Psych · intellectual-disability
Behavioural phenotypes and genetic syndromes
Also known as Behavioural phenotype genetic syndrome · Down syndrome psychiatry · Fragile X psychiatric phenotype · Prader-Willi psychosis · Angelman syndrome behaviour · 22q11.2 deletion syndrome psychiatry · Velocardiofacial syndrome schizophrenia · VCFS psychosis risk · DiGeorge psychiatric
Exam-exhaustive behavioural phenotypes of genetic syndromes for FRANZCP and global boards: probabilistic phenotype concept; Down syndrome depression and Alzheimer dementia; fragile X full mutation and premutation spectrum; Prader-Willi hyperphagia and UPD-associated psychosis; Angelman severe ID and seizures; 22q11.2DS/VCFS highest monogenic CNV psychosis risk; genetics work-up; psychopharmacology caveats in ID.
On this page & tools
Your progress
Saved locally on this device.
10 MCQs with explanations
Target exams
Red flags
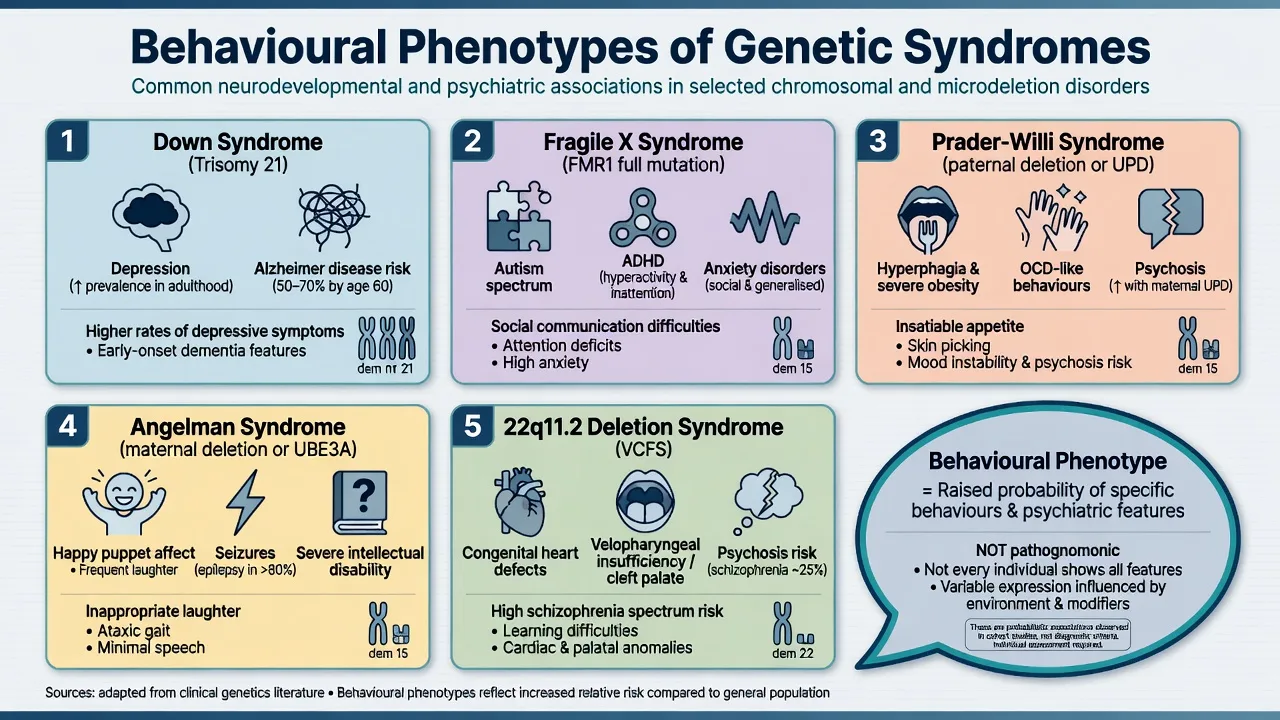
1. Definition and classification
Behavioural phenotype (Dykens). The modern exam definition is probabilistic: a genetic syndrome confers a heightened likelihood of particular cognitive strengths/weaknesses and behavioural or psychiatric features. Features are neither universal nor exclusive to that syndrome, and severity of intellectual disability alone does not fully explain the pattern.[1][2]
What a phenotype is not. It is not a DSM-5-TR or ICD-11 code. People with the same microdeletion can meet criteria for ADHD, anxiety, ASD, psychosis, or none of these at a given time point. You still diagnose the psychiatric syndrome, then use the genetic context for prognosis, medical surveillance and family counselling.[1][11]
Major genetic mechanisms examiners map (aneuploidy, FMR1 expansion, 15q imprinting, 22q11 microdeletion):[3][6][7][9][13][14]
| Mechanism | Exam examples | Psychiatric relevance |
|---|---|---|
| Aneuploidy | Trisomy 21 (Down syndrome) | Depression; early-onset Alzheimer dementia risk |
| Trinucleotide expansion | FMR1 CGG (fragile X full mutation) | ID, ASD, ADHD, anxiety; premutation: FXPOI, FXTAS, FXAND |
| Genomic imprinting | 15q11-q13 paternal vs maternal gene loss | PWS vs Angelman — opposite parental origin, opposite phenotype |
| Microdeletion CNV | 22q11.2DS (VCFS/DiGeorge spectrum) | Highest known single CNV risk for schizophrenia-spectrum illness |
| [3][6][7][9][13][14] |
Naming trap. Velocardiofacial syndrome (VCFS), DiGeorge syndrome and 22q11.2 deletion syndrome describe overlapping presentations of the same microdeletion continuum — in viva, state that modern preferred term is 22q11.2DS while recognising older VCFS literature (including Murphy) underpins psychosis risk teaching.[10][12][13]
2. Epidemiology and risk
Order-of-magnitude numbers for exams
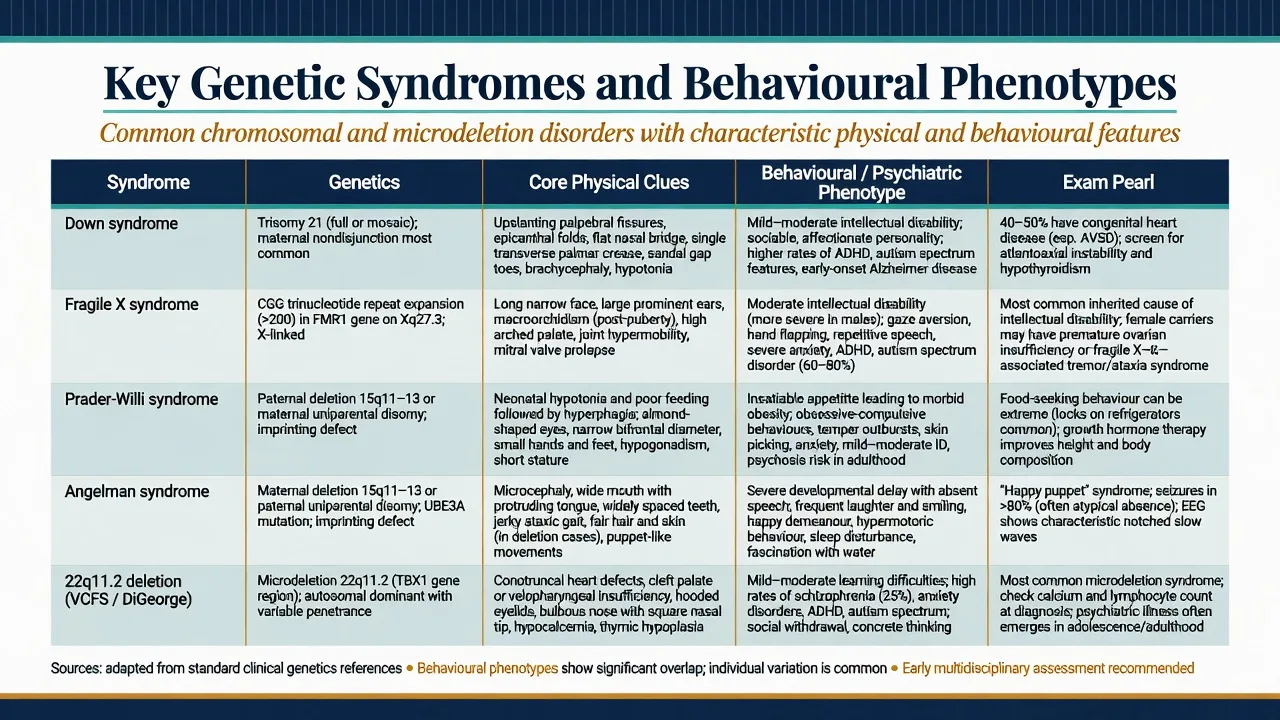
Down syndrome. Trisomy 21 is among the commonest chromosomal causes of intellectual disability. Almost all adults develop Alzheimer-type neuropathology with age; clinical dementia prevalence rises steeply from mid-life, with population-based work documenting substantial rates and characteristic presentations in adults with DS.[3][4][5]
Fragile X. Full mutation silencing of FMR1 is the commonest inherited cause of ID and a major monogenic contributor to autism spectrum presentations. Inheritance is X-linked with expansion dynamics; maternal premutation carriers may have family history of premature ovarian insufficiency or late-onset ataxia/tremor (FXTAS) that examiners use as cascade clues.[6]
Prader-Willi and Angelman. Reciprocal imprinting disorders at 15q11-q13. Incidence for each is on the order of 1 in 10,000–20,000. PWS genetic subtypes (paternal deletion, maternal uniparental disomy, imprinting defect) matter because psychotic illness clusters with maternal UPD in landmark population work.[7][8][9]
22q11.2DS. Among the commonest pathogenic microdeletions. Murphy and colleagues demonstrated high rates of schizophrenia in adults with VCFS — a foundational psychiatry paper. The International Consortium (Schneider et al.) mapped high rates of ADHD, anxiety, ASD features and psychotic disorders across childhood to adulthood, making longitudinal psychiatric surveillance mandatory in exam answers.[10][11][13]
3. Pathophysiology
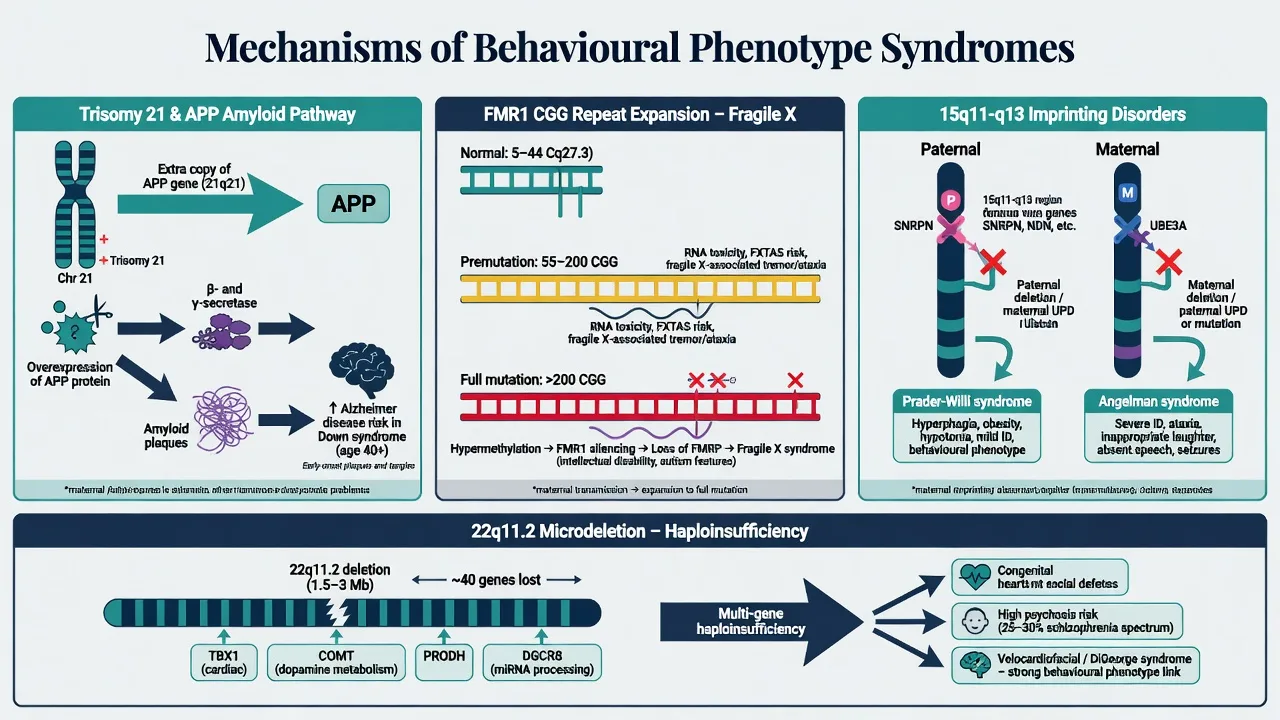
Down syndrome. Extra chromosome 21 includes the APP locus; lifelong amyloid overproduction drives early Alzheimer neuropathology and elevated clinical dementia risk — the neurobiological backbone of DS–Alzheimer teaching.[3][4]
Fragile X. Full mutation (greater than 200 CGG repeats) leads to FMR1 hypermethylation and loss of FMRP, disrupting synaptic plasticity. Premutation alleles (classically 55–200 repeats) can produce RNA toxicity phenotypes (FXTAS) and fragile X-associated neuropsychiatric disorders (FXAND) without classic full-mutation FXS.[6]
Imprinting at 15q11-q13 — classic viva pair. Same cytogenetic region, opposite parental origin, opposite syndromes: Prader-Willi from lack of paternally expressed genes (paternal deletion, maternal UPD, or imprinting centre defect) with hypothalamic hyperphagia drive; Angelman from loss of maternal UBE3A function in neurons (maternal deletion, paternal UPD, UBE3A mutation, or imprinting defect) with severe developmental encephalopathy.[7][9]
22q11.2DS. Heterozygous deletion of contiguous genes produces multi-system developmental disruption. Single-gene COMT stories are oversimplified for fellowship answers; prefer multi-gene haploinsufficiency within broader CNV risk architecture for schizophrenia and neurodevelopmental disorders.[13][14]
4. Clinical presentation by syndrome
Down syndrome
Psychiatric and behavioural. Depression (withdrawal, irritability, functional drop), anxiety, obsessive-compulsive-like behaviours, and behavioural change that may herald dementia. Social engagement is often a relative strength earlier in life, which can make later apathy more conspicuous to carers.[3][5]
Dementia interface. Alzheimer-type dementia presents with memory loss, executive decline, personality change, seizures later, and loss of adaptive skills. Age-related risk is extreme compared with the general population, but every new decline still needs a reversible-factor work-up.[4][5]
Fragile X syndrome
Males with full mutation. Variable ID; high rates of ASD features, ADHD, hyperarousal, gaze aversion, social anxiety; physical clues after puberty include long face, prominent ears, macroorchidism. Females are more variable because of X-inactivation — learning difficulties, anxiety and social communication problems without frank ID are common exam stems.[6]
Prader-Willi syndrome
Developmental arc. Neonatal hypotonia and poor feeding → later insatiable hyperphagia with life-threatening obesity risk if food is unrestricted; skin picking; rigidity; temper outbursts; high rates of compulsive/OCD-like behaviours.[7]
Psychosis pearl. Boer and colleagues linked psychotic illness particularly to the maternal uniparental disomy genetic subtype — always ask which molecular mechanism is known when PWS presents with psychosis.[8]
Angelman syndrome
Severe ID, minimal or absent speech, ataxic gait, seizures, sleep disturbance, and a characteristic happy demeanour with inappropriate laughter and hand-flapping motor patterns. Consensus diagnostic criteria (Williams et al.) remain the clinical scaffold while molecular testing confirms mechanism.[9]
22q11.2 deletion / velocardiofacial spectrum
Medical. Conotruncal cardiac defects, palatal anomalies, hypocalcaemia, immunodeficiency, characteristic facial features — any one may be subtle in mild cases.[12][13]
Psychiatric across ages. Childhood ADHD, anxiety disorders, ASD traits and learning disability are common; adolescence and adulthood bring a very high risk of schizophrenia-spectrum psychosis. Murphy's adult VCFS series and the Schneider consortium data are the named evidence examiners expect.[10][11]
5. Differential diagnosis
Win differentials on tempo, medical confounders, developmental history and form of psychopathology — not syndrome shopping lists.[3][5]
DS decline
- Depression: often subacute, guilt/tearfulness, diurnal mood, treatment-responsive
- Alzheimer: progressive adaptive loss, memory, late seizures
- Always exclude thyroid disease, B12, hearing/vision, sleep apnoea, drugs
Psychosis
- 22q11: developmental + medical red flags + high base rate
- PWS UPD: genotype-linked psychosis risk
- Primary schizophrenia without syndrome still possible — do not force genetics
ASD-like
- Fragile X and 22q11 both raise ASD-feature rates
- Angelman motor/affect/seizure package differs from idiopathic ASD
- Dual labels allowed when criteria met
Challenging behaviour
- Communication failure, pain, sensory load, environment
- PWS food-seeking is drive + environment, not 'naughty'
- Functional analysis before antipsychotic
Diagnostic overshadowing is the cardinal pitfall: assuming every new symptom is "just the syndrome" and missing depression, psychosis, abuse, constipation, dental pain, or hypocalcaemia.[2][12]
6. Assessment
Structure a fellowship ID-psychiatry assessment as follows: (1) three-generation pedigree for ID, ASD, psychosis, congenital anomalies, infertility/POI, tremor/ataxia; (2) developmental and adaptive history with communication method (speech, AAC, gesture); (3) syndrome-specific behavioural inventory using phenotype probabilities as hypotheses; (4) MSE adapted for ID — simple language, visual supports, longer time, carer collateral, careful psychotic form; (5) risk including self-injury, aggression, exploitation, PWS food-seeking, choking, suicide when mood or psychosis present; (6) decision-specific capacity with supported decision-making under local statute; (7) carer system, restrictive practice review and cultural formulation.[1][12]
7. Investigations
| Clinical clue | Priority testing | Note |
|---|---|---|
| Unexplained developmental disability / congenital anomalies | Chromosomal microarray (first-tier principle) | Detects many CNVs including 22q11.2 |
| ID/ASD, FX physical clues, maternal POI family history | FMR1 CGG repeat analysis | Full mutation vs premutation interpretation |
| Suspected PWS or Angelman | DNA methylation analysis at 15q11-q13 | Then subtype mechanism (deletion/UPD/imprinting/UBE3A) |
| Cardiac, palate, hypocalcaemia, immunodeficiency + psych | 22q11.2 evaluation (often via microarray) | Multisystem guidelines apply |
| Adult DS cognitive decline | TSH, B12, medication review, hearing/vision, consider sleep study | Do not jump to Alzheimer alone |
| Before antipsychotics | Weight/BMI, glucose/lipids, FBC, U&E, LFT, ECG as indicated | Hypocalcaemia vigilance in 22q11 |
MRI/EEG are for neurological red flags (seizure, focal signs, atypical regression), not routine fishing for a "phenotype scan."[9][12]
8. Acute and emergency management
De-escalate with sparse language, familiar carers, low sensory load. Exclude medical drivers first: pain, constipation, UTI, seizure, delirium, hypocalcaemia in 22q11, thyroid crisis mimics, medication toxicity.[12][13]
Psychotic emergency (especially 22q11 adolescence/adulthood or PWS maternal UPD): treat as first-episode or relapse psychosis with medical work-up, least restrictive legal framework under local law, and antipsychotic therapy with ID-appropriate dosing vigilance.[8][10][11]
9. Definitive management
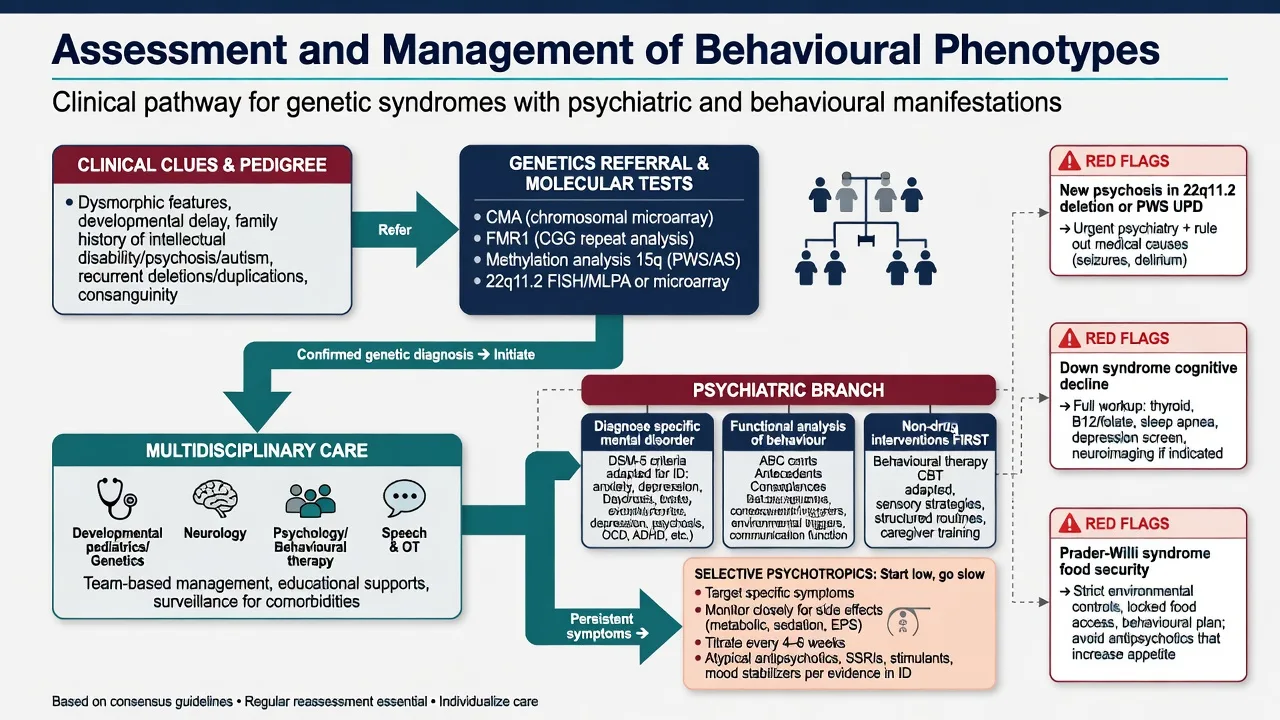
Core principles
- Multidisciplinary syndrome care — clinical genetics, cardiology, endocrinology, immunology, speech/OT, special education, ID psychiatry as relevant.[7][12][13]
- Non-pharmacological first for challenging behaviour — functional analysis, positive behaviour support, communication supports, sensory and environmental design.
- Pharmacotherapy targets diagnosed psychiatric illness or severe risk behaviour, not the genetic label. In ID: start low, go slow, minimise polypharmacy, monitor metabolic and neurological adverse effects carefully.
- Genetic counselling is non-directive and mechanism-specific (de novo vs inherited translocation vs maternal FMR1 premutation expansion risk).[6][14]
Syndrome-specific psychiatric notes
Down syndrome. Treat major depression with an SSRI when indicated after medical work-up — e.g. sertraline oral starting low (often 25 mg daily in sensitive adults with ID, titrate toward usual adult range as tolerated) with review of activation, hyponatraemia risk in older adults, and suicide risk where capacity allows self-report; always individualise and check local product information.[3][5] For established Alzheimer dementia: carer education, environmental supports, cholinesterase inhibitors only under specialist protocols where used regionally; avoid antipsychotics for mild BPSD given stroke/mortality risk framing from general dementia literature adapted cautiously to DS.
Fragile X. No standard disease-modifying drug is required as the exam "first line" answer. Treat ADHD (stimulant or non-stimulant with careful monitoring), anxiety (adapted CBT where feasible; SSRI when moderate–severe), and severe aggression only after behavioural formulation.[6]
Prader-Willi. Cornerstones are food security, specialist endocrine care including growth hormone under protocol, skin-picking management, and psychiatric treatment of mood, OCD-like and psychotic disorders. For psychosis, use an antipsychotic with metabolic monitoring — e.g. risperidone oral start often 0.25–0.5 mg daily in ID/youth contexts, slow titration, or alternatives with lower metabolic burden when appropriate; monitor weight obsessively given baseline obesity drive.[7][8]
Angelman. Seizure management with neurology; sleep supports; behavioural and communication strategies; limited role for psychotropics beyond specific target symptoms.[9]
22q11.2DS. Follow practical multisystem guidelines (calcium, cardiac, immune, speech). For emerging psychosis use standard early-intervention principles with added medical complexity (QTc, metabolic syndrome, hypocalcaemia). ADHD and anxiety are highly prevalent and treatable; coordinate stimulant or SSRI use with cardiology when structural heart disease is present.[11][12][13]
10. Subtypes and high-yield scenarios
- Adult DS with apathy — depression vs Alzheimer pathway; reversible work-up first.[4][5]
- Boy with ID, large ears, maternal relative with early menopause — FMR1 testing cascade.[6]
- Adolescent PWS with first-episode psychosis — confirm maternal UPD status if unknown; treat psychosis; reinforce food security.[8]
- Young adult with repaired tetralogy of Fallot, learning disability, new paranoia — 22q11.2DS until proven otherwise.[10][13]
- Non-verbal child with seizures and happy affect — Angelman molecular pathway.[9]
- Transition at 18 — written multiagency plan; adult ID/psychosis services; genetics letter in the file.
11. Complications and pitfalls
- Diagnostic overshadowing of depression, psychosis, pain, abuse.[2]
- Diagnosing Alzheimer dementia in DS without reversible work-up.[5]
- Ignoring PWS genetic subtype when psychosis appears.[8]
- Antipsychotics as first response to communication-driven behaviour.
- Missing hypocalcaemia, hypothyroidism, sleep apnoea as behaviour drivers.[12]
- Genetic essentialism, stigma, reproductive coercion in counselling.[14]
- Failing cascade testing for relatives when mechanism implies familial risk (e.g. maternal FMR1 premutation).[6]
12. Prognosis and disposition
Outcomes are highly variable and modifiable by supports, comorbidity control and environment. DS requires planning for dementia-capable care in mid-to-late adulthood.[3][4] 22q11.2DS requires lifelong psychiatric vigilance because psychosis risk persists into adult years.[11][13] PWS morbidity and mortality track obesity and respiratory complications when food security fails.[7] Disposition is shared care: specialist ID psychiatry where available, general adult/CAMHS with genetics liaison, carer support and clear crisis plans.
13. Special populations
Children — early molecular diagnosis changes medical surveillance and school planning.[15] Adolescents — psychosis prodrome vigilance especially in 22q11 and PWS UPD.[8][11] Older adults with DS — dementia pathways and end-of-life planning.[4][5] Premutation carriers — FXPOI counselling for women; FXTAS risk discussion for ageing men with premutation.[6] Cultural care — equitable access to genetics and ID services; avoid deficit-only framing of Indigenous and minority families.[12]
14. Evidence and guidelines
Named landmarks for answers: Dykens 1995 probabilistic behavioural phenotype; Di Nuovo 2011 comparative profiles; Antonarakis 2020, Zigman 2007, Holland 1998 for DS biology and dementia; Hagerman 2017 fragile X; Cassidy 2012 and Boer 2002 for PWS care and UPD psychosis; Williams 2006 Angelman criteria; Murphy 1999, Schneider 2014, Bassett 2011, McDonald-McGinn 2015 for 22q11 psychiatry and multisystem care; Malhotra 2012 and Miller 2010 for CNV architecture and microarray first-tier testing.[1][2][3][4][5][6][7][8][9][10][11][12][13][14][15]
15. Exam pearls
- Behavioural phenotype = raised probability, not destiny or pathognomonic list.[1]
- 15q11-q13: paternal expression loss → PWS; maternal UBE3A loss → Angelman.[7][9]
- PWS maternal UPD → highest psychosis risk among PWS subtypes.[8]
- 22q11.2DS / VCFS: adult schizophrenia-spectrum risk is order-of-magnitude very high (Murphy landmark; consortium multi-disorder burden) — quote carefully by age band.[10][11]
- DS decline: depression and medical causes first; Alzheimer risk is real but not automatic in every apathetic spell.[4][5]
- Fragile X: full mutation greater than 200 CGG; premutation 55–200 with different adult phenotypes.[6]
- Psychotropics: start low, go slow; treat the diagnosed mental disorder, not the karyotype.[11][12]
PHENO-GENES (board memory hook)
References
- [1]Dykens EM Measuring behavioral phenotypes: provocations from the "new genetics" Am J Ment Retard, 1995.PMID 7779347
- [2]Di Nuovo S, Buono S Behavioral phenotypes of genetic syndromes with intellectual disability: comparison of adaptive profiles Psychiatry Res, 2011.PMID 21507490
- [3]Antonarakis SE, Skotko BG, Rafii MS, et al. Down syndrome Nat Rev Dis Primers, 2020.PMID 32029743
- [4]Zigman WB, Lott IT Alzheimer's disease in Down syndrome: neurobiology and risk Ment Retard Dev Disabil Res Rev, 2007.PMID 17910085
- [5]Holland AJ, Hon J, Huppert FA, Stevens F, Watson P Population-based study of the prevalence and presentation of dementia in adults with Down's syndrome Br J Psychiatry, 1998.PMID 9828989
- [6]Hagerman RJ, Berry-Kravis E, Hazlett HC, et al. Fragile X syndrome Nat Rev Dis Primers, 2017.PMID 28960184
- [7]Cassidy SB, Schwartz S, Miller JL, Driscoll DJ Prader-Willi syndrome Genet Med, 2012.PMID 22237428
- [8]Boer H, Holland A, Whittington J, Butler J, Webb T, Clarke D Psychotic illness in people with Prader Willi syndrome due to chromosome 15 maternal uniparental disomy Lancet, 2002.PMID 11809260
- [9]Williams CA, Beaudet AL, Clayton-Smith J, et al. Angelman syndrome 2005: updated consensus for diagnostic criteria Am J Med Genet A, 2006.PMID 16470747
- [10]Murphy KC, Jones LA, Owen MJ High rates of schizophrenia in adults with velo-cardio-facial syndrome Arch Gen Psychiatry, 1999.PMID 10530637
- [11]Schneider M, Debbané M, Bassett AS, et al. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome Am J Psychiatry, 2014.PMID 24577245
- [12]Bassett AS, McDonald-McGinn DM, Devriendt K, et al. Practical guidelines for managing patients with 22q11.2 deletion syndrome J Pediatr, 2011.PMID 21570089
- [13]McDonald-McGinn DM, Sullivan KE, Marino B, et al. 22q11.2 deletion syndrome Nat Rev Dis Primers, 2015.PMID 27189754
- [14]Malhotra D, Sebat J CNVs: harbingers of a rare variant revolution in psychiatric genetics Cell, 2012.PMID 22424231
- [15]Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies Am J Hum Genet, 2010.PMID 20466091