Psych · Old age psychiatry — neurocognitive disorders
Frontotemporal dementia
Also known as Frontotemporal lobar degeneration · FTD · FTLD · Behavioural variant FTD · bvFTD · Primary progressive aphasia · Pick disease · Semantic dementia
Exam-exhaustive fellowship reference on frontotemporal dementia — Rascovsky bvFTD criteria; Gorno-Tempini PPA variants (nfv/sv/lv); proteinopathies (tau, TDP-43, FUS); genetics (C9orf72, MAPT, GRN); psychiatric misdiagnosis; ALS-FTSD overlap; work-up; non-pharmacological care; SSRI/trazodone symptomatic options; avoid routine AChEI; antipsychotic mortality caution; capacity and risk. FRANZCP-primary, globally tagged.
On this page & tools
Your progress
Saved locally on this device.
10 MCQs with explanations
Target exams
Red flags
Frontotemporal dementia (FTD) is a high-yield old-age and neuropsychiatry examination topic. FRANZCP MEQs demand Rascovsky features, a named genetic cause, differentiation from bipolar or late-onset psychosis, and a management plan that does not blindly transplant Alzheimer pharmacotherapy. MRCPsych CASC stations test communication with families about diagnosis, genetics, and behaviour. ABPN items test PPA variants, C9orf72–ALS overlap, and treatment limits. This topic is written so a candidate who has read nothing else can answer at consultant depth.[9][10][14]
Overview and definition
Frontotemporal dementia is a clinical umbrella for progressive syndromes dominated by early changes in social behaviour, executive control, and/or language, reflecting degeneration of frontal and/or anterior temporal networks. Frontotemporal lobar degeneration (FTLD) is the pathologic umbrella — proteinopathies that may present as FTD clinically.[3][9][16]
Historical Pick disease terminology is often reserved for a specific tauopathy (Pick bodies) or used loosely in older literature; prefer modern clinical (bvFTD/PPA) and pathologic (FTLD-tau/TDP/FUS) language in exams.[9][16]
DSM-5-TR codes major (or mild) frontotemporal neurocognitive disorder with behavioural or language variants. ICD-11 retains frontotemporal dementia terminology. State which manual you are using when classification language is examined.[9]
Classification and clinical constructs
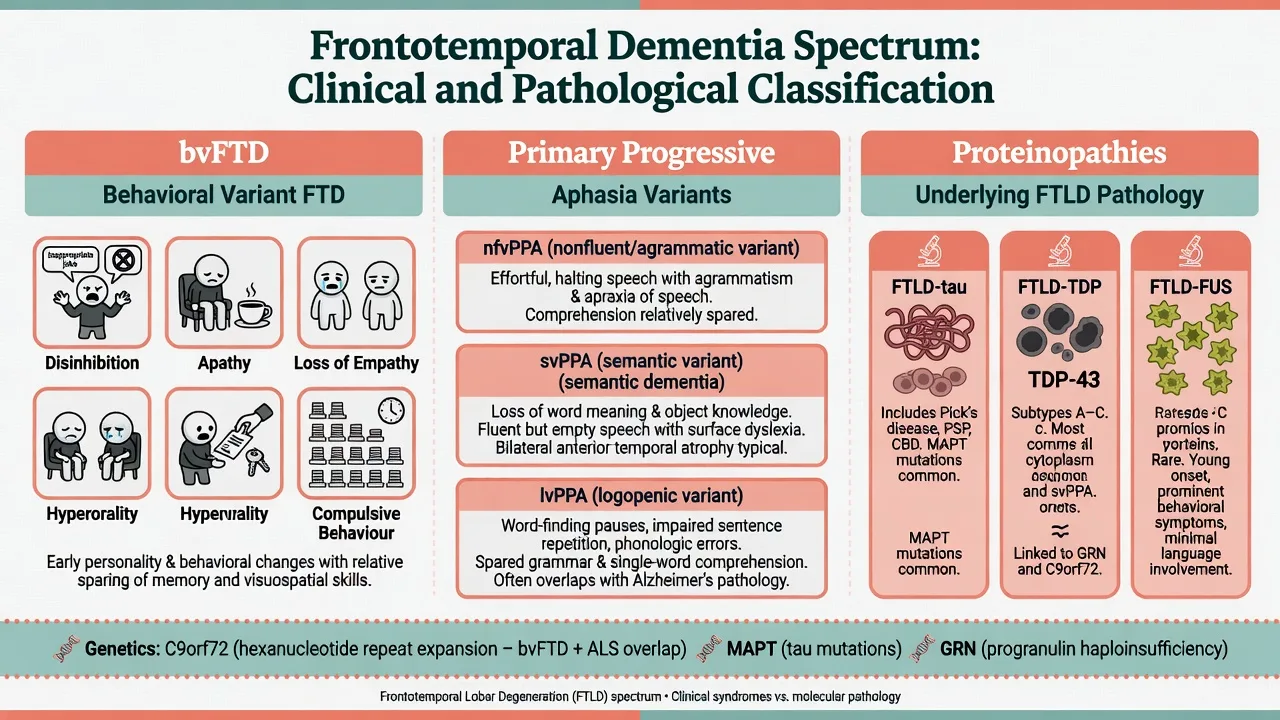
Behavioural variant FTD (Rascovsky 2011)
Revised international criteria stratify possible, probable, and definite bvFTD.[1]
Core behavioural/cognitive features (need progressive deterioration of behaviour and/or cognition):
- Early behavioural disinhibition (socially inappropriate behaviour, loss of manners, impulsive/rash actions)
- Early apathy or inertia
- Early loss of sympathy or empathy
- Early perseverative, stereotyped, or compulsive/ritualistic behaviour
- Hyperorality and dietary changes
- Neuropsychological profile: executive/generation deficits with relative sparing of memory and visuospatial functions These six domains are the Rascovsky core feature set used to score possible and probable bvFTD.[1]
Possible bvFTD: three of the six features plus functional decline / caregiver dependence pattern as specified in the criteria framework.[1]
Probable bvFTD: meets possible criteria plus significant functional decline and imaging consistent with frontotemporal involvement (frontal and/or anterior temporal atrophy, hypoperfusion, or hypometabolism — educational pattern language, not invented patient scan reports).[1][10]
Definite bvFTD: clinical syndrome plus histopathologic FTLD or a known pathogenic mutation.[1]
Neary 1998 FTLD consensus criteria remain historically important examinable background for FTD, progressive nonfluent aphasia, and semantic dementia prototypes.[3]
Primary progressive aphasia (Gorno-Tempini 2011)
PPA requires language difficulty as the principal cause of impaired daily living at onset, with aphasia the most prominent deficit at onset and in early stages.[2]
| Variant | Core clinical signature | Typical imaging association (educational) | Common pathology tendency |
|---|---|---|---|
| nfvPPA (nonfluent/agrammatic) | Effortful, agrammatic speech; apraxia of speech common | Left posterior fronto-insular | Often FTLD-tau |
| svPPA (semantic) | Impaired confrontation naming and single-word comprehension; loss of object knowledge; surface dyslexia/dysgraphia | Anterior temporal (often left greater) | Often TDP-43 (type C) |
| lvPPA (logopenic) | Impaired single-word retrieval and sentence repetition; phonological errors | Left temporoparietal | Often Alzheimer pathology |
| PPA variant diagnosis is clinical first; imaging and biomarkers refine etiology, especially when logopenic features raise Alzheimer pathway likelihood.[2][11] |
Semantic dementia is the classic clinicopathologic language-led syndrome of progressive conceptual knowledge loss; it maps largely to svPPA in modern PPA classification.[11]
ALS–frontotemporal spectrum (ALS-FTSD)
Motor neuron disease and FTD form a continuum. Revised Strong criteria formalise ALS-frontotemporal spectrum disorder, from pure ALS through behavioural/cognitive impairment to FTD with ALS.[15] Always examine for fasciculations, wasting, bulbar signs, and upper motor neuron findings when FTD is suspected.
Epidemiology and risk
FTD is a major cause of young-onset dementia (commonly midlife, often 45–65 years, with wider range). It is less common than Alzheimer disease among the oldest-old but disproportionately devastating because of working-age onset, dependent children, employment loss, and behavioural risk.[9][10]
A positive family history is common (order-of-magnitude teaching: roughly one-third to one-half of series show familial clustering; a smaller fraction show clear autosomal dominant monogenic disease). Among Mendelian causes, C9orf72 hexanucleotide expansion is the most frequent genetic cause of familial FTD and familial ALS in many populations; MAPT and GRN follow as the classic exam triad.[4][5][6][7][8][9]
Psychiatric misdiagnosis is a signature trap: a substantial minority of patients later confirmed to have FTD received a prior primary psychiatric diagnosis, especially for behavioural presentations.[14]
Pathophysiology
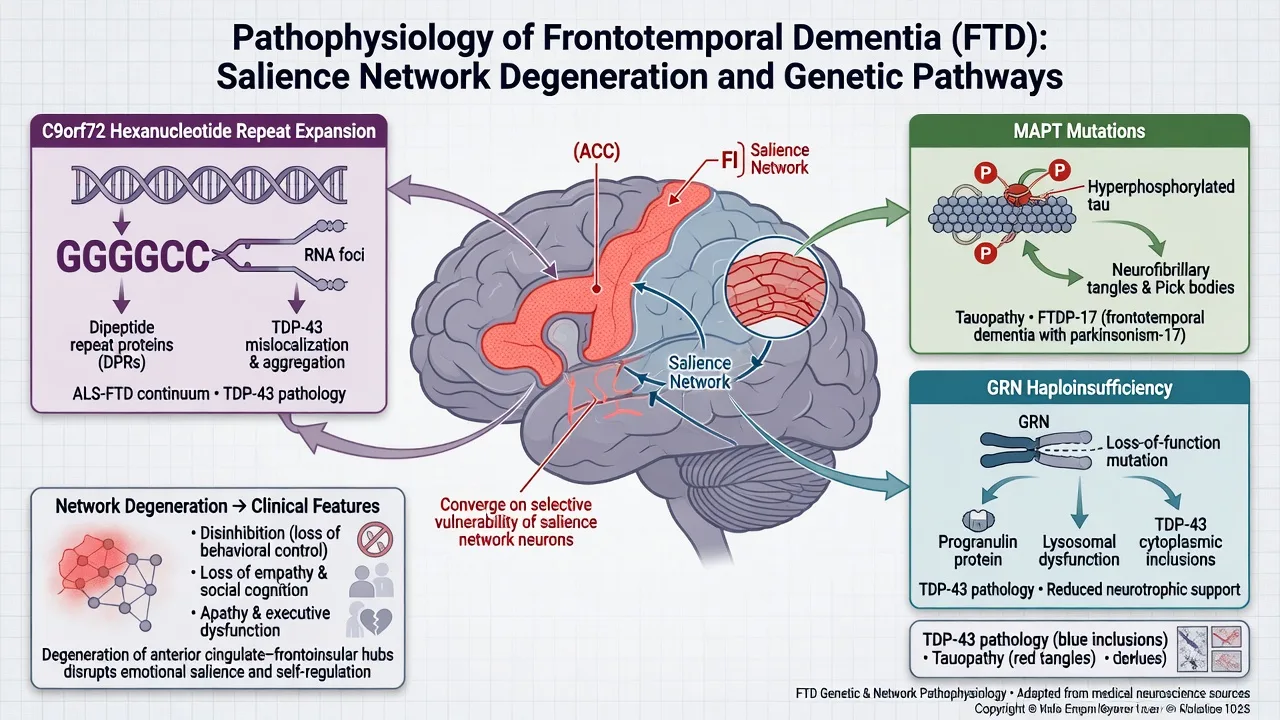
Proteinopathies (Mackenzie update). FTLD is classified by the dominant pathologic protein: FTLD-tau, FTLD-TDP (with subtypes), and FTLD-FUS, among rarer entities. Clinical syndrome imperfectly predicts protein type — hence the need for careful phenotype–pathology humility in viva answers.[16]
Networks. bvFTD preferentially disrupts salience and frontoinsular systems supporting social cognition, empathy, and behavioural control; language variants track language-network topography (left peri-Sylvian, anterior temporal, temporoparietal).[9][10]
Genetics — the exam triad
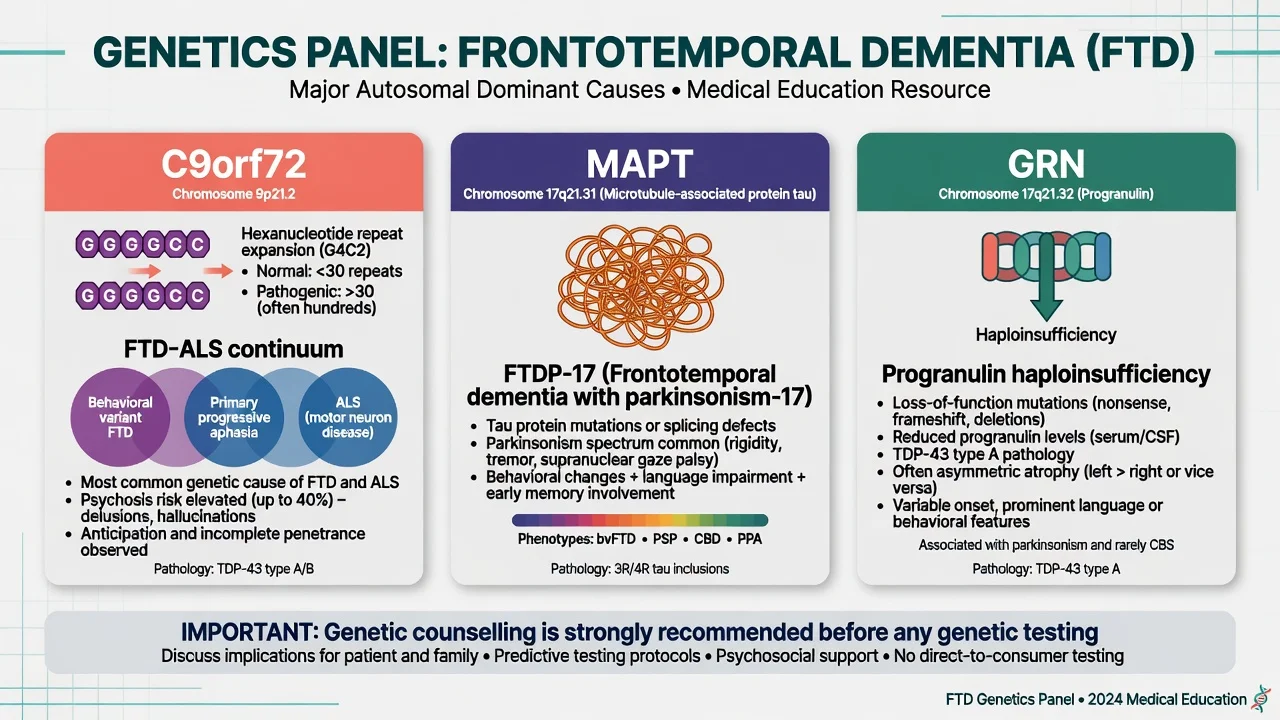
C9orf72. Expanded GGGGCC hexanucleotide repeat in a noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS — independently established in landmark 2011 studies.[4][5] Clinical pearls: FTD, ALS, or both in the same pedigree; psychosis and bizarre behaviour can be prominent; anticipation-like family stories may appear but mechanism teaching should stay with expansion biology rather than classic trinucleotide dogma.
MAPT. Missense and splice-site mutations in the tau gene cause FTDP-17 and related tauopathies — the classic molecular bridge between hereditary FTD and tau biology.[6] Parkinsonism and PSP/CBD-like features may co-travel in MAPT families.
GRN (progranulin). Mutations causing progranulin haploinsufficiency produce tau-negative, ubiquitin/TDP-positive FTLD linked to chromosome 17 — established in parallel 2006 Nature reports.[7][8] Often asymmetric atrophy clinically; plasma progranulin can be a research/clinical adjunct in specialist pathways (do not treat as a standalone community screening test).
Clinical presentation
bvFTD
Early social disinhibition (tactless remarks, traffic risk, sexual comments), apathy that families call "laziness," coldness or loss of empathy, rigid rituals, clock-watching, and new sweet-food preference or hyperorality are classic. Insight is often poor. Episodic memory and visuospatial skills may be relatively preserved early — a key discriminator from typical amnestic Alzheimer disease.[1][10]
MSE may show fatuous affect, utilization behaviour, echolalia, stimulus-bound behaviour, or bland indifference to deficits. Collateral is usually more informative than the patient's self-report.[10][14]
Language variants
nfvPPA: effortful, halting, agrammatic output; sound distortions of apraxia of speech; relatively better single-word comprehension early.[2]
svPPA / semantic dementia: fluent but empty speech, anomia, loss of word meaning ("What is a zebra?"), surface dyslexia; behavioural features of the FTD spectrum may emerge over time.[2][11]
lvPPA: word-finding pauses, impaired phrase/sentence repetition, phonological errors; often slower progression of "behavioural FTD" features and higher likelihood of Alzheimer biomarkers.[2]
Motor and neuropsychiatric overlays
Parkinsonism, falls, gaze palsy spectrum features, or clear MND signs expand the differential into PSP/CBD-related tauopathies and ALS-FTSD.[9][15] Psychosis is not rare in genetic FTD, especially C9orf72-related disease, and drives misclassification as primary late-onset psychosis.[4][5][14]
Differential diagnosis
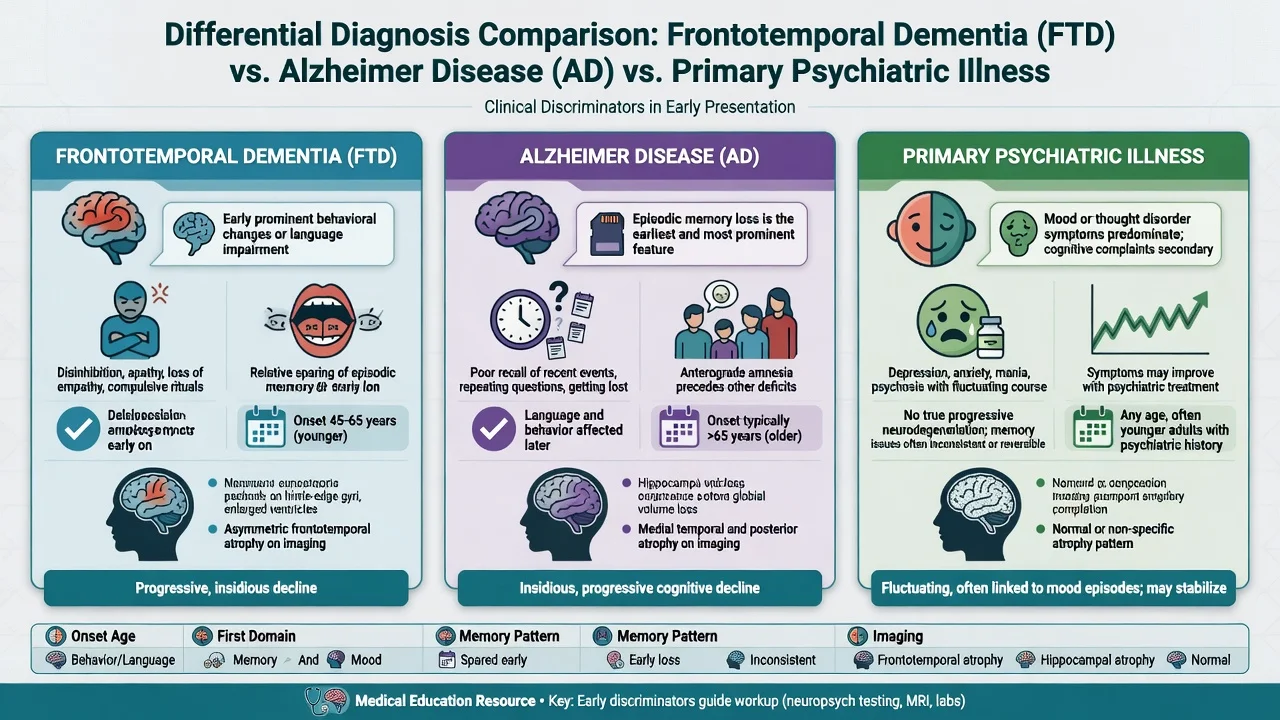
Longitudinal decline is the practical discriminator — many patients labelled "late bipolar" or "personality disorder" are only reclassified after relentless progression and imaging support.[10][14]
Other key differentials. Typical and frontal-variant Alzheimer disease; DLB (fluctuations, RBD, well-formed visual hallucinations, neuroleptic sensitivity); vascular frontal-subcortical syndromes; alcohol-related brain injury; TBI; frontal tumours; autoimmune encephalitis; rapidly progressive dementias (including CJD); adult-onset leukodystrophies in selected young-onset work-ups.[9]
bvFTD phenocopy syndrome: non-progressive or minimally progressive behavioural syndrome that mimics bvFTD clinically without clear neurodegenerative trajectory — requires longitudinal follow-up and restraint in definitive labelling.[10]
Assessment
History structure. Onset and tempo; first symptom domain (behaviour vs language vs motor); work and relationship collapse; legal/financial events; dietary change; hygiene; empathy anecdotes; rituals; sleep; substances; head injury; family history of dementia, ALS, parkinsonism, or psychiatric institutionalisation; driving and weapon access; dependent children at home.[10][14]
MSE and cognition. Full behavioural and language sampling. Standard MMSE may be deceptively high early in bvFTD; use broader screens (e.g. MoCA or ACE-style batteries) and dedicated executive/social-cognition probes. Formal neuropsychology is often decisive for PPA subtyping and for medico-legal capacity reports.[1][2][10]
Neurologic exam. Always: eye movements, extrapyramidal signs, fasciculations, bulk/power/reflexes, primitive reflexes, gait, bulbar function.[15]
Risk and capacity. Assess driving, finances, exploitation vulnerability, aggression, fire/cooking, sexual disinhibition, and child safety. Capacity is decision-specific: understand, appreciate, reason, communicate a choice (Appelbaum framework). Use supported decision-making and least-restrictive legal pathways; statutes are jurisdiction-specific — name principles, not invented section numbers for other countries.[18]
Investigations
Bloods. Standard dementia panel (FBC, U&E, LFT, B12, folate, TSH, glucose; consider HIV/syphilis if risk or atypical). These exclude co-contributors; they do not diagnose FTD.[9]
Imaging. Structural MRI once to exclude mass lesions and to support pattern diagnosis (frontal/anterior temporal predominance in bvFTD; language-network topography in PPA). Discuss patterns educationally — never fabricate a patient's scan description. FDG-PET/SPECT can support hypometabolism patterns when structural imaging is equivocal in specialist pathways.[1][2][10]
Biomarkers. CSF or PET Alzheimer biomarkers help when lvPPA or mixed amnestic features raise AD likelihood. They are not a community screen for pure bvFTD.[2][9]
Genetics. Offer testing with pre- and post-test counselling when there is family history, young onset, ALS-FTD spectrum features, or clear patient/family goals. Do not force predictive testing on asymptomatic relatives without formal genetic counselling pathways.[4][5][6][7][9]
EMG / neurology. When MND is clinically possible.[15]
Acute and emergency management
If imminent risk of harm persists after non-drug measures, a time-limited lowest-effective-dose antipsychotic trial may be considered with explicit documentation of the increased mortality signal associated with atypical antipsychotics in elderly patients with dementia, and a plan to review and stop.[17]
Definitive management
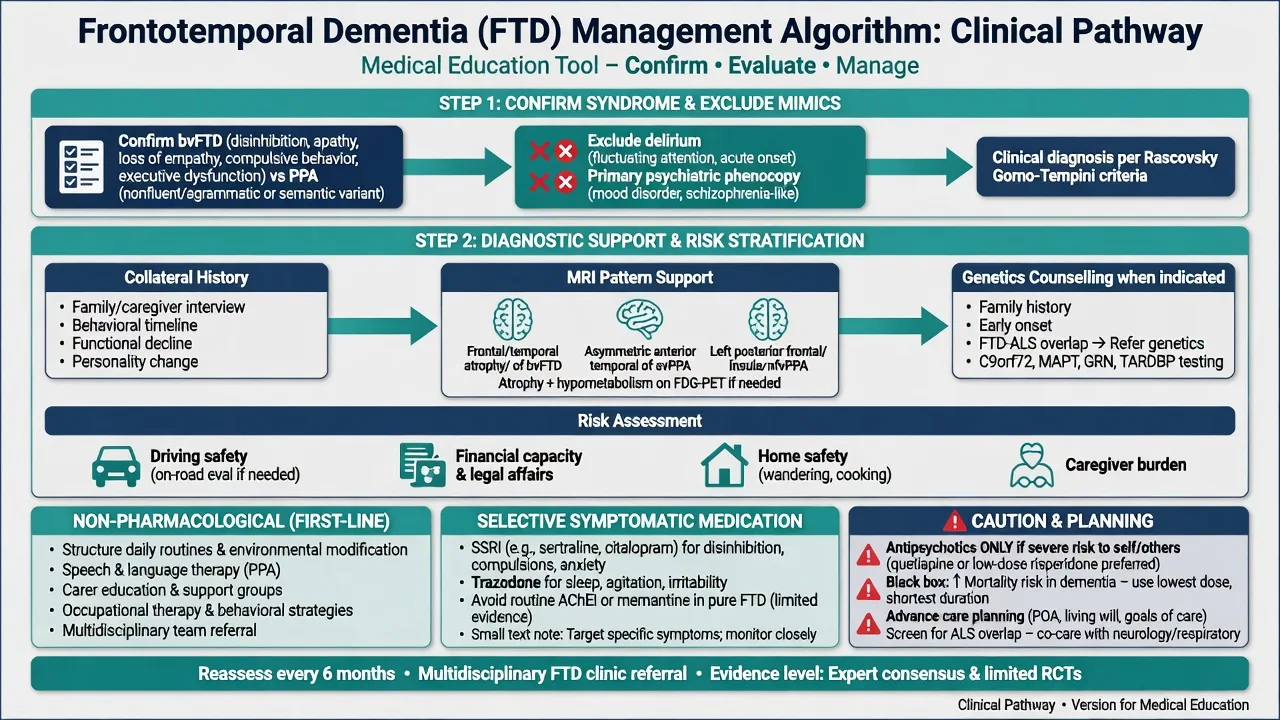
Non-pharmacological care (first line)
Structure the day; simplify choices; redirect rather than confront; secure the environment; speech-language therapy for PPA (communication aids, script training concepts); occupational therapy for safety; social work for benefits, guardianship interfaces, and respite; carer education and peer support. Expect anosognosia to limit "insight-oriented" psychotherapy as the primary disease treatment.[9][10]
Pharmacotherapy — know the limits
There is no established disease-modifying pharmacotherapy for FTD in standard clinical practice; drug development remains an active research frontier.[9][12]
Serotonergic agents. A systematic review of neurotransmitter deficits and treatments supports exploring serotonergic strategies for behavioural symptoms; SSRIs are commonly used in practice for disinhibition, irritability, compulsive behaviours, and hyperorality, with a limited rigorous evidence base — set modest expectations and monitor hyponatraemia, falls, and activation in older/frail patients.[12]
Named symptomatic example (individualise; specialist context): sertraline 25–50 mg orally daily, titrate cautiously (e.g. toward 50–100 mg) while monitoring behaviour with carer scales, sodium in at-risk older adults, GI effects, and sleep. This is symptomatic behavioural care, not disease modification.[12]
Trazodone. A randomised controlled trial in FTD found trazodone improved behavioural symptoms versus placebo; sedation and falls risk require caution. Example specialist-style dosing discussion often starts low (e.g. 50 mg orally at night, titrate as tolerated — confirm local product information and comorbidity).[13]
Cholinesterase inhibitors and memantine. Unlike mild–moderate Alzheimer disease, these are not first-line for pure FTD and may lack benefit or worsen behaviour in some patients. Exception thinking: if the syndrome is lvPPA with Alzheimer biomarkers, an AD symptomatic trial may be considered under cognitive-disorder specialist care — do not extend that logic automatically to bvFTD or svPPA.[2][9][12]
Antipsychotics. Reserve for severe psychosis or dangerous aggression after behavioural measures. Counsel on the increased mortality risk demonstrated in meta-analysis of atypical antipsychotics for dementia; use lowest dose, shortest duration, and review stop dates.[17]
Stimulants, mood stabilisers, and benzodiazepines. Not routine first-line disease therapy; benzos add falls/delirium risk; any trial must be highly individualised and time-limited.[12]
Multidisciplinary and genetic care
Co-manage with neurology for MND, parkinsonism, or complex language syndromes; involve palliative care early when ALS-FTSD or advanced behavioural FTD emerges; offer genetic counselling for families.[9][15]
Prognosis and disposition
Course is progressive. Survival is variable by syndrome, co-MND, and genetics; FTD often shortens survival relative to late-onset amnestic AD on average, but avoid single-number dogma in exams — emphasise trajectory, MND status, and support needs.[9][10]
Disposition ladder. Memory/behaviour clinic; old-age psychiatry or neuropsychiatry; speech pathology; MND clinic; community packages; residential care with behavioural expertise; involuntary pathways only when risk and incapacity meet local legal thresholds. Advance care planning should start while residual capacity may still allow participation.[10][18]
Regional guideline notes
ANZ (FRANZCP / RANZCP context). Frame FTD within old-age and CL neuropsychiatry services, dementia care pathways, and jurisdiction-specific guardianship/mental health law. Genetic testing access varies by state/service — counsel first.[9][10]
UK (NICE dementia principles). Person-centred care, carer support, non-drug approaches for behaviour, and careful antipsychotic use align with FTD management even when disease-specific FTD pharmacotherapy evidence is thin.[9][17]
US (APA / ABPN). Emphasise Rascovsky/Gorno-Tempini criteria literacy, C9orf72 spectrum, and limits of AChEI in pure FTD; research trials may be discussed for eligible patients.[1][2][9]
India (MD/DNB / NEET-SS). Same clinical criteria and genetic triad apply; adapt drug availability and family-based care structures; still name agents you can monitor.[9][12]
Exam pearls
FTD exam checklist
This checklist compresses the high-yield FTD viva axes: criteria, genetics, misdiagnosis, treatment limits, MND overlap, and medicolegal risk.[1][9][12][15][17]
High-yield traps for MCQ/MEQ
- Diagnosing bipolar disorder from progressive disinhibition without longitudinal or imaging thinking.[14]
- Starting donepezil automatically because "it is dementia."[9][12]
- Missing fasciculations in a "psychiatric" middle-aged patient with new apathy.[15]
- Forgetting that lvPPA is often AD pathologically.[2]
- Using long-term antipsychotics for wandering without behavioural formulation or mortality counselling.[17]
- Offering predictive genetic testing to relatives without counselling infrastructure.[4][7]
References
- [1]Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia Brain, 2011.PMID 21810890
- [2]Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants Neurology, 2011.PMID 21325651
- [3]Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria Neurology, 1998.PMID 9855500
- [4]DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS Neuron, 2011.PMID 21944778
- [5]Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD Neuron, 2011.PMID 21944779
- [6]Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17 Nature, 1998.PMID 9641683
- [7]Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17 Nature, 2006.PMID 16862116
- [8]Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21 Nature, 2006.PMID 16862115
- [9]Bang J, Spina S, Miller BL Frontotemporal dementia Lancet, 2015.PMID 26595641
- [10]Piguet O, Hornberger M, Mioshi E, Hodges JR Behavioural-variant frontotemporal dementia: diagnosis, clinical staging, and management Lancet Neurol, 2011.PMID 21147039
- [11]Hodges JR, Patterson K Semantic dementia: a unique clinicopathological syndrome Lancet Neurol, 2007.PMID 17945154
- [12]Huey ED, Putnam KT, Grafman J A systematic review of neurotransmitter deficits and treatments in frontotemporal dementia Neurology, 2006.PMID 16401839
- [13]Lebert F, Stekke W, Hasenbroekx C, Pasquier F Frontotemporal dementia: a randomised, controlled trial with trazodone Dement Geriatr Cogn Disord, 2004.PMID 15178953
- [14]Woolley JD, Khan BK, Murthy NK, et al. The diagnostic challenge of psychiatric symptoms in neurodegenerative disease: rates of and risk factors for prior psychiatric diagnosis in patients with frontotemporal dementia J Clin Psychiatry, 2011.PMID 21382304
- [15]Strong MJ, Abrahams S, Goldstein LH, et al. Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria Amyotroph Lateral Scler Frontotemporal Degener, 2017.PMID 28054827
- [16]Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update Acta Neuropathol, 2010.PMID 19924424
- [17]Schneider LS, Dagerman KS, Insel P Risk of death with atypical antipsychotic drug treatment for dementia: meta-analysis of randomized placebo-controlled trials JAMA, 2005.PMID 16234500
- [18]Appelbaum PS Clinical practice. Assessment of patients' competence to consent to treatment N Engl J Med, 2007.PMID 17978292