Psych · Intellectual disability — neurodevelopmental
Fragile X syndrome
Also known as FXS · Fragile X · FMR1-related disorder · Martin-Bell syndrome · Fragile X-associated tremor/ataxia syndrome · FXTAS · Fragile X-associated primary ovarian insufficiency · FXPOI · FMR1 premutation
Exam-exhaustive fellowship reference on fragile X syndrome and the FMR1 spectrum — CGG allele classes, FMRP loss and premutation RNA toxicity, X-linked inheritance and Sherman paradox, behavioural phenotype (ID, ASD features, anxiety, ADHD), female variability, FXTAS/FXPOI, ACMG molecular diagnosis, cascade counselling, and comorbidity-focused management without a proven disease-modifying core drug. FRANZCP-primary, globally tagged.
On this page & tools
Your progress
Saved locally on this device.
10 MCQs with explanations
Target exams
Red flags
Fragile X syndrome (FXS) is a high-yield interface topic for intellectual disability, child and adolescent, dual-diagnosis and consultation-liaison psychiatry. Examiners test whether you can name allele classes and molecular mechanisms, separate full-mutation FXS from premutation phenotypes, recognise the behavioural phenotype (ID, social anxiety, ADHD, ASD features, sensory hyperarousal), order the correct molecular test, manage without inventing a core cure, and deliver genetic counselling that protects the wider family.[1][2][3]
Overview and definition
FXS is an X-linked neurodevelopmental disorder caused by pathogenic variation in FMR1 (fragile X messenger ribonucleoprotein 1; historically fragile X mental retardation 1) at Xq27.3. The classic mechanism is expansion of a CGG trinucleotide repeat in the 5-prime untranslated region beyond about 200 repeats (full mutation), with CpG island hypermethylation, transcriptional silencing and near-absent FMRP.[1][5][6]
GeneReviews and primer-level reviews frame FMR1 disorders as a spectrum: full-mutation FXS; premutation-associated conditions including FXTAS and FXPOI; and rarer non-expansion FMR1 loss-of-function variants that can mimic FXS.[2][1]
Classification and allele classes
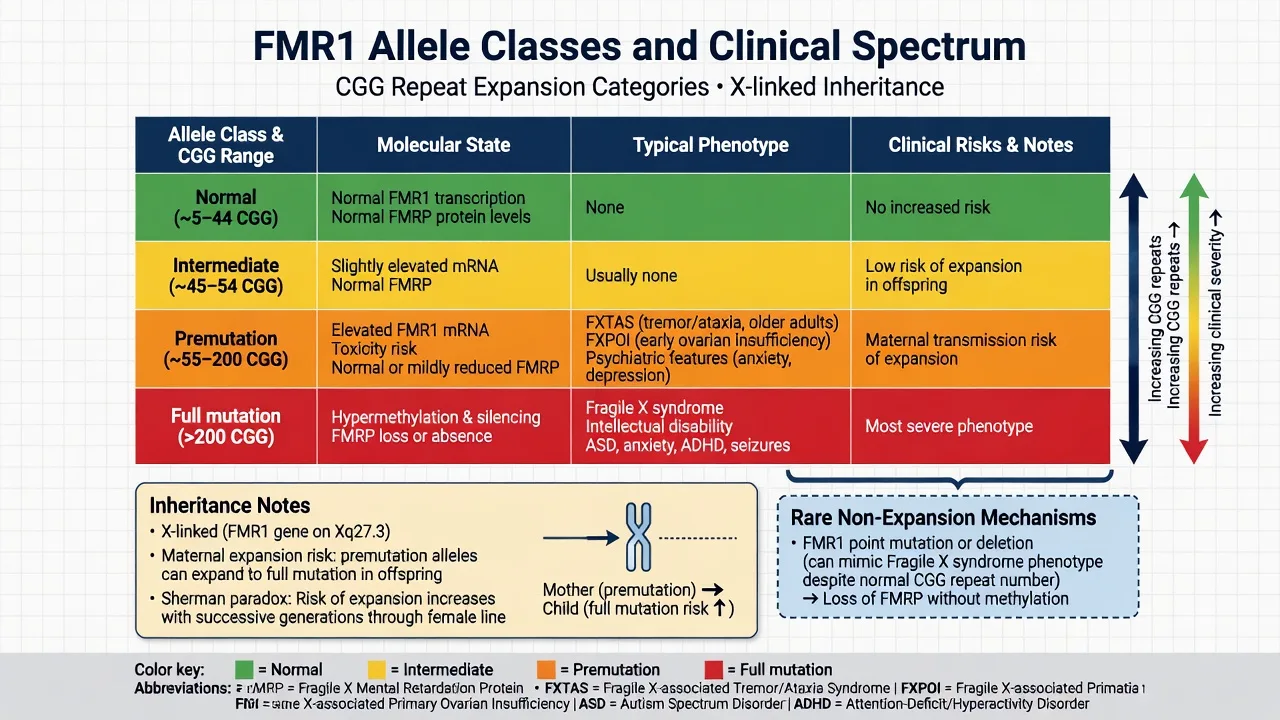
Standard clinical ranges (exam-working numbers; laboratory cut-points follow ACMG technical standards):[2][12][3]
| Allele class | Approximate CGG range | Molecular gist | Clinical exam pearl |
|---|---|---|---|
| Normal | about 5–44 | Normal FMRP | No FXS risk from this allele |
| Intermediate (gray zone) | about 45–54 | Usually asymptomatic | Small expansion risk over generations |
| Premutation | about 55–200 | Elevated FMR1 mRNA; toxic RNA pathway | FXTAS, FXPOI, psychiatric risk; maternal expansion risk |
| Full mutation | greater than 200 | Methylation silencing; FMRP loss | Classic FXS phenotype |
| [2][12][3] |
Inheritance. FXS is X-linked. Full-mutation males are almost always clinically affected. Full-mutation females show highly variable expression related to X-inactivation. Full mutations are almost always transmitted through a carrier mother (premutation or full mutation); paternal premutation alleles rarely expand to full mutation in offspring — a classic viva discriminator.[1][2][6]
Sherman paradox. Earlier pedigree observations of increasing penetrance in successive generations were resolved by documenting intergenerational CGG instability, especially maternal expansion from premutation toward full mutation.[6]
Epidemiology and risk
Headline FXS numbers for exams
A systematic review and meta-analysis by Hunter and colleagues provides fellowship-usable prevalence estimates on the order of about 1.4 full-mutation males per 10 000 and about 0.9 full-mutation females per 10 000, with premutation carrier frequencies substantially higher — meaning cascade testing finds many asymptomatic or differently affected relatives.[4]
Male full-mutation individuals nearly always have cognitive impairment; female full-mutation phenotypes range from learning difficulty and anxiety to frank intellectual disability.[1][3] Premutation carriers are not "mild FXS"; they have a different molecular pathophysiology and a different risk set.[2][10]
Pathophysiology
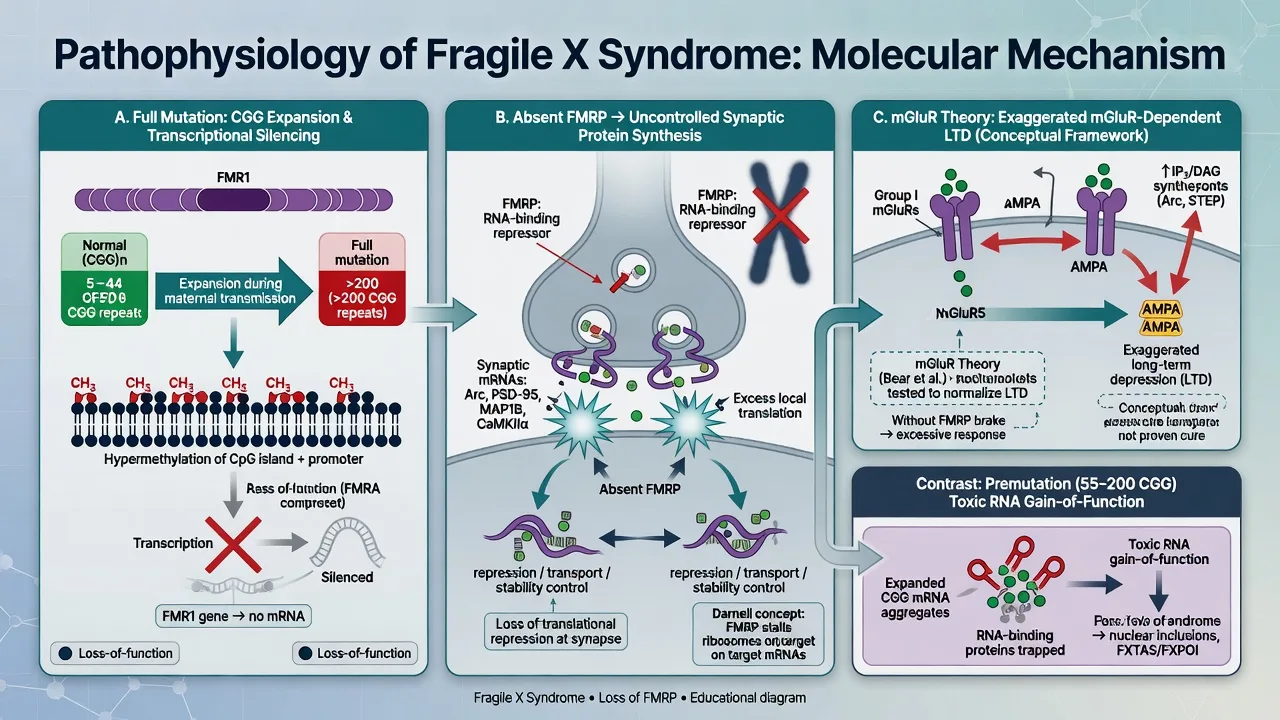
Full mutation. Expanded CGG repeats trigger DNA methylation of the FMR1 promoter, silencing transcription so FMRP is absent or severely reduced.[1][5] FMRP is an RNA-binding translational repressor enriched at synapses; its loss dysregulates local protein synthesis of transcripts linked to synaptic plasticity and autism-related pathways.[1]
mGluR theory. Bear, Huber and Warren proposed that loss of FMRP exaggerates group I metabotropic glutamate receptor-dependent long-term depression, providing a mechanistic rationale for mGluR5-targeted experimental therapies. Know the theory and the sobering translation gap: these agents have not become routine disease-modifying standard of care.[7][1]
Premutation. Pathophysiology is dominated by toxic gain-of-function RNA (elevated FMR1 mRNA, RNA foci, RAN translation products), not simple FMRP absence — explaining why premutation adults can develop neurodegeneration (FXTAS) despite having largely preserved FMRP early on.[2][10]
Clinical presentation and behavioural phenotype
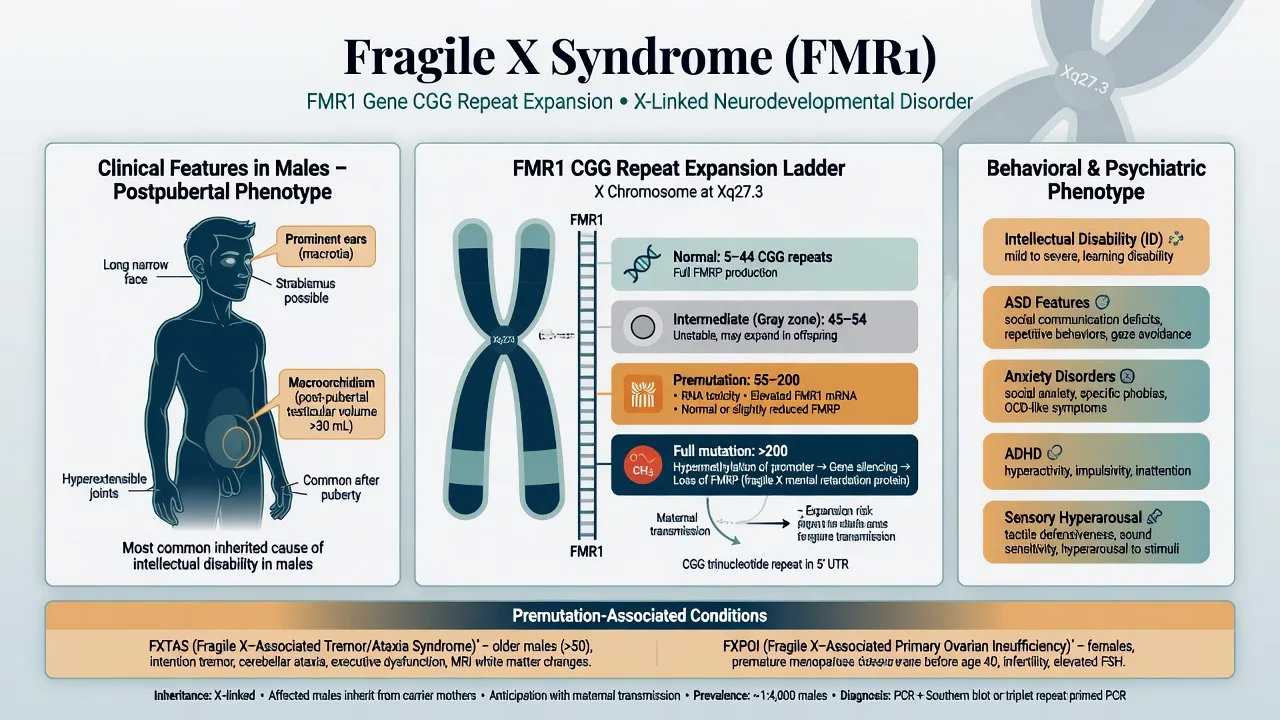
Full-mutation FXS (especially males)
Neurodevelopmental. Developmental delay, intellectual disability (often mild–moderate), language delay, poor eye contact, social avoidance, hyperactivity, impulsivity, sensory hyperarousal, perseveration, and in some individuals hand-flapping or self-injurious behaviour under stress.[1][3]
Physical clues. Long narrow face, prominent ears, soft connective tissue with joint hypermobility, flat feet, and macroorchidism after puberty in males. Mitral valve prolapse and other connective-tissue findings may occur — examine and refer when indicated.[1][3]
Psychiatric comorbidity. Structured assessment shows very high rates of anxiety disorders (social anxiety, specific phobia, and related presentations are especially common).[8] ADHD-like symptoms and autism spectrum features frequently co-occur; FXS is a classic monogenic model within the autism differential, but not every person with FXS meets full ASD criteria and not every autistic person has FXS.[9][1] Seizures occur in a minority and need standard epilepsy pathways.[1]
Female full mutation
Phenotype is variable: anxiety, social and learning difficulties, or ID depending on residual FMRP and X-inactivation. Never dismiss significant psychiatric morbidity because the patient is female or "only a carrier" without checking allele class.[1][2]
Premutation phenotypes
- FXTAS: progressive intention tremor, cerebellar ataxia, parkinsonism, executive and memory decline, and psychiatric morbidity in ageing premutation carriers; penetrance rises with age and is higher in males (Jacquemont 2004 landmark penetrance study).[10][2]
- FXPOI: premutation females have a substantially elevated risk of premature ovarian insufficiency / premature ovarian failure — international collaborative data established premutation as a significant risk factor.[11]
- Psychiatric symptoms in carriers: anxiety, depression and related symptoms are clinically important even without FXTAS/FXPOI.[2][1]
MSE adaptations. Reduce sensory load, use concrete language, allow processing time, and do not misread gaze aversion or social anxiety as simple oppositionality.[1][8]
Differential diagnosis
Win differentials on molecular allele class + pedigree + phenotype, not on a single behaviour.[1][2]
Idiopathic ASD / ADHD
- May look similar at school age
- Family pedigree and physical clues raise FXS
- Molecular FMR1 test when ID/ASD plus maternal-line clues
- Dual labels allowed when criteria met
Other genetic ID syndromes
- Down, Angelman, Prader–Willi, Rett, tuberous sclerosis
- Different dysmorphology and molecular tests
- Microarray and syndrome-specific genes as indicated
- FXS testing is targeted molecular, not 'karyotype only'
Premutation vs full mutation
- Premutation is not mild FXS
- RNA toxicity vs FMRP loss
- FXTAS/FXPOI vs childhood ID phenotype
- Counselling content differs
FXTAS mimics
- Parkinson disease, essential tremor, MSA, vascular/AD dementia
- Family FXS history and MCP-type imaging clues help
- Still a clinical-molecular diagnosis
- Treatable psychiatric comorbidity remains
Clinical and bedside assessment
Structure the fellowship assessment as follows:[1][2][3]
- Developmental and educational history — milestones, language, school supports, adaptive skills.[1]
- Behavioural and psychiatric review — anxiety, ADHD, ASD features, aggression, self-injury, sleep, seizures.[1][8]
- Physical examination — facial features, ears, connective tissue, post-pubertal testicular volume when appropriate, cardiac signs.[1][3]
- Three-generation pedigree with attention to maternal-line ID/ASD, premature menopause/infertility, and late-onset tremor/ataxia.[2][11]
- Risk — exploitation, carer burnout, self-harm when mood/anxiety severe, behavioural crisis triggers (sensory overload).[1]
- Capacity — decision-specific; support decision-making with plain language and trusted supporters.[2]
- Family system — who needs cascade testing and reproductive counselling now.[2]
Investigations
| Indication | Test / action | Note |
|---|---|---|
| Suspected FXS or unexplained ID/ASD with clues | Molecular FMR1 CGG sizing ± methylation analysis | Follow ACMG laboratory technical standards |
| Broader ID work-up | Chromosomal microarray ± other genes as indicated | Complements, does not replace FMR1 when FXS suspected |
| Seizure suspicion | EEG; neurology | Seizures are a minority but treatable |
| Before psychotropics | Weight/BMI, metabolic panel, BP; ECG as indicated | Same physical-health culture as other neurodevelopmental care |
| Suspected FXTAS | Neurology review; MRI when indicated | Classic middle cerebellar peduncle T2 change is supportive, not mandatory |
| Premutation woman with menstrual/fertility issues | FSH/endocrine/reproductive assessment | FXPOI pathway |
| [12][2][1] |
Critical pearl: modern diagnosis is molecular, not the historical fragile site on routine karyotype alone.[12][3]
Management — acute crisis
Use low-arousal environments, single-step concrete instructions, familiar carers and sensory reduction. Exclude pain, otitis media, constipation, dental disease, seizure and medication side effects before assuming "behaviour only." Least-restrictive safety measures; avoid reflexive high-dose polypharmacy.[1]
Management — definitive and stepwise
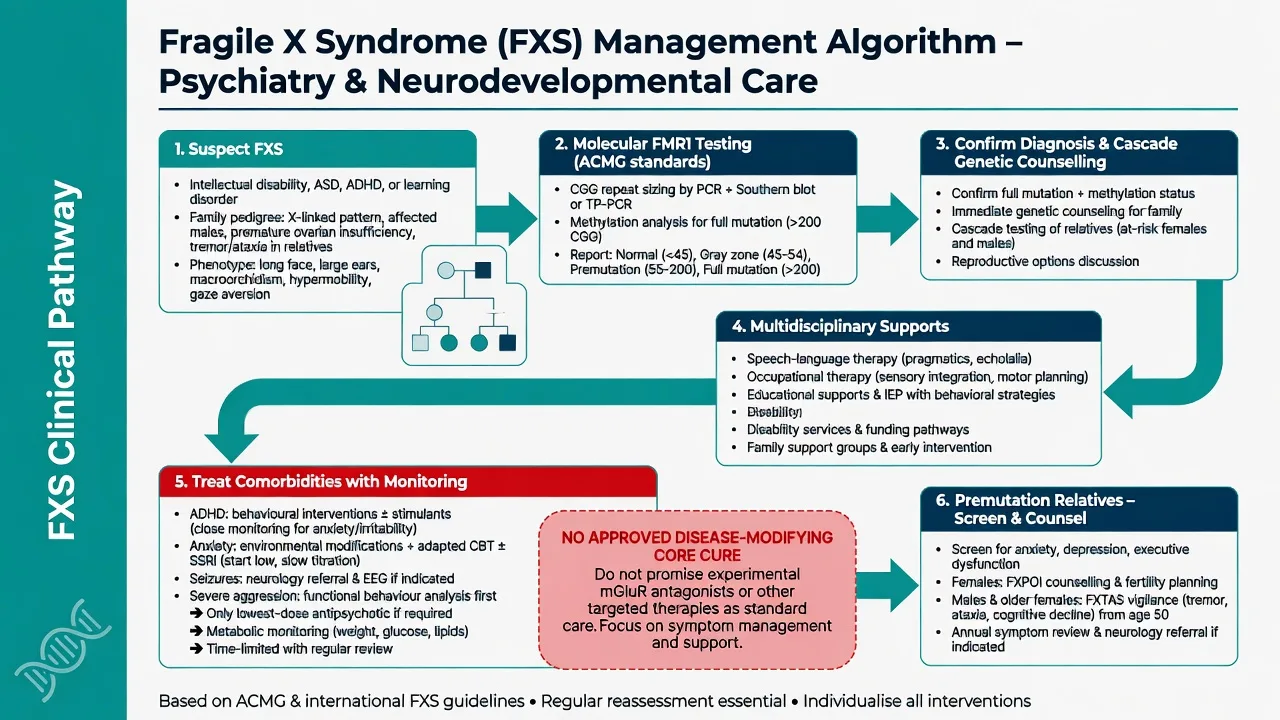
Core principle
There is no approved disease-modifying therapy that restores FMRP or cures core FXS in routine practice. Standard care is multidisciplinary support plus treatment of comorbidities, with rigorous physical-health monitoring when medicines are used.[1][2]
Developmental and environmental supports
Speech and language therapy, occupational therapy (including sensory strategies), special education, behavioural supports, and disability funding pathways (e.g. NDIS interface in Australia) are the backbone of care. Structure, predictability and reduced sensory load are clinical interventions, not optional soft extras.[1][3]
Comorbidity-targeted psychiatry
- Anxiety: environmental scaffolding and adapted psychological approaches when cognitive profile allows; SSRIs are widely used clinically for social anxiety and hyperarousal (e.g. sertraline starting at low paediatric/ID-adapted doses such as 25 mg oral daily in many youth pathways, titrating slowly with monitoring for activation, sleep change, agitation and self-harm) — individualise to age, weight and local formulary.[8][1]
- ADHD symptoms: behavioural strategies plus stimulant therapy when criteria are met (e.g. methylphenidate starting low, such as 5 mg oral morning test dose in stimulant-naive school-age children under specialist/paediatric protocols, with monitoring of appetite, sleep, growth, blood pressure and anxiety) or non-stimulant alternatives when stimulants worsen anxiety or tics.[1][2]
- ASD features: autism-informed supports; do not withhold ASD services because a genetic diagnosis exists.[9]
- Sleep: hygiene, rule out OSA and environmental chaos; consider melatonin pathways under local guidance when appropriate.[1]
- Seizures: neurology-led anticonvulsant care.[1]
- Severe aggression: functional behaviour analysis and medical screen first; if an atypical antipsychotic is required for safety after behavioural optimisation, use the lowest effective dose, define time-limited goals, and perform metabolic monitoring (weight, glucose, lipids) — the indication is risk behaviour, not "treating FXS."[1]
Experimental and targeted therapies
mGluR5 negative allosteric modulators and other targeted approaches grew from the mGluR theory and FMRP biology. Fellowship answer: know the mechanism, do not claim proven routine disease modification.[7][1]
Genetic counselling and cascade testing
Every new diagnosis is a family diagnosis. Offer genetic counselling for parents and at-risk relatives; discuss reproductive options; screen premutation relatives for psychiatric symptoms, FXPOI risks in women, and age-related FXTAS symptoms in adults.[2][11][10]
Specific subtypes and scenarios
Toddler male with delay and maternal uncle with ID. High pre-test probability — early FMR1 testing and early intervention in parallel.[1][2]
School-age ADHD/ASD labels. Re-open pedigree and physical exam; molecular test when clues accumulate.[9][3]
Adolescent female full mutation. Anxiety and learning support may dominate; monitor mood and self-harm risk; avoid under-treatment because IQ is "borderline."[8][1]
Premutation mother after child's diagnosis. Address guilt without blame theatre; screen her anxiety/depression; offer FXPOI and reproductive counselling; document cascade plan.[11][2]
Middle-aged man with tremor and family FXS history. Think FXTAS; neurology referral; do not attribute everything to "stress" or essential tremor without evaluation.[10]
Capacity, safeguarding and multiagency care
Capacity is decision-specific. Support decision-making; involve disability advocates and family as appropriate. Guardianship and mental health statutes are jurisdiction-specific — state principles (support first, least restrictive) rather than inventing foreign section numbers.[2]
Safeguarding issues include exploitation of socially anxious people with ID, carer breakdown, and school exclusion. Report under local child protection / adult safeguarding duties when thresholds are met.[1]
Complications and pitfalls
- Using karyotype alone and missing molecular FXS.[12]
- Calling premutation carriers "mild fragile X."[2]
- Assuming females are unaffected.[1]
- Diagnostic overshadowing: missing treatable anxiety, ADHD, seizures or pain.[8][1]
- Promising experimental drugs as standard cures.[7][1]
- Failing cascade counselling after the index diagnosis.[2]
- Ignoring FXPOI reproductive risk in premutation women.[11]
Prognosis and disposition
FXS is lifelong. Adaptive outcomes improve with early diagnosis, educational scaffolding, treated comorbidities and stable caregiving. Life expectancy is often near population norms when medical problems are managed; quality of life tracks supports more than allele size alone.[1][2] Premutation carriers need a different long-term watch for FXTAS and FXPOI.[10][11] Disposition is multiagency: developmental paediatrics/genetics, education, disability services, dual-diagnosis psychiatry as needed, and family genetic follow-up.
Special populations
Females with full mutation. Variable expression; anxiety and learning supports often central.[1]
Premutation carriers across the lifespan. Psychiatric, reproductive and neurodegenerative surveillance as age-appropriate.[2][10][11]
Cultural and reproductive contexts. Genetic testing decisions are values-laden; offer non-directive counselling and interpreter-supported discussion.[2]
Transition to adult services. Plan dual-diagnosis handovers before paediatric services drop off.[1]
Evidence, guidelines and regional differences
FRANZCP candidates should integrate DSM/ICD neurodevelopmental formulation, arrange molecular testing via clinical genetics pathways, interface with NDIS/disability supports in Australia, and manage comorbidities with physical-health monitoring. Cascade counselling is part of the psychiatric care plan after any new syndromic diagnosis in dual-diagnosis settings.[1][2]
Landmark list to name in viva: Verkerk 1991 FMR1 identification; Fu 1991 CGG instability and Sherman paradox; Bear 2004 mGluR theory; Hagerman 2017 primer; Hunter 2014 epidemiology; Garber 2008 clinical overview; Cordeiro 2011 anxiety; Yu and Berry-Kravis 2014 autism interface; Jacquemont 2004 FXTAS penetrance; Allingham-Hawkins 1999 POF/FXPOI; ACMG laboratory standards (Spector 2021).[5][6][7][1][4][12]
Exam pearls
[1] [2] [6] [12]FRAGILEX
Self-test: 7-year-old boy with ID, social anxiety, hyperactivity; maternal uncle in special school; mother had early menopause
Suspect FXS and FMR1 spectrum in the family. Order molecular FMR1 testing for the child; arrange genetic counselling. Mother may be a premutation carrier with FXPOI history — do not attribute early menopause to chance alone. Treat anxiety and ADHD with environmental supports and carefully monitored medicines if indicated. Offer cascade testing and document multiagency school/disability plan. Do not promise a drug that cures core FXS.[1][2][8][11]
References
- [1]Hagerman RJ, Berry-Kravis E, Hazlett HC, et al. Fragile X syndrome Nat Rev Dis Primers, 2017.PMID 28960184
- [2]Hunter JE, Berry-Kravis E, Hipp H, Todd PK FMR1 Disorders GeneReviews, 1993.PMID 20301558
- [3]Garber KB, Visootsak J, Warren ST Fragile X syndrome Eur J Hum Genet, 2008.PMID 18398441
- [4]Hunter J, Rivero-Arias O, Angelov A, et al. Epidemiology of fragile X syndrome: a systematic review and meta-analysis Am J Med Genet A, 2014.PMID 24700618
- [5]Verkerk AJ, Pieretti M, Sutcliffe JS, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome Cell, 1991.PMID 1710175
- [6]Fu YH, Kuhl DP, Pizzuti A, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox Cell, 1991.PMID 1760838
- [7]Bear MF, Huber KM, Warren ST The mGluR theory of fragile X mental retardation Trends Neurosci, 2004.PMID 15219735
- [8]Cordeiro L, Ballinger E, Hagerman R, Hessl D Clinical assessment of DSM-IV anxiety disorders in fragile X syndrome: prevalence and characterization J Neurodev Disord, 2011.PMID 21475730
- [9]Yu TW, Berry-Kravis E Autism and fragile X syndrome Semin Neurol, 2014.PMID 25192504
- [10]Jacquemont S, Hagerman RJ, Leehey MA, et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population JAMA, 2004.PMID 14747503
- [11]Allingham-Hawkins DJ, Babul-Hirji R, Chitayat D, et al. Fragile X premutation is a significant risk factor for premature ovarian failure: the International Collaborative POF in Fragile X study Am J Med Genet, 1999.PMID 10208170
- [12]Spector E, Behlmann A, Kronquist K, et al. Laboratory testing for fragile X, 2021 revision: a technical standard of the American College of Medical Genetics and Genomics (ACMG) Genet Med, 2021.PMID 33795824