovery, and physical health monitoring (metabolic, endocrine adverse effects). They do not require routine HPA dynamic testing or cytokine panels to diagnose MDD or schizophrenia. Use regional deltas when discussing resource-limited testing or clozapine/metabolic pathways, not when inventing biomarker mandates. [2][6][15]
Subtypes and high-yield scenarios
| Scenario | Mechanism pearl | Clinical move |
|---|---|---|
| Melancholic MDD | Relative HPA hyperdrive; GR feedback issues | Standard severe-depression care; not DST diagnosis [2][11] |
| Atypical features | Contrasting stress-system organisation narrative | Phenotype-informed formulation [11] |
| PTSD / early trauma | Developmental HPA programming | Trauma-focused care + safety [12] |
| Medical illness / IFN | Cytokine-driven sickness–depression overlap | Coordinate with medical team; treat depression [5][7] |
| Schizophrenia-spectrum | Mild inflammatory signals; infection/autoimmune risk elevation at population level | Organic red-flag screen; standard psychosis care [8][16] |
| Risperidone / FGA / amisulpride | High prolactin liability | Symptom enquiry + prolactin strategy [15] |
Complications and pitfalls
- Diagnosing depression with DST or a single cortisol. [14][2]
- Equating raised CRP with antibiotic need or mandatory anti-inflammatory psychiatric prescription. [6]
- Ignoring symptomatic hyperprolactinaemia until infertility or fracture risk accumulates. [15]
- Missing primary Cushing, Addison, thyroid disease, or autoimmune encephalitis by over-attributing to functional stress. [16]
- Claiming inflammation causes all depression. [6]
- Replacing suicide risk assessment with biomarkers. [6]
Prognosis and disposition
Chronic allostatic load and untreated medical comorbidity contribute to multi-morbidity and poorer function. Disposition still follows risk, function, supports, and syndrome severity. Physical health monitoring (metabolic syndrome, prolactin symptoms, thyroid on lithium) is part of psychiatric outcome, not optional extras. [1][15]
Special populations
- Child/adolescent. Developmental stress programming; risperidone prolactin effects; avoid speculative cytokine treatment claims. [12][15]
- Perinatal. Marked HPG/HPA change; stick to established perinatal psychiatry and psychotropic evidence bases. [2]
- Older adults. Multimorbidity, inflammatory burden, delirium risk; interpret cortisol/CRP in medical context. [1][4]
- Intellectual disability. Behavioural change may signal pain, infection, or endocrine disease — examine and investigate. [16]
- Cultural. Integrate explanatory models of stress, spirit, and immunity without dismissing either biology or meaning. [1]
Evidence and guidelines
Landmarks to name in viva. McEwen on stress mediators and allostatic framing; Sapolsky on glucocorticoid actions; Holsboer GR hypothesis; Pariante–Lightman HPA in depression; Gold–Chrousos stress-system organisation; Dantzer sickness behaviour; Raison–Capuron–Miller and Miller–Raison inflammation–depression syntheses; Capuron–Miller immune-to-brain signalling; Felger–Lotrich mechanisms; Irwin–Cole reciprocal neural–immune regulation; Heim–Nemeroff early trauma; Khandaker childhood IL-6/CRP longitudinal risk; Benros autoimmune/infection and schizophrenia risk; Carroll DST history; Haddad hyperprolactinaemia management. [1][2][3][4][5][6][7][8][9][10][11][12][13][14][15][16]
Exam pearls
CORTISOL exam anchors
- DST ≠ depression diagnosis in contemporary practice. [14][2]
- Sickness behaviour ≠ MDD, but explains medical illness depression risk and symptom overlap. [4]
- High-prolactin agents: risperidone, paliperidone, amisulpride, many FGAs; lower liability: aripiprazole, quetiapine (agent-specific). [15]
- Childhood IL-6/CRP associate with later psychiatric risk — epidemiology, not destiny. [8]
- Never say "antidepressants work only by fixing cortisol" or "depression is just inflammation." [2][6]
References
- [1]McEwen BS Protective and damaging effects of stress mediators N Engl J Med, 1998.PMID 9428819
- [2]Pariante CM, Lightman SL The HPA axis in major depression: classical theories and new developments Trends Neurosci, 2008.PMID 18675469
- [3]Holsboer F The corticosteroid receptor hypothesis of depression Neuropsychopharmacology, 2000.PMID 11027914
- [4]Dantzer R, O'Connor JC, Freund GG, Johnson RW, et al. From inflammation to sickness and depression: when the immune system subjugates the brain Nat Rev Neurosci, 2008.PMID 18073775
- [5]Raison CL, Capuron L, Miller AH Cytokines sing the blues: inflammation and the pathogenesis of depression Trends Immunol, 2006.PMID 16316783
- [6]Miller AH, Raison CL The role of inflammation in depression: from evolutionary imperative to modern treatment target Nat Rev Immunol, 2016.PMID 26711676
- [7]Capuron L, Miller AH Immune system to brain signaling: neuropsychopharmacological implications Pharmacol Ther, 2011.PMID 21334376
- [8]Khandaker GM, Pearson RM, Zammit S, Lewis G, et al. Association of serum interleukin 6 and C-reactive protein in childhood with depression and psychosis in young adult life: a population-based longitudinal study JAMA Psychiatry, 2014.PMID 25133871
- [9]Irwin MR, Cole SW Reciprocal regulation of the neural and innate immune systems Nat Rev Immunol, 2011.PMID 21818124
- [10]Felger JC, Lotrich FE Inflammatory cytokines in depression: neurobiological mechanisms and therapeutic implications Neuroscience, 2013.PMID 23644052
- [11]Gold PW, Chrousos GP Organization of the stress system and its dysregulation in melancholic and atypical depression: high vs low CRH/NE states Mol Psychiatry, 2002.PMID 11920153
- [12]Heim C, Nemeroff CB The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies Biol Psychiatry, 2001.PMID 11430844
- [13]Sapolsky RM, Romero LM, Munck AU How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions Endocr Rev, 2000.PMID 10696570
- [14]Carroll BJ, Feinberg M, Greden JF, Tarika J, et al. A specific laboratory test for the diagnosis of melancholia. Standardization, validation, and clinical utility Arch Gen Psychiatry, 1981.PMID 7458567
- [15]Haddad PM, Wieck A Antipsychotic-induced hyperprolactinaemia: mechanisms, clinical features and management Drugs, 2004.PMID 15456328
- [16]Benros ME, Nielsen PR, Nordentoft M, Eaton WW, et al. Autoimmune diseases and severe infections as risk factors for schizophrenia: a 30-year population-based register study Am J Psychiatry, 2011.PMID 22193673