Psych · Psychopharmacology — first-generation antipsychotics
First-generation antipsychotics
Also known as Typical antipsychotics · Conventional antipsychotics · Neuroleptics · FGA · Haloperidol · Chlorpromazine · Perphenazine · Phenothiazines · Butyrophenones
Exam-exhaustive fellowship monograph on first-generation antipsychotics — chemical and potency classification, D2 occupancy and EPS/prolactin pathways, CATIE/CUtLASS/EUFEST evidence vs SGAs, oral and depot dosing scaffolds, acute EPS and NMS emergencies, TD epidemiology (Carbon), monitoring, special populations. FRANZCP-primary, globally tagged.
On this page & tools
Your progress
Saved locally on this device.
10 MCQs with explanations
Target exams
Red flags
First-generation antipsychotics (FGAs, typical/conventional neuroleptics) are a high-yield fellowship monograph because examiners test class literacy, landmark comparative effectiveness, receptor–side-effect mapping, and emergency EPS/NMS management. CATIE and CUtLASS dismantled the myth that every SGA is always superior; EUFEST shows first-episode patients tolerate low-dose high-potency FGA poorly; Carbon meta-analyses quantify the TD differential; TRRIP stops endless FGA switches in treatment resistance.[1][2][3][8][9][12]
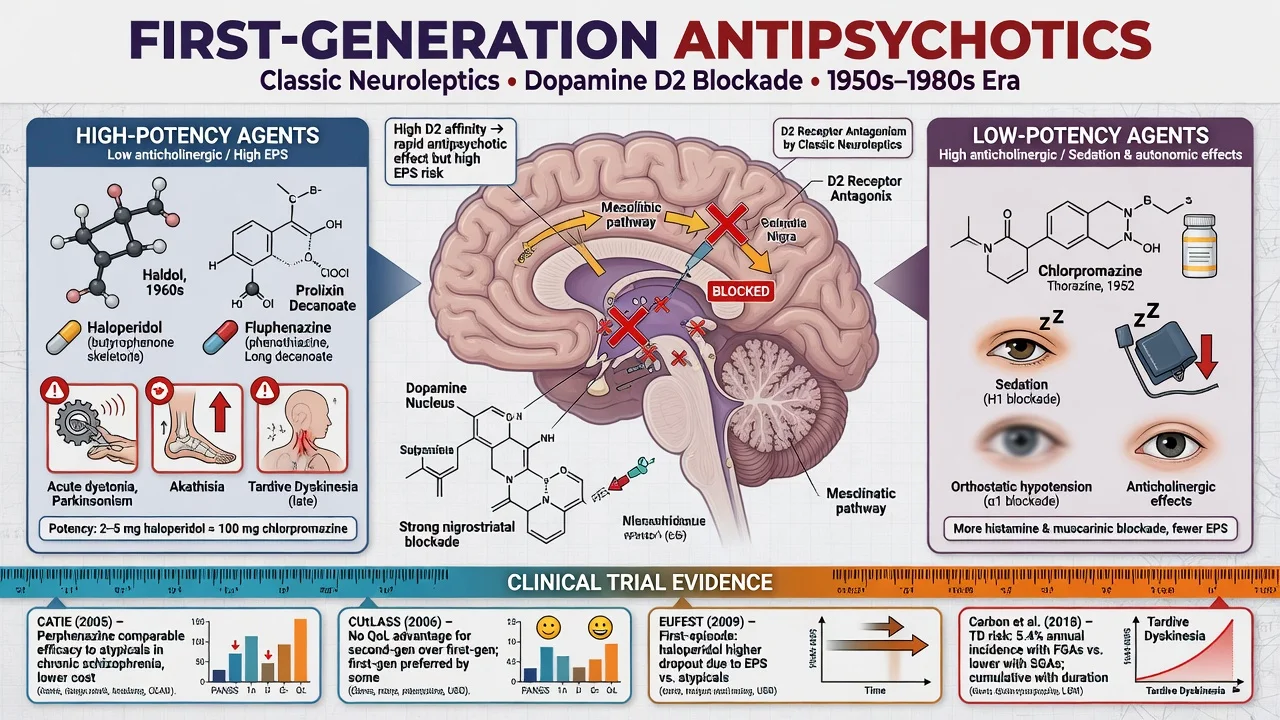
Definition and classification
FGAs are antipsychotics whose primary clinical action is dopamine D2 receptor antagonism, historically developed before the multi-receptor clozapine-inspired SGA era. They are not a single homogeneous class: chemical family and potency determine the adverse-effect map more than the “typical” label alone.[4][5][6]
Chemical families (exam examples): phenothiazines, thioxanthenes, butyrophenones and related agents map onto the same D2-antagonist class with different potency and side-effect loads.[4]
| Family | Exam agents | Notes |
|---|---|---|
| Phenothiazines — aliphatic | Chlorpromazine | Low potency; sedation, hypotension, anticholinergic |
| Phenothiazines — piperidine | Thioridazine (historical caution) | Anticholinergic; QT and pigmentary retinopathy warnings at high dose |
| Phenothiazines — piperazine | Fluphenazine, trifluoperazine, perphenazine | Higher potency; more EPS |
| Thioxanthenes | Flupentixol, zuclopenthixol | Oral and classic oil depots |
| Butyrophenones | Haloperidol | Prototype high-potency FGA; oral, short-acting IM, depot |
| Diphenylbutylpiperidines | Pimozide | Specialist use; QT teaching |
| Chemical family and formulation differences matter clinically for depot choice and adverse-effect prediction.[4][14] |
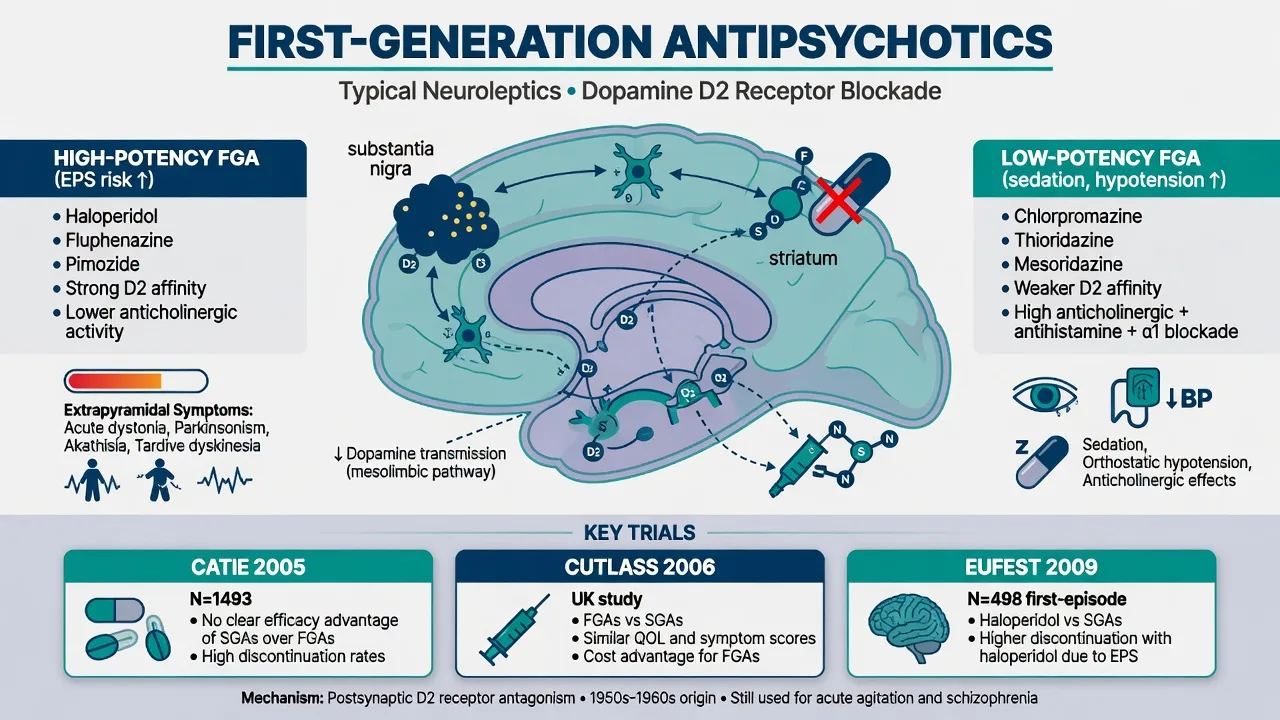
Potency axis (non-negotiable viva language): high-potency FGAs (haloperidol, fluphenazine, trifluoperazine) → more EPS and hyperprolactinaemia, less sedation/hypotension/anticholinergic load. Low-potency FGAs (chlorpromazine, thioridazine) → more sedation, postural hypotension, anticholinergic effects, less pure EPS at equieffective antipsychotic doses. Mid-potency agents (perphenazine — the CATIE FGA arm) sit between.[1][4]
Formulations: oral maintenance; short-acting IM for acute agitation (haloperidol teaching staple in many rapid-tranquillisation protocols); oil-based LAI depots (flupentixol, zuclopenthixol, fluphenazine, haloperidol decanoate where available). Depot is not chemical restraint and is not a substitute for shared decision when capacity allows.[14]
Epidemiology, use and the SGA myth
FGAs remain widely used for cost, depot familiarity, involuntary settings, and metabolic-sparing niches. The fellowship trap is binary thinking (“SGA always better”) — CATIE and CUtLASS both undercut automatic SGA superiority claims.[1][2]
CATIE (Lieberman 2005). Pragmatic effectiveness trial in chronic schizophrenia: olanzapine, quetiapine, risperidone, ziprasidone, and the FGA perphenazine. All-cause discontinuation was high across arms. Olanzapine had longer time to discontinuation but more metabolic burden. Perphenazine was broadly comparable to several SGAs on effectiveness when patients with tardive dyskinesia were excluded from randomisation to perphenazine — the exam one-liner that still surprises candidates.[1]
CUtLASS 1 (Jones 2006). UK pragmatic RCT: no advantage of SGAs over FGAs on quality of life at one year — cost-utility and class-superiority claims need evidence, not marketing.[2]
EUFEST (Kahn 2008). First-episode schizophrenia/schizophreniform open randomised trial: low-dose haloperidol versus several SGAs. Discontinuation of treatment was higher with haloperidol despite similar efficacy signals — FEP patients are EPS-sensitive; high-potency FGA first-line is hard to defend when alternatives exist.[3]
Leucht 2013 NMA. Hierarchical comparative efficacy and tolerability of 15 antipsychotics including classic FGAs: all beat placebo on symptoms; agents differ more on side-effect profiles than on tiny efficacy gaps (clozapine remains efficacy outlier for resistance, not a routine FGA).[4]
Mortality context (FIN11). Long-term antipsychotic treatment associated with lower mortality than no antipsychotic use in schizophrenia — arguing against nihilistic non-treatment; agent choice still balances metabolic vs neurological toxicity.[7]
Numbers and names every candidate must own
Landmark comparative and occupancy teaching anchors for viva speed.[1][2][3][4][6][8]
Pathophysiology and receptor map
Howes and Kapur’s dopamine hypothesis version III frames psychosis as a final common pathway of striatal dopamine dysregulation; antipsychotics reduce D2 transmission in that pathway.[5]
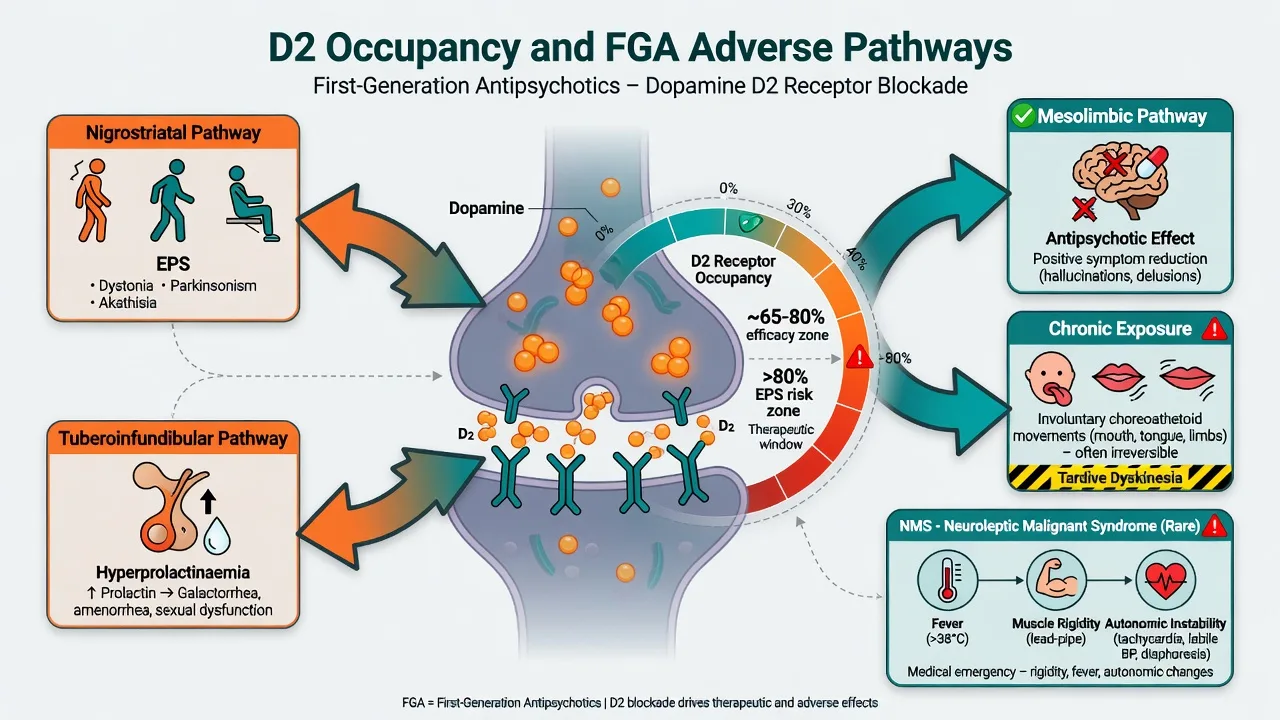
Occupancy teaching model (viva staple): antipsychotic efficacy often associates with roughly 65–80% striatal D2 occupancy; EPS risk rises as occupancy pushes higher (classically toward and beyond about 80%). High-potency FGAs reach high occupancy at relatively low milligram doses — which is why “low milligrams” can still produce dystonia.[5][6]
Kapur–Seeman fast-off hypothesis: many atypicals (especially clozapine/quetiapine) dissociate more rapidly from D2 than classic FGAs such as haloperidol, accommodating physiological dopamine transmission and reducing EPS/prolactin liability — a teaching model, not the whole story of atypicality.[6]
Pathway map: circuit-level teaching links mesolimbic D2 block to antipsychotic effect and nigrostriatal/tuberoinfundibular block to EPS and prolactin effects.[5][6]
| Circuit teaching | Clinical consequence of D2 block |
|---|---|
| Mesolimbic | Antipsychotic effect on positive symptoms |
| Nigrostriatal | Acute dystonia, parkinsonism, contribution to akathisia |
| Tuberoinfundibular | Hyperprolactinaemia |
| Mesocortical (hypothesised) | Secondary negative/cognitive dulling |
| These pathway associations are teaching models used to organise adverse-effect prediction in viva settings.[5][13] |
Chronic D2 antagonism underpins tardive dyskinesia risk models (including receptor supersensitivity teaching language). Severe dopamine blockade plus systemic dysregulation produces neuroleptic malignant syndrome (rigidity, hyperthermia, autonomic instability, altered consciousness).[8][11][13]
Clinical effects and adverse-effect phenotypes
Therapeutic effect. Positive psychotic symptoms often begin to settle over days to a few weeks; full functional recovery needs psychosocial care, not milligrams alone. FGAs are not superior for primary negative symptoms — do not promise that.[4][5]
Acute dystonia. Hours to days after start or dose rise: torticollis, oculogyric crisis, opisthotonus; laryngeal dystonia is an airway emergency. High-potency FGAs and young men are classic risk groups.[13]
Akathisia. Subjective restlessness plus objective inability to keep still — use Barnes Akathisia Rating Scale language. Mislabeling as anxiety or “agitation from psychosis” leads to the lethal error of raising the antipsychotic.[10][13]
Drug-induced parkinsonism. Bradykinesia, rigidity, tremor, shuffling gait — dose-related; distinguish from idiopathic Parkinson disease and from negative symptoms (Ward and Citrome review teaching).[13]
Tardive dyskinesia. Delayed, often irreversible choreoathetoid movements (orofacial classic). Schooler–Kane research diagnostic criteria still structure research language; clinical practice documents with AIMS and acts early.[8][15]
Hyperprolactinaemia. Amenorrhoea, galactorrhoea, sexual dysfunction, long-term bone health concerns — common with high-potency FGAs and some SGAs (risperidone/paliperidone family); counsel and monitor symptoms.[4]
Low-potency load. Sedation, postural hypotension (alpha-1), dry mouth/constipation/urinary retention (anticholinergic), photosensitivity (chlorpromazine teaching), weight/metabolic effects less headline than olanzapine/clozapine but not absent.[4]
Differentials that save lives
Mislabeling akathisia as psychosis, dystonia as “functional,” or NMS as simple infection is a classic exam and clinical trap.[10][13]
| Presentation on FGA | Prefer | Do not miss |
|---|---|---|
| Restless, pacing, “can’t sit” | Akathisia; reduce/switch | Escalating antipsychotic dose |
| Stiff, slow, expressionless | Drug-induced parkinsonism | Pure “negative symptoms only” |
| Sudden neck/eye spasm | Acute dystonia; anticholinergic | “Functional” without treatment trial |
| Fever + rigidity + autonomic storm | NMS pathway | Serotonin syndrome, infection, catatonia differentials |
| New tongue/orofacial movements | TD work-up (AIMS) | Ignoring because “SGA era = no TD” |
| Syncope on thioridazine/high-dose IV haloperidol teaching | QTc, electrolytes, ECG | Anxiety faint only |
| Carbon meta-analyses: TD remains more prevalent and incident with FGAs than SGAs, but SGAs are not TD-free — exam killers love this nuance.[8][9][10][13] |
Assessment and consent
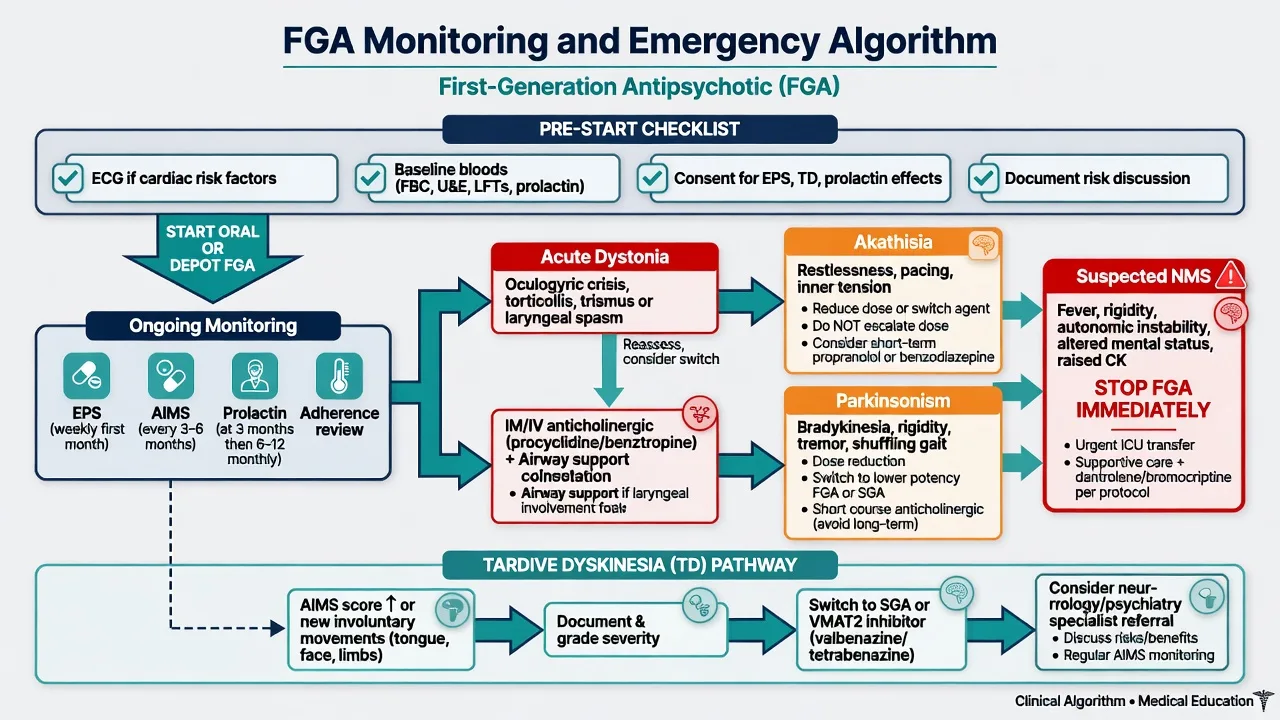
Before first dose: confirm indication (schizophrenia-spectrum, mania with psychosis, short-term agitation, other psychoses as justified); past EPS/TD; falls and Parkinson disease/DLB (FGAs generally avoid); cardiac history; pregnancy intent; substances; capacity/legal status; concurrent QT drugs.[11][12]
Consent points: expected benefits; EPS (including dystonia), akathisia, TD risk over time, prolactin effects, NMS rarity but seriousness, need for movement monitoring, and why this FGA rather than an SGA or depot alternative. For depots: oral same-molecule (or same-class) tolerability first where feasible, injection interval, missed-dose rules, and that LAI is an adherence tool not a punishment.[14]
Investigations and monitoring
Baseline (teaching scaffold): FBC, U&E, LFT, fasting glucose and lipids (cardiometabolic context of severe mental illness), weight/BMI, pregnancy test when relevant; ECG if cardiac risk, older adult, high-risk agent/dose (historical thioridazine; IV or high-dose haloperidol teaching), or QT co-drugs.[4][16]
Ongoing: clinical EPS review every visit early; serial AIMS for chronic use; symptom-directed prolactin; adherence; metabolic co-monitoring as for any long-term antipsychotic population. AGNP-style TDM is not routine for every FGA but plasma levels may help for selected agents when adherence or toxicity is in question (haloperidol among agents with TDM literature).[16]
Acute NMS labs: CK, renal function, infection screen, exclude differentials — treat clinically while results return.[13]
Initiation, dosing scaffolds and definitive use
Exact licensed doses and products are local — state principles and product information. Teaching scaffolds examiners expect modern low-dose high-potency practice rather than historical megadose culture.[3][4]
Oral high-potency (haloperidol). Modern psychosis practice uses low doses (often in the low single-digit to low double-digit mg/day range total, titrated to response and EPS), not the historical 20–40+ mg/day culture that drove EPS. Divide or night-load per product; reassess EPS early.[3][4]
Oral low-potency (chlorpromazine). Higher milligram ranges for antipsychotic effect (often hundreds of mg/day teaching); watch BP, sedation, anticholinergic burden. Chlorpromazine-equivalent tables are comparison tools, not rigid laws.[4]
Mid-potency (perphenazine). CATIE’s FGA arm — reminds examiners that not every FGA is “haloperidol-strength EPS.”[1]
Short-acting IM. Haloperidol IM for acute agitation within local rapid-tranquillisation protocols; monitor for dystonia and QT risk; know local bans/cautions on dangerous combinations (e.g. some services avoid pairing certain IM antipsychotics with parenteral benzodiazepines).[4]
Oil depots. After oral tolerability of the same agent where possible: agent-specific test dose and interval (commonly 1–4 weeks). FGA depots retain a role when adherence is the barrier and the patient has responded to oral FGA; real-world depot data (Tiihonen) support LAI strategies after first hospitalisation as a population-level adherence tool — still shared decision when capacity allows.[14]
Anticholinergic prophylaxis. Not automatic lifelong co-prescription. Short courses when high dystonia risk (young, high-potency start, history of dystonia); review and stop — chronic anticholinergics add cognitive burden and raise TD-risk concerns in teaching literature.[11][13]
When FGA is reasonable: prior good response and tolerability; metabolic catastrophe on SGAs; cost/access; established stable depot; patient preference after informed discussion. When to prefer SGA/other: FEP default pathways in most services; high TD risk (older age, prior TD, female sex teaching factors from Solmi); Parkinson disease/DLB; patient EPS-phobic after education.[3][11]
Treatment resistance. Two adequate antipsychotic trials with confirmed adherence → TRRIP language and clozapine offer — do not rotate FGAs indefinitely.[12]
Emergency management
- Laryngeal dystonia / airway threat: oxygen/airway support + parenteral anticholinergic (benztropine or procyclidine per local product) immediately; stop or change offending FGA after stabilisation.
- Non-airway acute dystonia: IM/IV anticholinergic; oral continuation short-term; reduce dose or switch agent.
- Akathisia: reduce dose or switch class; beta-blocker or other evidence-informed options per local practice; never increase antipsychotic as first move.[10][13]
- Drug-induced parkinsonism: dose reduction, switch to lower EPS-liability agent, short anticholinergic when needed — do not permanently mask high occupancy with anticholinergics alone.[13]
- NMS: stop all dopamine antagonists; ABC; cooling; IV fluids; ICU; investigate differentials; dantrolene/bromocriptine as specialist adjuncts in selected protocols — not a ward “wait and see.”
- TD pathway: document AIMS; minimise unnecessary dopamine antagonists; consider switch (including clozapine if TRS); VMAT2 inhibitors (valbenazine/deutetrabenazine) where available — detail lives in the EPS/TD leaf topic.[8][9][12][15]
Subtypes and clinical scenarios
First-episode psychosis. EUFEST argues against default high-potency FGA when SGA options exist; if FGA used, low dose and intense EPS monitoring.[3]
Chronic multi-episode. CATIE/CUtLASS support individualised FGA use when effective and tolerated.[1][2]
Depot after non-adherence. Choose agent matching prior oral response; plan clinic DNA and re-initiation.[14]
Agitation/mania short cover. Time-limited FGA with clear stop/step-down plan once the acute behavioural risk settles.[4]
TRS on FGA. TRRIP → clozapine pathway, not another phenothiazine.[12]
Special populations
Older adults. High EPS, TD, fall, and delirium risk — avoid high-potency FGA when possible; start very low, go slow.[11]
Youth/FEP. Prefer lower EPS-liability agents; if FGA unavoidable, modern low dose and carer education on dystonia.[3]
Pregnancy and lactation. Individualised perinatal psychiatry plan; FGAs have longer human exposure history than some new SGAs but neonatal EPS/withdrawal and maternal side effects still matter — lowest effective dose, do not stop abruptly without a relapse plan.[4]
Parkinson disease / DLB. FGAs generally worsen motor function — avoid; use specialist pathways (often quetiapine/clozapine themes, not exam-safe FGA defaults).[13]
Intellectual disability. Carers must recognise dystonia and akathisia; capacity and consent frameworks apply because movement toxicity is easy to miss without self-report.[10][13]
Hepatic impairment. Phenothiazines require caution; prefer agents with safer hepatic profile when possible and monitor closely.[4][16]
Prognosis and disposition
Continuous antipsychotic exposure prevents relapse for most people with schizophrenia; an FGA that is taken and tolerated can outperform an SGA that is abandoned. Shared care must specify who checks movement, prolactin symptoms, metabolic co-morbidity, and depot attendance. After NMS, rechallenge is specialist, delayed, and often uses a different lower-risk agent — never casual same-day restart.[1][7][14]
Evidence, guidelines and regional differences
| Source | Exam take-home |
|---|---|
| CATIE 2005 | Perphenazine comparable to many SGAs on effectiveness; olanzapine more durable but metabolic |
| CUtLASS 2006 | No QoL superiority of SGAs over FGAs |
| EUFEST 2008 | FEP: more discontinuation with low-dose haloperidol |
| Leucht 2013 | Efficacy hierarchy modest; tolerability differs widely |
| Howes & Kapur 2009 | Dopamine final common pathway framework |
| Kapur & Seeman 2001 | Fast-off D2 model of atypicality |
| Tiihonen FIN11 | Antipsychotic treatment linked to lower mortality vs none |
| Carbon 2017/2018 | Higher TD with FGAs; SGAs not TD-free |
| Landmark synthesis for viva speed.[1][2][3][4][5][6][7][8][9] | |
| Barnes 1989 | Akathisia rating structure |
| Schooler–Kane 1982 | TD research diagnostic language |
| TRRIP 2017 | Define resistance; path to clozapine |
| AGNP TDM | Selected FGA level use for adherence/toxicity |
ANZ: SGA-first culture in many early-psychosis services; FGAs remain in acute IM and depot formularies — follow local rapid-tranquillisation and depot protocols. UK: NICE schizophrenia guidance historically emphasises individualised choice and monitoring rather than blanket FGA ban; CUtLASS is British exam culture. US: APA supports individualised antipsychotic selection; CATIE is core ABPN teaching. Europe: strong depot traditions (zuclopenthixol/flupentixol) and AGNP TDM culture for selected agents. Product availability and QT warnings differ — principles travel; labels do not.[1][2][3][16]
Exam pearls
TYPICAL non-negotiables
TYPICAL
References
- [1]Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia N Engl J Med, 2005.PMID 16172203
- [2]Jones PB, Barnes TR, Davies L, et al. Randomized controlled trial of the effect on Quality of Life of second- vs first-generation antipsychotic drugs in schizophrenia: Cost Utility of the Latest Antipsychotic Drugs in Schizophrenia Study (CUtLASS 1) Arch Gen Psychiatry, 2006.PMID 17015810
- [3]Kahn RS, Fleischhacker WW, Boter H, et al. Effectiveness of antipsychotic drugs in first-episode schizophrenia and schizophreniform disorder: an open randomised clinical trial Lancet, 2008.PMID 18374841
- [4]Leucht S, Cipriani A, Spineli L, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis Lancet, 2013.PMID 23810019
- [5]Howes OD, Kapur S The dopamine hypothesis of schizophrenia: version III--the final common pathway Schizophr Bull, 2009.PMID 19325164
- [6]Kapur S, Seeman P Does fast dissociation from the dopamine d(2) receptor explain the action of atypical antipsychotics?: A new hypothesis Am J Psychiatry, 2001.PMID 11229973
- [7]Tiihonen J, Lönnqvist J, Wahlbeck K, et al. 11-year follow-up of mortality in patients with schizophrenia: a population-based cohort study (FIN11 study) Lancet, 2009.PMID 19595447
- [8]Carbon M, Hsieh CH, Kane JM, Correll CU Tardive Dyskinesia Prevalence in the Period of Second-Generation Antipsychotic Use: A Meta-Analysis J Clin Psychiatry, 2017.PMID 28146614
- [9]Carbon M, Kane JM, Leucht S, Correll CU Tardive dyskinesia risk with first- and second-generation antipsychotics in comparative randomized controlled trials: a meta-analysis World Psychiatry, 2018.PMID 30192088
- [10]Barnes TR A rating scale for drug-induced akathisia Br J Psychiatry, 1989.PMID 2574607
- [11]Solmi M, Pigato G, Kane JM, Correll CU Clinical risk factors for the development of tardive dyskinesia J Neurol Sci, 2018.PMID 29439776
- [12]Howes OD, McCutcheon R, Agid O, et al. Treatment-Resistant Schizophrenia: Treatment Response and Resistance in Psychosis (TRRIP) Working Group Consensus Guidelines on Diagnosis and Terminology Am J Psychiatry, 2017.PMID 27919182
- [13]Ward KM, Citrome L Antipsychotic-Related Movement Disorders: Drug-Induced Parkinsonism vs. Tardive Dyskinesia-Key Differences in Pathophysiology and Clinical Management Neurol Ther, 2018.PMID 30027457
- [14]Tiihonen J, Haukka J, Taylor M, et al. A nationwide cohort study of oral and depot antipsychotics after first hospitalization for schizophrenia Am J Psychiatry, 2011.PMID 21362741
- [15]Schooler NR, Kane JM Research diagnoses for tardive dyskinesia Arch Gen Psychiatry, 1982.PMID 6121550
- [16]Hiemke C, Bergemann N, Clement HW, et al. Consensus Guidelines for Therapeutic Drug Monitoring in Neuropsychopharmacology: Update 2017 Pharmacopsychiatry, 2018.PMID 29390205