Cardiology · Cardiology
Cardiogenic Shock
Also known as Cardiac shock · Cardiogenic circulatory collapse · Acute circulatory failure · Shock secondary to cardiac pump failure
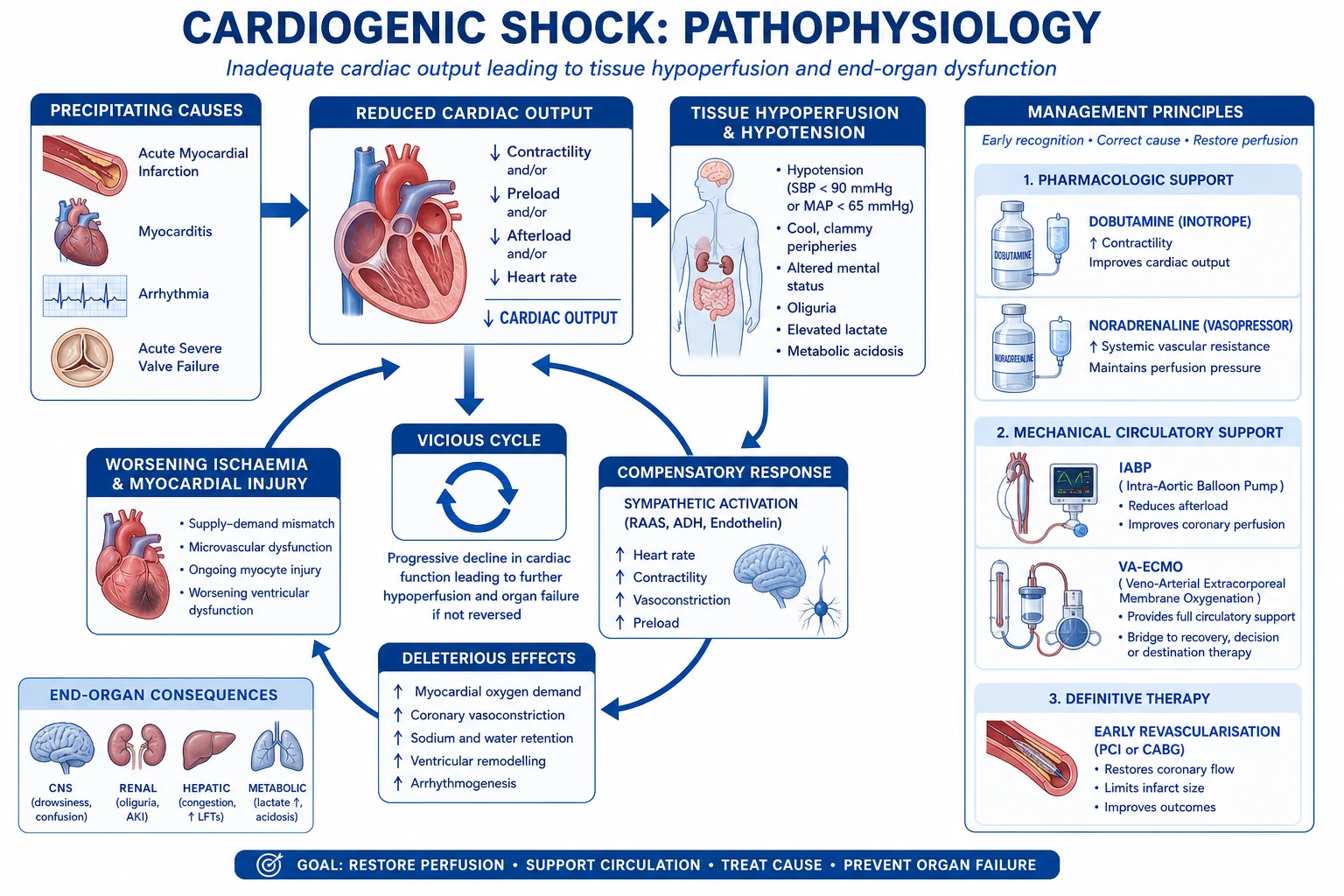
Cardiogenic shock (CS) is a state of end-organ hypoperfusion due to cardiac pump failure, defined for acute MI as systolic blood pressure below 90 mmHg for at least 30 minutes (or needing inotropes/vasopressors to keep it above this), cardiac index below 2.2 L/min/m2, and signs of hypoperfusion (cool peripheries, oliguria under 0.5 mL/kg/hour, altered mentation, raised lactate), with pulmonary capillary wedge pressure over 18 mmHg (confirming a cardiac cause). The commonest cause is acute MI with left ventricular failure (about 80 percent) — typically large anterior STEMI, but also right ventricular infarction and mechanical complications (papillary muscle rupture, ventricular septal rupture, free-wall rupture) at a mean of 3 to 6 days post-MI. Other causes: acute decompensated heart failure, fulminant myocarditis, end-stage cardiomyopathy, arrhythmia, valvular catastrophe (acute severe MR/AR), massive pulmonary embolism, tamponade, drug toxicity (beta-blocker, CCB, digoxin). Pathophysiology is a vicious downward spiral: myocardial injury reduces stroke volume, falling cardiac output drops systemic and coronary perfusion pressure, which worsens ischaemia, which further reduces contractility — the spiral is broken only by early reperfusion and circulatory support. The SCAI SHOCK stages (A to E) stratify severity from 'at risk' through 'extremis' and predict mortality (A roughly 4 percent, E over 80 percent). Diagnosis is clinical (hypoperfusion) plus echo (cardiac cause) plus invasive haemodynamics (PA catheter); raised lactate and low cardiac power output (CPO below 0.6 W) confirm severity. Management is the SHOCK-funnel / National Cardiogenic Shock Initiative protocol: ABCDE and oxygen; early escalation to a shock centre; bed-side echo; arterial line and PA catheter; pharmacological haemodynamic support (inotrope — dobutamine or milrinone; vasopressor — noradrenaline); prompt revascularisation (PCI within 90 minutes; culprit-lesion-only PCI per CULPRIT-SHOCK); mechanical circulatory support (IABP, Impella, VA-ECMO) as bridge to recovery, decision, transplant or LVAD. Early revascularisation reduced 6-month and 1-year mortality in the SHOCK trial (benefit confined to under-75s). Routine IABP did NOT reduce mortality in IABP-SHOCK II (demoted from Class I). Despite modern care, in-hospital mortality remains 40 to 50 percent.
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags
Overview & Definition
Cardiogenic shock is a clinical syndrome of inadequate tissue perfusion due to cardiac pump dysfunction. The heart cannot deliver sufficient cardiac output to meet the metabolic demands of the body, despite adequate or even raised intravascular volume.[6][13]
The most widely used diagnostic criteria are those of the SHOCK trial registry, refined for acute MI complicated by shock:[1]
- Systolic blood pressure below 90 mmHg for at least 30 minutes, OR requiring inotropes or vasopressors to maintain SBP at least 90 mmHg;
- Cardiac index below 2.2 L/min/m2 of body surface area;
- Signs of end-organ hypoperfusion: cool, mottled, clammy peripheries, oliguria (urine output below 0.5 mL/kg/hour), altered mental status, raised serum lactate;
- Pulmonary capillary wedge pressure over 18 mmHg when measured (this criterion confirms the cause is cardiac rather than hypovolaemic, and is the single feature that distinguishes cardiogenic shock from other shock states);
- Exclusion of hypovolaemia, sepsis, and other non-cardiac causes as the primary driver. [1]
The clinical importance of cardiogenic shock is its lethality: it complicates roughly 6 to 10 percent of acute MIs, is the leading cause of in-hospital death in MI patients under 75, and — even with modern revascularisation, inotropes and mechanical circulatory support — carries an in-hospital mortality of 40 to 50 percent. The exam skill lies in (1) recognising shock early, before SBP collapses (perfusing organs fail before blood pressure falls — watch lactate, urine output, mentation, skin); (2) distinguishing cardiogenic shock from its mimics (septic, hypovolaemic, obstructive, distributive), because the management is the opposite; (3) identifying the cardiac cause and the reversible subgroup (MI, tamponade, massive PE, mechanical complication, drug toxicity); and (4) applying the shock-funnel protocol with rapid escalation from inotropes to mechanical circulatory support and early revascularisation.[13]
Classification
Cardiogenic shock is classified by SCAI staging (the modern prognostic standard), by Forrester haemodynamic subset (the bedside physiological model), and by aetiology (which determines treatment). [1]
SCAI A — At risk
- Patient at risk of CS but not yet hypoperfused
- e.g. large STEMI, acute severe HF without hypoperfusion
- In-hospital mortality roughly 3 to 4 percent
- Vigilance, monitor closely
SCAI B — Beginning
- Beginning CS — hypoperfusion starting; may still be normotensive
- Tachycardia, falling urine output, mild lactate rise, vasoconstriction
- Haemodynamically 'warm' but with subtle hypoperfusion
- The B to C boundary is the appearance of overt hypoperfusion
- In-hospital mortality roughly 7 to 9 percent
SCAI C — Classic
- Classic CS — the textbook form
- Hypoperfusion now present with hypotension requiring inotrope or vasopressor
- Wedge over 18, CI below 2.2, lactate raised
- The form on which most trials and criteria are built
- In-hospital mortality roughly 12 to 20 percent
SCAI D — Deteriorating
- Deteriorating CS — getting worse despite escalating inotropes or vasopressors
- Worsening hypoperfusion, rising lactate, end-organ dysfunction
- Trigger for mechanical circulatory support
- In-hospital mortality roughly 40 percent
SCAI E — Extremis
- Extremis — circulatory collapse; cardiac arrest or peri-arrest
- Requiring CPR or on multiple pressors plus MCS
- VA-ECMO or emergent cannulation
- In-hospital mortality over 80 percent
The SCAI staging was published in 2019 (Baran et al.) and updated in 2022 (Naidu et al.); the B-to-C boundary is the appearance of hypoperfusion (a verbatim concept from the consensus statement), and stages track in-hospital mortality in a steep gradient that has been validated in multiple registries (Jentzer et al., Naidu et al.).[6][7][8]
The older Forrester haemodynamic subsets (Forrester, Diamond, Chatterjee; NEJM 1976) classify the patient at the bedside using two numbers: pulmonary capillary wedge pressure (proxy for left-sided filling) and cardiac index.[10][11]
- Subset I — warm and dry: wedge below 18 mmHg, CI above 2.2 L/min/m2. Normal perfusion, no congestion. Mortality about 1 percent.
- Subset II — warm and wet: wedge over 18 mmHg, CI above 2.2 L/min/m2. Pulmonary oedema, good peripheral perfusion. Mortality about 10 percent.
- Subset III — cold and dry: wedge below 18 mmHg, CI below 2.2 L/min/m2. Hypoperfusion without congestion — usually relative hypovolaemia; treat with cautious fluid challenge. Mortality about 20 percent.
- Subset IV — cold and wet: wedge over 18 mmHg, CI below 2.2 L/min/m2. This is classic cardiogenic shock. Mortality about 60 percent untreated. [1]
By aetiology (the classification that drives treatment) — see Differential Diagnosis and Specific Subtypes for the full list, but the headline categories are: acute MI (LV failure — the commonest), acute mechanical complication of MI (papillary muscle rupture, VSR, free-wall rupture), right ventricular infarction, acute decompensated chronic heart failure / cardiomyopathy, fulminant myocarditis, acute severe valvular disease (MR, AR, MS), arrhythmia (VT, bradyarrhythmia, AF with rapid rate), cardiac tamponade, massive pulmonary embolism (the right ventricle failing against acute afterload), post-cardiotomy shock, drug toxicity (beta-blocker, calcium-channel blocker, digoxin), and rejection of a transplanted heart.[9][13]
Epidemiology & Risk Factors
Cardiogenic shock complicates roughly 6 to 10 percent of acute MIs (down from 8 to 12 percent in the pre-reperfusion era but unchanged over the last two decades despite routine primary PCI, because patients are older and sicker). It is the leading cause of in-hospital death after MI.[1][9]
Demographic and clinical risk factors (the high-yield list): [1]
- Age — patients in shock are on average older (about 65 to 70 years); the SHOCK-trial survival benefit of early revascularisation was confined to those under 75, so age is both a risk factor and a treatment-decision modifier.[2]
- Female sex — women are over-represented in CS-MI cohorts (later presentation, smaller body size, atypical symptoms); the SHOCK trial was roughly one-third female.
- Anterior STEMI — large anterior (especially proximal LAD) infarcts are the single most common infarct location to cause CS, because of the large area of myocardium at risk.
- Large infarct size — peak CK / troponin, LV ejection fraction below 40 percent, wall-motion score index, and lack of collateral circulation or incomplete reperfusion (TIMI flow below 3, no-reflow).
- Prior MI or known LV dysfunction — the second hit on an already-compromised ventricle tips the patient into shock.
- Diabetes mellitus, chronic kidney disease, peripheral arterial disease — common comorbidities that worsen outcome.
- Delay to presentation or reperfusion — every 30-minute delay in door-to-balloon increases 1-year mortality.
- Multivessel coronary disease — supply-side failure; downstream culprit-only versus multivessel PCI is a key decision (CULPRIT-SHOCK).[4]
- Mechanical complications of MI — occur most often on day 3 to 6 post-MI, in patients with smaller infarcts (subendocardial or first MI): papillary muscle rupture (inferior MI — posteromedial papillary muscle has single blood supply), ventricular septal rupture, free-wall rupture.[9]
Non-ischaemic causes — fulminant myocarditis (young patients with recent viral illness, sudden severe LV dysfunction — high recovery rate if supported); peripartum cardiomyopathy; acute severe valvular regurgitation (endocarditis, traumatic); drug overdose (beta-blocker, calcium-channel blocker, digoxin); advanced cardiomyopathy with a precipitant.[12]
Pathophysiology
The pathophysiology of cardiogenic shock is best understood as a self-perpetuating downward spiral in which cardiac dysfunction, neurohormonal activation, systemic inflammation and end-organ failure reinforce one another.[1][13]
1. The initiating insult — pump failure. Acute MI kills roughly 40 percent of the LV myocardium before shock supervenes; loss of functioning muscle reduces stroke volume and cardiac output (CO = heart rate × stroke volume). When the remaining viable myocardium cannot compensate, cardiac index falls below 2.2 L/min/m2. The same injury — through the Frank-Starling mechanism — raises left ventricular end-diastolic pressure (LVEDP), which transmits back to the left atrium and pulmonary capillaries, raising wedge pressure above 18 mmHg and producing pulmonary congestion and oedema.[10]
2. The vicious spiral — coronary hypoperfusion. Reduced cardiac output lowers diastolic blood pressure and hence coronary perfusion pressure (diastolic BP minus wedge pressure). The subendocardium is most vulnerable (it is perfused in diastole only, against the highest intramyocardial pressure). Worsened subendocardial ischaemia further reduces contractility of the surviving myocardium, which drops stroke volume further — a spiral that terminates in asystole or pulseless electrical activity unless interrupted. This spiral is the biological reason early reperfusion works.[1]
3. Compensatory neurohormonal activation — initially helpful, ultimately harmful. Baroreceptors sense falling blood pressure and activate the sympathetic nervous system (tachycardia, vasoconstriction, increased contractility — raising myocardial oxygen demand in an ischaemic ventricle) and the renin-angiotensin-aldosterone system (vasoconstriction, sodium and water retention). Vasopressin (ADH) rises. These responses initially maintain central organ perfusion, but in established shock they increase afterload, worsen subendocardial ischaemia, and drive further decompensation — the rationale for vasodilator (GTN) and beta-blockade strategies in the chronic phase, and the danger of indiscriminate fluids in the acute phase.[12]
4. The systemic inflammatory response syndrome (SIRS) in CS. In established shock the gut becomes hypoperfused, the endothelium is injured, and a pro-inflammatory cytokine cascade (TNF-alpha, IL-1, IL-6) is released from ischaemic and necrotic myocardium. This produces nitric oxide synthase (iNOS) induction, excess nitric oxide and peroxynitrite, vasoplegia (pathological vasodilatation on top of pump failure), capillary leak, and myocardial depression — the so-called "septic-like" phenotype of late cardiogenic shock. This is why some CS patients become warm and vasoplegic rather than cold and vasoconstricted, and why noradrenaline rather than dobutamine alone is needed. The NOS inhibitor tilarginine (L-NMMA) was tested in the TRIUMPH trial (NEJM/JAMA 2007) and did NOT reduce mortality, with a trend to harm — an important negative trial.[5]
5. End-organ hypoperfusion — the clinical face of CS. [1]
- Kidney — falling renal perfusion activates tubuloglomerular feedback, dropping GFR; oliguria (under 0.5 mL/kg/hour) is one of the earliest bedside signs; type 1 cardiorenal syndrome and acute tubular necrosis follow.
- Brain — confusion, agitation, obtundation.
- Liver — ischaemic hepatitis ("shock liver") with transaminases over 1000 IU/L and centrilobular necrosis; cardiac cirrhosis in chronic congestion.
- Gut — hypoperfusion, bacterial translocation, ileus; mesenteric ischaemia in extreme cases.
- Skin — cool, clammy, mottled, delayed capillary refill (over 2 seconds) — vasoconstriction shunts blood to vital organs.
- Skeletal muscle — lactic acidosis from anaerobic metabolism; lactate above 2 mmol/L (and rising) is a marker of severity and a target of therapy. [1]
6. The role of the right ventricle. RV infarction (with inferior MI) is a special pathophysiology: the dilated, failing RV causes systemic venous congestion (raised JVP, hepatomegaly) with clear lung fields (the LV is underfilled because the RV cannot deliver preload), and hypotension that is exquisitely preload-sensitive — nitrates, diuretics and beta-blockers precipitate collapse; volume challenge and reperfusion are the answer. RV failure on top of LV failure (biventricular CS) is the most lethal phenotype and demands biventricular mechanical support (VA-ECMO rather than isolated LV assist). [1]
7. The macro- and microcirculation dissociate. In late CS, macro-haemodynamics may improve on inotropes while the microcirculation remains failed — the so-called "lost in translation" phenomenon. This underlies the search for microcirculatory monitoring (sublingual SDF imaging) and the failure of pure pressure-based endpoints.[13]
Clinical Presentation
The clinical face of cardiogenic shock is the triad of hypoperfusion plus congestion plus a cardiac cause.[13]
Symptoms (the patient is usually too sick to give a long history): [1]
- Cardiac symptoms of the underlying cause — chest pain of acute MI (crushing central, radiation to arm/jaw, autonomic features), palpitation (VT, AF), syncope (arrhythmia, PE, tamponade), breathlessness of pulmonary oedema.
- Hypoperfusion symptoms — dizziness, light-headedness, syncope, fatigue, confusion, oliguria or anuria, cold extremities.
- Pulmonary congestion — orthopnoea, paroxysmal nocturnal dyspnoea, frothy pink sputum (frank pulmonary oedema) — unless the cause is RV infarct, massive PE, or tamponade, in which case the lungs are clear despite shock. [1]
Vital signs (the first clue): [1]
- Hypotension — SBP below 90 mmHg, or MAP below 65 mmHg, or a fall of more than 30 mmHg from baseline, or only sustained on inotropes/vasopressors. A normal BP does not exclude shock — compensatory vasoconstriction can mask hypoperfusion; assess perfusion not pressure.
- Tachycardia — compensatory (under 100 is a sinister sign — severe pump failure or conduction disease/AV block).
- Tachypnoea — from pulmonary oedema, metabolic acidosis (Kussmaul breathing), or anxiety.
- Hypoxaemia — pulmonary oedema, V/Q mismatch.
- Cool peripheries, delayed capillary refill (over 2 s), mottled skin — vasoconstriction. But in late, vasoplegic shock the skin may be warm (NO-mediated).
- Oliguria — catheterise early; urine output under 0.5 mL/kg/hour.
- Altered mental status — confusion, agitation, obtundation. [1]
Examination (organ by organ): [1]
- JVP — typically raised (biventricular failure, RV infarct, tamponade, PE); a flat JVP suggests hypovolaemia or vasodilatory shock and prompts a fluid challenge.
- Precordium — diffuse, weak apex (LV failure); RV heave (RV infarct, PE, pulmonary hypertension); right parasternal heave in massive PE; absent apex with muffled sounds in tamponade.
- Auscultation — S3 gallop (the auscultatory hallmark of severe LV failure), bibasal crackles (pulmonary oedema), new pansystolic murmur at apex radiating to axilla (papillary muscle rupture — acute severe MR) or at lower left sternal edge with thrill (VSR); diastolic murmur of acute severe AR (dissection, endocarditis); pericardial rub (tamponade/myopericarditis); silent precordium (large effusion/tamponade, cardiac arrest).
- Abdomen — tender pulsatile hepatomegaly with hepatojugular reflux (right-heart failure).
- Periphery — cool, clammy, mottled, peripheral cyanosis; pulse is weak and thready; narrow pulse pressure (low stroke volume). [1]
Atypical presentations (high-yield): [1]
- Elderly / diabetic — painless MI with confusion, falls, breathlessness, fatigue as the only clues; shock may be the first sign of infarction.
- Right ventricular infarction (with inferior MI) — the clear-lung-fields shock: raised JVP, hypotension, Kussmaul sign (JVP rises paradoxically with inspiration), clear chest; worsens catastrophically with nitrates or diuretics.[9]
- Mechanical complication at day 3 to 6 post-MI — sudden new murmur with haemodynamic collapse in a patient who had been recovering; papillary muscle rupture (pansystolic apical, often no thrill because LA pressure equalises with LV) or ventricular septal rupture (loud pansystolic lower-left-sternal-edge murmur with thrill).
- Cardiac tamponade — Beck triad (hypotension, raised JVP, muffled heart sounds), pulsus paradoxus (drop in SBP over 10 mmHg on inspiration), electrical alternans on ECG; emergency pericardiocentesis.
- Massive pulmonary embolism — sudden syncope, pleuritic pain, right-heart failure pattern on ECG (S1Q3T3, right-axis deviation, T-wave inversion V1 to V3, RBBB), hypoxaemia with clear CXR; right ventricular strain on echo.
- Drug overdose (beta-blocker, calcium-channel blocker, digoxin) — bradycardia in shock is the clue; specific antidotes (glucagon, high-dose insulin euglycaemic therapy, digoxin Fab, calcium, lipid emulsion).
Differential Diagnosis
The first task is to separate cardiogenic shock from its mimics, because the management is the opposite (cardiogenic needs inotrope, vasopressor and decongestion; hypovolaemic and distributive need fluid; obstructive needs needle/treatment of the obstruction).[13]
- Hypovolaemic shock — blood loss (GI bleed, trauma, ruptured AAA, postpartum haemorrhage), fluid loss (vomiting, diarrhoea, burns). Distinguish: flat JVP, dry peripheries initially then cold, wedge below 18 mmHg, high SVR, no chest pain. Treat with balanced crystalloid 30 mL/kg then blood if bleeding.
- Septic / distributive shock — infection, warm peripheries early (vasodilatation), wedge normal or low, high cardiac output (until late), SVR low, often leucocytosis and source. Treat with fluids, antibiotics within 1 hour, noradrenaline, source control. A common pitfall: late sepsis can become cold with low-output myocardial depression, mimicking CS — the bedside echo (looking for a primary cardiac problem) and the wedge help.
- Obstructive shock — massive PE, tension pneumothorax, cardiac tamponade. Each is immediately reversible:
- Tamponade — Beck triad, pulsus paradoxus, equalisation of diastolic pressures, swinging heart on echo; pericardiocentesis.
- Tension pneumothorax — tracheal deviation, hyper-resonance, absent breath sounds on one side; needle decompression (5th intercostal space, mid-axillary line) then chest drain.
- Massive PE — sudden syncope, RV strain on ECG and echo, D-dimer raised, CT pulmonary angiogram; systemic thrombolysis (alteplase 100 mg over 2 hours) or catheter-directed thrombolysis / embolectomy.
- Anaphylactic shock — exposure, urticaria, angioedema, bronchospasm, hypotension within minutes; treat with IM adrenaline 0.5 mg (0.5 mL of 1 in 1000), IV fluids, chlorphenamine, hydrocortisone.
- Acute adrenal crisis (Addisonian crisis) — weakness, abdominal pain, hyponatraemia, hyperkalaemia, hypoglycaemia, pigmentation; treat with IV hydrocortisone 100 mg and fluids.
- Cardiogenic mimics of shock — severe aortic stenosis with low-output failure, hypertrophic obstructive cardiomyopathy (LVOT obstruction, worsened by inotropes and diuresis — beware!), severe acidosis, drug overdose. [1]
The single best discriminator is the bedside echocardiogram combined with wedge pressure / cardiac index measurement (where available) — a globally hypokinetic, dilated LV with raised wedge and low CI confirms cardiogenic shock; a normal LV with low wedge points to hypovolaemia; a dilated RV with raised JVP and clear lungs points to RV infarct or PE. [1]
Clinical & Bedside Assessment
ABCDE first. Cardiogenic shock is a perfusion diagnosis, so assess perfusion, not just pressure:[13]
Bedside perfusion assessment (the earliest signs): [1]
- Capillary refill time (press sternum or finger pad 5 s, release) — delayed if over 2 seconds; mottled skin in advanced shock.
- Urine output — catheterise early; oliguria under 0.5 mL/kg/hour is one of the earliest signs of failing renal perfusion, preceding BP fall.
- Mental status — confusion, agitation, obtundation.
- Lactate — arterial or venous; over 2 mmol/L is raised, over 4 mmol/L severe; a rising lactate despite therapy is a marker of failure to resuscitate.
- Skin temperature — cool peripheries (cold shock) vs warm (vasoplegic late shock). [1]
JVP assessment at 45°: raised in CS (biventricular failure, RV infarct, PE, tamponade); flat JVP challenges the diagnosis and prompts a cautious fluid challenge. [1]
Pulsus paradoxus — drop in SBP over 10 mmHg on inspiration — tamponade, severe asthma/COPD, tension pneumothorax, massive PE. [1]
The "shock bundle" at the bedside (the National Cardiogenic Shock Initiative / Tehrani et al. 2020 protocol):[13]
- Arterial line (continuous BP) — the brachial/femoral route; radial under-reads in severe vasoconstriction.
- Two large-bore IV cannulae and central venous access (right internal jugular or femoral, given the need for PA catheter and possible ECMO cannulation).
- Bedside echo within minutes — confirm the cardiac cause, identify mechanical complications, RV function, valve lesions, effusion/tamponade.
- Pulmonary artery (Swan-Ganz) catheter — for the deteriorating or vasopressor-dependent patient: gives wedge, cardiac output (thermodilution), mixed venous saturation (SvO2), and calculates cardiac power output (CPO = MAP × CO / 451, in Watts). CPO below 0.6 W is the most powerful single haemodynamic predictor of mortality in CS.
- Lactate, ABG, full blood count, U&E, LFT, troponin, BNP/NT-proBNP, group-and-save, cross-match, coagulation — baseline labs.
- ECG — STEMI (drive to emergency PCI), tachy/bradyarrhythmia, PE pattern, tamponade (electrical alternans).
- CXR — pulmonary oedema, cardiomegaly, widened mediastinum (dissection), pneumothorax. [1]
Investigations
First-line (in parallel with resuscitation):[9][13]
- 12-lead ECG — STEMI drives emergency revascularisation (door-to-balloon within 90 min); ST elevation in II/III/aVF with RV4 suggests RV infarct; S1Q3T3, right-axis deviation, RBBB, T-wave inversion V1 to V3 suggests massive PE; electrical alternans suggests tamponade; wide QRS / hyperkalaemia — beware acidosis-induced arrhythmia.
- Chest X-ray — pulmonary oedema (Bat's-wing alveolar shadowing, Kerley B lines, cardiomegaly, pleural effusions); clear lung fields with shock suggests RV infarct, PE, tamponade, hypovolaemia.
- Echocardiography (transthoracic, emergency bedside) — the single most important diagnostic test. Look for: global LV hypokinesis (large MI, myocarditis, cardiomyopathy), regional wall-motion abnormality in a coronary distribution (acute MI), dilated failing RV with preserved LV (RV infarct, massive PE), severe MR with flail leaflet (papillary muscle rupture), ventricular septal defect with high-velocity left-to-right jet on colour Doppler (VSR), pericardial effusion with diastolic RV collapse and swinging heart (tamponade), regional AKI of aortic valve (acute severe AR), LV apical thrombus. Echo also estimates LVEF and pulmonary artery systolic pressure (TR jet).
- Arterial blood gas — metabolic acidosis (low pH, low bicarbonate, raised lactate), hypoxaemia, raised base deficit (correlates with lactate and outcome).
- Lactate — arterial or venous; clearance is a target of therapy (failing to clear lactate is an indication to escalate).
- Troponin — confirms myocardial injury (but may already be high in chronic HF or renal failure; trend the rise).
- BNP / NT-proBNP — high in CS (cardiac cause); low BNP argues against a cardiac cause.
- Renal function, electrolytes, liver function, coagulation, full blood count — baseline and end-organ function.
- Drug levels — digoxin, salicylate, paracetamol (overdose screen), beta-blocker/CCB (history). [1]
Invasive haemodynamics (PA catheter) — for the deteriorating or refractory patient:[10][13]
- Pulmonary capillary wedge pressure (PCWP) — over 18 mmHg in cardiogenic shock (the defining haemodynamic criterion; below 18 mmHg with low CI is "Forrester III — cold and dry" and should prompt cautious fluid challenge rather than diuretic).
- Cardiac index (CI) — below 2.2 L/min/m2 in CS.
- Mixed venous saturation (SvO2) — below 65 percent indicates inadequate oxygen delivery; goal is 70 percent.
- Systemic vascular resistance (SVR) — typically high (compensatory vasoconstriction) in early CS, low in vasoplegic late CS.
- Cardiac Power Output (CPO) = (MAP × CO) / 451 (in Watts) — CPO below 0.6 W identifies severe CS and is the strongest single haemodynamic predictor of mortality (Fincke et al., cited in the SCAI update).[7]
Coronary angiography — mandatory in MI-related CS; identifies the culprit lesion, drives immediate revascularisation (culprit-lesion-only PCI per CULPRIT-SHOCK), and identifies the surgical or mechanical problem.[4]
Advanced (case-by-case): CT coronary angiography (if non-ischaemic cause suspected and stable enough), cardiac MRI (myocarditis, cardiomyopathy, infiltrative disease — usually once stabilised), CT pulmonary angiogram (massive PE), endomyocardial biopsy (giant-cell myocarditis, sarcoidosis, transplant rejection), drug screen (overdose). [1]
Named scores / criteria — reproduced verbatim
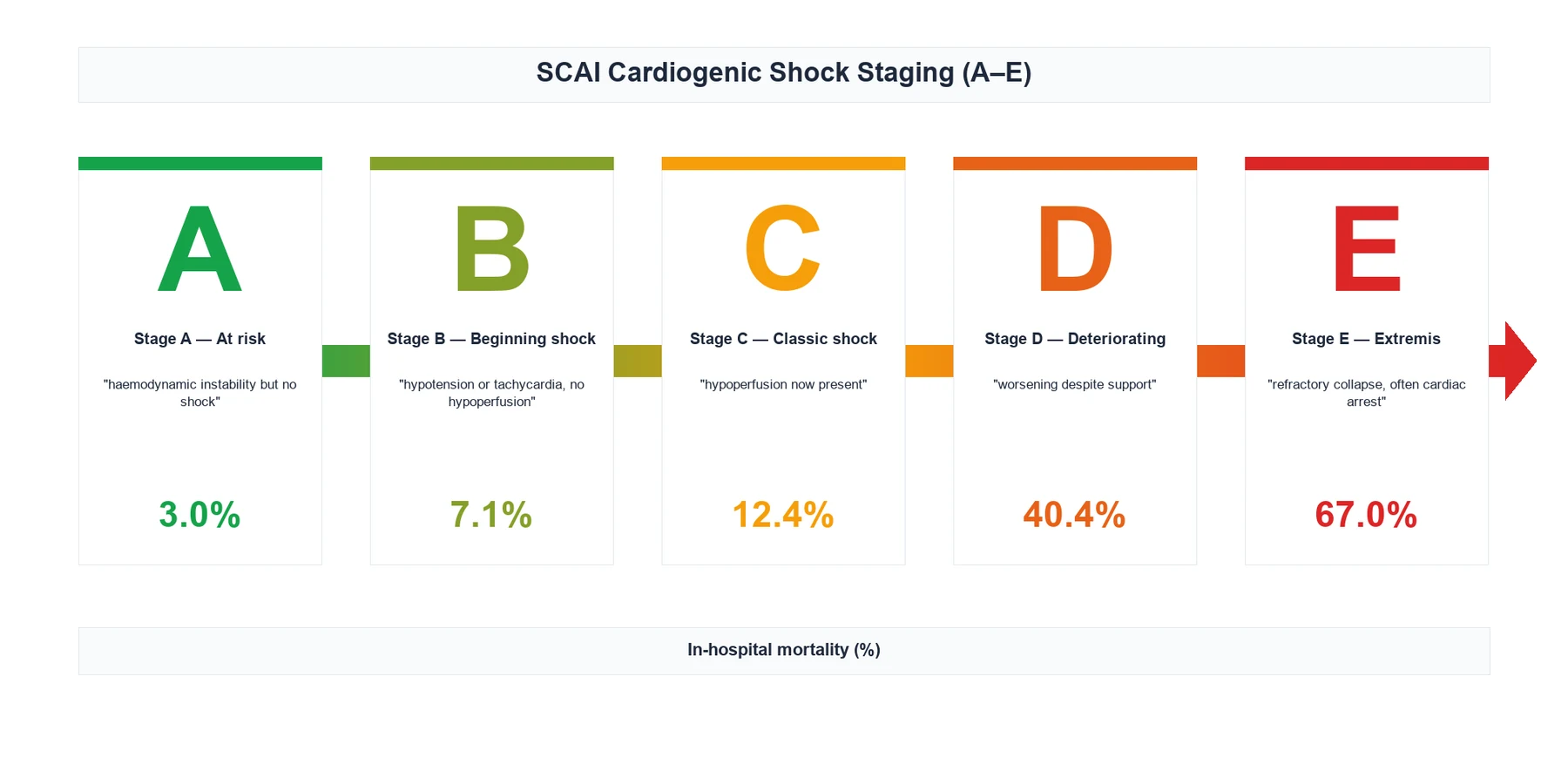
SCAI SHOCK stages (Baran 2019, Naidu 2022)[6][7]
| Stage | Definition | Approximate in-hospital mortality |
|---|---|---|
| A — At risk | Predisposing condition, no hypoperfusion | 3 to 4 percent |
| B — Beginning | Hypoperfusion starting; may be normotensive; tachycardia, mild lactate rise, vasoconstriction | 7 to 9 percent |
| C — Classic | Hypoperfusion with hypotension needing inotrope/vasopressor; wedge over 18, CI below 2.2 | 12 to 20 percent |
| D — Deteriorating | Worsening despite escalating inotropes/vasopressors; rising lactate, end-organ dysfunction | about 40 percent |
| E — Extremis | Circulatory collapse; cardiac arrest or peri-arrest; CPR or multiple pressors plus MCS | over 80 percent |
The B-to-C boundary is the appearance of overt hypoperfusion. [1]
Forrester haemodynamic subsets (Forrester, Diamond, Chatterjee; NEJM 1976)[10][11]
| Subset | PCWP | CI | Phenotype | Mortality (historical) |
|---|---|---|---|---|
| I | below 18 | above 2.2 | Warm and dry | about 1 percent |
| II | over 18 | above 2.2 | Warm and wet (pulmonary oedema) | about 10 percent |
| III | below 18 | below 2.2 | Cold and dry (hypovolaemic) | about 20 percent |
| IV | over 18 | below 2.2 | Cold and wet (classic CS) | about 60 percent |
SHOCK trial entry criteria (Hochman 1999)[1]
- SBP below 90 mmHg for at least 30 minutes OR requiring inotropes/vasopressors to maintain SBP at least 90 mmHg;
- CI below 2.2 L/min/m2 AND PCWP over 15 mmHg (the original trial used 15);
- plus clinically evident hypoperfusion (oliguria, cold peripheries, altered mentation). [1]
Cardiogenic shock — key numbers
Management — Resuscitation

ABCDE first, in parallel with diagnostic work-up. Cardiogenic shock is a time-critical emergency — every minute of hypoperfusion deepens the spiral.[9][13]
Airway and breathing: [1]
- High-flow oxygen to target SpO2 94 to 98 percent (88 to 92 percent in COPD / CO2 retainers); escalate to high-flow nasal cannula or non-invasive ventilation (CPAP/BiPAP) for pulmonary oedema — NIV reduces work of breathing, recruits alveoli and lowers preload/afterload.
- Intubate and mechanically ventilate if the patient is tiring, comatose, or in cardiac arrest — positive-pressure ventilation also reduces LV afterload (a useful haemodynamic effect in CS) but reduces venous return, so titrate carefully and ensure adequate preload. [1]
Circulation — early vascular access and monitoring: [1]
- Two large-bore cannulae, arterial line (continuous BP — radial under-reads in vasoconstriction), central venous line (right IJ preferred, leave femoral for possible ECMO), urinary catheter.
- PA catheter for the deteriorating, vasopressor-dependent, or MCS-considered patient.
- Fluid challenge only if hypovolaemic or wedge below 15 mmHg — 250 mL balanced crystalloid over 10 minutes, reassess; avoid fluid overload in CS with raised wedge (it worsens pulmonary oedema and RV strain). [1]
Circulation — pharmacological haemodynamic support (the core of early therapy):[13]
The goal is MAP at least 65 mmHg and CI at least 2.2 L/min/m2 while preserving coronary perfusion, without worsened arrhythmia or ischaemia. Choice of agent depends on the haemodynamic phenotype: [1]
-
Cold, hypotensive CS (low CI, low MAP, raised wedge — the classic form) — start an inotrope to raise CI and a vasopressor to defend MAP:
- Dobutamine 2 to 20 micrograms/kg/min IV — beta-1 agonist; positive inotrope and chronotrope; raises CO but also myocardial oxygen demand and may worsen ischaemia and arrhythmia; tachyphylaxis; first-line inotrope in most CS. Titrated to CI and lactate.
- Milrinone 0.125 to 0.75 micrograms/kg/min IV (loading 50 micrograms/kg over 10 min) — PDE-3 inhibitor, "inodilator" (inotropy plus vasodilatation); preferred when pulmonary hypertension or RV failure coexist, and in beta-blocker-related CS; vasodilator effect may worsen hypotension — combine with a vasopressor.
- Levosimendan — calcium-sensitiser (inodilator without raising intracellular calcium); loading 6 to 12 micrograms/kg over 10 min then 0.05 to 0.2 micrograms/kg/min; used in Europe for refractory CS, especially post-cardiotomy and takotsubo; can cause hypotension.
- Noradrenaline 0.05 to 1 microgram/kg/min IV — alpha and beta-1 agonist; the preferred first-line vasopressor in CS (more potent vasoconstrictor than dopamine, fewer arrhythmias); titrate to MAP at least 65 mmHg.
- Adrenaline 0.05 to 1 microgram/kg/min IV — for severe or refractory CS; powerful inotrope and vasoconstrictor; raises lactate (anaerobic skeletal-muscle glycolysis) — confounds lactate monitoring; high arrhythmia risk.
- Vasopressin 0.01 to 0.04 units/min IV — catecholamine-sparing; useful in vasoplegic late CS or beta-blocker/CCB overdose. [1]
-
Warm vasoplegic CS (low SVR despite inotrope — the late, SIRS-like phenotype) — noradrenaline first-line, add vasopressin if catecholamine-resistant; consider steroids (hydrocortisone 200 mg/day) in refractory vasoplegia (controversial). [1]
-
Dopamine (2 to 20 micrograms/kg/min) — no longer recommended as first-line in CS: the SOAP-II trial showed more arrhythmia and higher mortality than noradrenaline in cardiogenic shock; reserve for symptomatic bradycardia where pacing is not available.[13]
The "SHOCK funnel" — escalation triggers: escalate when the patient is SCAI stage D/E, has rising lactate despite inotrope, worsening end-organ function, high inotrope/vasopressor doses, or cardiac arrest / peri-arrest.[13]
Adjunctive therapy in MI-related CS:[9]
- Dual antiplatelet therapy — aspirin 300 mg loading then 75 mg daily; P2Y12 inhibitor — ticagrelor 180 mg loading then 90 mg twice daily (preferred in CS-PCI; non-competitive, fast onset), prasugrel 60 mg loading then 10 mg daily (avoid in stroke history, age over 75, weight under 60 kg), or clopidogrel 600 mg loading then 75 mg daily (if high bleeding risk).
- Parenteral anticoagulation — unfractionated heparin (preferred in CS for rapid reversibility and renal-independent clearance); bivalirudin in heparin-induced thrombocytopenia.
- Statins — atorvastatin 80 mg high-intensity.
- Glycaemic control — maintain glucose 7.8 to 10 mmol/L (avoid hypoglycaemia; tight control is harmful in CS).
- Revascularisation — see Definitive Management. [1]
Management — Definitive & Stepwise
The definitive management of CS is cause-specific and team-based: identify and treat the cardiac cause, support the circulation to break the spiral, and escalate to mechanical circulatory support (MCS) as a bridge to recovery, decision, transplant or durable LVAD.[1][13]
Step 1 — Identify and treat the cause (within minutes to hours)
The single biggest mortality-reducing intervention in MI-CS is early coronary revascularisation.[1]
- STEMI-CS — primary PCI within 90 minutes of first medical contact (door-to-balloon), the gold standard; if PCI unavailable within 120 minutes, fibrinolysis (alteplase or tenecteplase weight-based) — but fibrinolysis is less effective in shock and carries bleeding risk.
- NSTEMI-CS — immediate invasive strategy (within 2 hours) with PCI.
- CULPRIT-lesion-only PCI is preferred over multivessel PCI in MI-CS (CULPRIT-SHOCK trial, Thiele 2017): culprit-lesion-only PCI reduced the composite of death OR severe renal failure needing RRT (45.9 percent vs 55.4 percent; relative risk 0.83, P equal to 0.01) compared with immediate multivessel PCI. Staged complete revascularisation is performed later once stabilised.[4]
- Mechanical complications of MI — emergency surgical repair for papillary muscle rupture, VSR, free-wall rupture (and for severe ischaemic MR not amenable to PCI); these are surgical emergencies, MCS as a bridge to surgery.
- Acute severe valvular disease — emergency valve surgery (acute severe MR, AR, MS).
- Cardiac tamponade — emergency pericardiocentesis (subxiphoid, echo-guided).
- Massive PE — systemic thrombolysis (alteplase 100 mg over 2 hours) or catheter-directed thrombolysis / surgical embolectomy.
- Arrhythmia — DC cardioversion if unstable tachyarrhythmia; temporary then permanent pacing for symptomatic bradyarrhythmia/AV block; amiodarone 300 mg over 1 hour then 900 mg over 24 hours for stable VT.
- Drug overdose — beta-blocker: glucagon 5 to 10 mg IV bolus then infusion 1 to 5 mg/hour, high-dose insulin euglycaemic therapy (1 unit/kg bolus then 0.5 to 1 unit/kg/hour with dextrose), lipid emulsion in refractory; calcium-channel blocker: calcium chloride 10 percent 10 to 20 mL or calcium gluconate, high-dose insulin, lipid emulsion; digoxin: digoxin-specific Fab antibody fragments (DigiFab) dose based on serum level or amount ingested.
- Fulminant myocarditis — supportive MCS (often VA-ECMO) ± IV corticosteroids and immunosuppression in giant-cell or autoimmune forms; high recovery rate if supported through the acute phase.
- End-stage cardiomyopathy or transplant rejection — MCS bridge to transplant or palliative pathway.
Step 2 — Mechanical circulatory support (MCS) — bridge to recovery or decision
MCS unloads the failing ventricle, restores end-organ perfusion, and buys time for definitive therapy. It is not a destination for most (post-cardiotomy support being the exception).[13]
IABP (intra-aortic balloon pump)
- Counterpulsation: balloon inflates in diastole (raises coronary perfusion), deflates in systole (reduces afterload, raises CO about 0.5 L/min)
- Inserted via femoral artery, positioned in descending thoracic aorta (distal to left subclavian)
- Contraindications: aortic regurgitation (moderate or worse), aortic dissection, severe peripheral vascular disease, uncontrolled sepsis
- Demoted from Class I after IABP-SHOCK II (Thiele 2012) — routine IABP did NOT reduce 30-day mortality (39.7 percent vs 41.3 percent, P equal to 0.69); reserved for select mechanical-complication and selected refractory cases
- Modest haemodynamic support; cannot match the univentricular axial pumps
Impella (percutaneous axial-flow LV assist)
- Catheter across the aortic valve, aspirates LV blood and expels into ascending aorta — direct LV unloading
- Impella 2.5 delivers 2.5 L/min; CP (3.5) up to 3.5 to 4.0 L/min; 5.0 up to 5.0 L/min (surgical cut-down)
- Best for LV-dominant CS, before or during high-risk PCI; ISAR-SHOCK suggested lower lactate and trend to lower 30-day mortality vs IABP
- Contraindications: severe aortic stenosis or regurgitation, LV thrombus, VSD, severe PAD; complications: haemolysis, limb ischaemia, device malposition
- Active area of trial evidence (DANGER SHOCK, Recover IV)
TandemHeart (percutaneous LA-to-femoral artery)
- Cannula across atrial septum into left atrium, extracorporeal pump, returns to femoral artery
- Up to 4 to 5 L/min flow; powerful LV unloading
- More complex insertion (trans-septal puncture); higher bleeding and limb-ischaemia rates
- Largely supplanted by Impella and VA-ECMO at most centres
VA-ECMO (veno-arterial extracorporeal membrane oxygenation)
- Drains venous blood, oxygenates it, returns to arterial system — provides both circulatory and respiratory support
- Up to 4 to 6 L/min; supports biventricular failure and oxygenation — best for the sickest patients (SCAI E)
- Femoro-femoral route commonest; can deliver **retrograde flow competing with LV ejection — increases LV afterload and wedge**; consider adding an Impella or IABP for LV venting
- Indications: refractory CS, cardiac arrest (ECPR), fulminant myocarditis, massive PE, post-cardiotomy failure
- Complications: **limb ischaemia (the commonest — ipsilateral perfusion cannula mandatory), bleeding, haemolysis, thromboembolism, stroke, infection, LV distension, pulmonary oedema**
- Requires a centre with ECMO capability; transfer ('ECMO retrieval') increasingly used
Durable LVAD / surgical pumps (CentriMag)
- Surgically implanted; CentriMag for temporary biventricular support
- Bridge to transplant or destination therapy in end-stage HF; surgical RVAD, BiVAD options
- Reserved for those who survive the acute event but cannot be weaned
The choice of MCS depends on the primary failing chamber, oxygenation need, anticipated duration, and centre expertise. A practical algorithm: LV-dominant CS — Impella (or IABP if Impella unavailable); RV-dominant or biventricular CS, or CS with hypoxaemia or arrest — VA-ECMO; combine (ECMO + Impella = "ECPELLA") when LV distension is a problem.[13]
Step 3 — Decongestion, oxygen delivery and metabolic support
- Diuresis — furosemide 20 to 80 mg IV bolus (or continuous infusion 5 to 20 mg/hour for diuretic-resistant) to bring wedge toward 18 mmHg once MAP is defended; cautious in RV infarct (preload-dependent).
- Ventilation — see Resuscitation; NIV for pulmonary oedema, invasive ventilation for the tiring or comatose patient.
- Correct electrolytes and acidosis — potassium to 4 to 4.5 mmol/L, magnesium at least 1 mmol/L (arrhythmia prevention); renal replacement therapy for severe acidosis (pH below 7.1), hyperkalaemia, or fluid overload unresponsive to diuretics.
- Glucose 7.8 to 10 mmol/L, haemoglobin at least 80 to 90 g/L (controversial — too aggressive transfusion worsens outcome; the TRICC-equivalent principle applies), thromboprophylaxis (LMWH unless contraindicated), stress-ulcer prophylaxis, early enteral nutrition once stable. [1]
Step 4 — De-escalation and disposition
- Wean MCS and inotropes as the underlying cause recovers — guided by lactate clearance, CI, CPO, and echocardiographic recovery of EF. Wean VA-ECMO by reducing sweep gas and then flow over hours; decannulate surgically.
- Long-term HF therapy — once euvolaemic and stable, introduce the four pillars of HFrEF (ARNI/ACE-inhibitor, beta-blocker, MRA, SGLT2 inhibitor) cautiously and uptitrate.
- Bridge to transplantation or durable LVAD for those who cannot wean (end-stage cardiomyopathy); palliative pathway for those not candidates. [1]
Specific Subtypes & Scenarios
- Acute MI with LV failure (the commonest CS) — typically large anterior STEMI; primary PCI within 90 min; inotrope + noradrenaline + MCS as bridge; culprit-lesion-only PCI.[1][4]
- Right ventricular infarction (with inferior STEMI) — the clear-lung-fields shock; raised JVP with Kussmaul sign, hypotension, clear chest. Volume challenge (250 mL boluses to a target of raised JVP without pulmonary oedema — RV is preload-dependent); avoid nitrates, diuretics, beta-blockers; reperfusion (PCI of the RCA); dobutamine if hypotension persists despite adequate preload; maintain AV synchrony (atrial infarct and AV block common — consider atrioventricular sequential pacing). Inotrope and diuretic inappropriately given for "pulmonary oedema" that is actually RV infarct is a classic fatal error.
- Acute mechanical complications of MI (day 3 to 6 post-MI) — sudden haemodynamic collapse with a new murmur:
- Papillary muscle rupture — posteromedial papillary muscle (single blood supply from PDA) in inferior MI; acute severe MR, pansystolic apical murmur (may be soft if LA pressure equalises with LV), pulmonary oedema, shock. Echo shows flail leaflet and eccentric MR jet. Emergency surgery (MV repair/replacement); IABP or Impella as bridge; vasodilator (nitroprusside) to reduce afterload if BP tolerates.
- Ventricular septal rupture — loud pansystolic murmur at lower left sternal edge with thrill, biventricular failure. Echo with colour Doppler shows left-to-right shunt. Emergency surgical closure (or percutaneous device in selected cases); MCS as bridge.
- Free-wall rupture — sudden chest pain, pericardial tamponade, electromechanical dissociation, usually fatal; emergency surgery if recognised.[9]
- Fulminant myocarditis — young patient, viral prodrome, sudden severe LV dysfunction ± arrhythmia and AV block. VA-ECMO support through the acute phase; IV corticosteroids and immunosuppression for giant-cell, eosinophilic, or biopsy-proven autoimmune myocarditis; high recovery rate (over 70 percent survival if supported). Consider endomyocardial biopsy if giant-cell suspected (poor prognosis without immunosuppression).
- Acute decompensated heart failure / end-stage cardiomyopathy — chronic HF with a precipitant (ischaemia, arrhythmia, infection, anaemia, NSAID, non-adherence); treat the precipitant and decongest, do not start or uptitrate beta-blocker until euvolaemic; consider MCS or transplant.
- Acute severe valvular disease — acute severe MR (papillary muscle rupture, endocarditis, trauma), acute severe AR (aortic dissection, endocarditis), acute severe MS (rare, thrombosis of prosthetic valve). Emergency valve surgery.
- Massive pulmonary embolism — sudden syncope, RV strain on ECG and echo, hypoxaemia with clear CXR, raised D-dimer, confirmed on CTPA. Systemic thrombolysis (alteplase 100 mg over 2 hours) if haemodynamically unstable; catheter-directed thrombolysis or surgical embolectomy if contraindication or failure.
- Cardiac tamponade — Beck triad, pulsus paradoxus, electrical alternans, diastolic RV/RV collapse on echo. Emergency pericardiocentesis (subxiphoid, echo-guided); treat the cause (malignancy, uraemia, infection, post-procedural, autoimmune).
- Drug overdose — beta-blocker: glucagon + high-dose insulin + lipid emulsion + MCS; calcium-channel blocker: calcium + high-dose insulin + lipid emulsion; digoxin: DigiFab. Beware: standard inotropes often fail; MCS (VA-ECMO) may be needed as bridge.
- Post-cardiotomy shock — failure to wean from cardiopulmonary bypass; ** CentriMag or VA-ECMO** support; high mortality (over 50 percent).
- Takotsubo cardiomyopathy — stress-induced apical ballooning; may present with shock and LVOT obstruction — beware inotropes (worsen LVOT obstruction); treat with beta-blocker and careful fluid, MCS if refractory.
- Pregnancy-related CS — peripartum cardiomyopathy (HF with EF below 45 percent in last month of pregnancy to 5 months postpartum); teratogenic drugs avoided in pregnancy (ACEi/ARB/MRA), use hydralazine-nitrate; bromocriptine 2.5 mg twice daily for 8 weeks (stop breastfeeding); anticoagulate if EF below 30 percent; high recovery rate (50 to 70 percent).
Complications & Pitfalls
Cardiac: refractory ventricular arrhythmia (VT/VF) and cardiac arrest, worsening pump failure (downward spiral), mechanical complications of MI (VSR, papillary muscle rupture, free-wall rupture), right-heart failure (biventricular CS — highest mortality), pericardial effusion/tamponade (post-MI, post-procedural, uraemic). [1]
Systemic / end-organ: acute kidney injury / type 1 cardiorenal syndrome (often needing RRT), ischaemic hepatitis (shock liver) with transaminases over 1000, liver failure, mesenteric ischaemia, disseminated intravascular coagulation, stroke (thromboembolism from LV thrombus, haemorrhagic from anticoagulation/MCS), critical-illness myopathy and neuropathy, pressure injury, nosocomial infection (line, lung, urinary). [1]
Complications of therapy: [1]
- Inotrope-related — tachyarrhythmia, worsened ischaemia, increased mortality (especially high-dose dobutamine, dopamine, adrenaline).
- Vasopressor-related — peripheral and mesenteric ischaemia, worsened afterload.
- MCS-related — limb ischaemia (the commonest VA-ECMO complication — ipsilateral perfusion cannula mandatory), bleeding (systemic anticoagulation, platelet dysfunction from pumps), haemolysis, thromboembolism (pump thrombosis), stroke, infection (line, pump pocket), LV distension / pulmonary oedema from VA-ECMO retrograde flow (add Impella or IABP to vent).
- Bleeding from dual antiplatelet + anticoagulation + MCS — the leading non-cardiac cause of death. [1]
Classic pitfalls (the errors that kill): [1]
- Treating CS as if it were septic or hypovolaemic shock — flooding a high-wedge patient with fluid causes flash pulmonary oedema and RV strain. Always check wedge / JVP / echo before fluids.
- Failing to recognise RV infarct — giving nitrates, diuretics or beta-blockers to a preload-dependent RV infarct causes catastrophic collapse.
- Delaying revascularisation for investigations — every minute of delay deepens the spiral; the SHOCK trial is the proof.
- Using dopamine as first-line inotrope — SOAP-II showed more arrhythmia and higher mortality than noradrenaline; dobutamine (or milrinone) + noradrenaline is the modern combination.
- Relying on blood pressure alone — perfusion fails before pressure; monitor lactate, urine output, mentation, skin.
- Misclassifying a mechanical complication as "worsening LV failure" — a new murmur post-MI is papillary muscle rupture or VSR until proven otherwise; urgent echo.
- Forgetting the differential of shock — tamponade, tension pneumothorax, massive PE, anaphylaxis, adrenal crisis are all immediately reversible; missing them is fatal.
- Indiscriminate IABP use after IABP-SHOCK II — routine IABP does NOT improve mortality; reserve for selected cases.
- Giving fluids or inotropes in HOCM-induced shock — worsens LVOT obstruction; use beta-blocker and fluid instead.
- Over-transfusion aiming for high haemoglobin — worsens afterload and mortality in CS; target 80 to 90 g/L. [1]
Prognosis & Disposition
In-hospital mortality of CS remains 40 to 50 percent despite modern care, and over 80 percent for SCAI stage E. One-year mortality approaches 60 to 70 percent in MI-CS without revascularisation, halved by early revascularisation.[1][2]
Predictors of poor outcome: older age (especially over 75), lower EF, lower cardiac index, lower CPO (below 0.6 W), higher wedge, rising lactate, renal dysfunction, hyperlactataemia not clearing, biventricular failure, mechanical complications, delay to revascularisation, out-of-hospital cardiac arrest, SCAI stage D/E at presentation, multi-organ failure.[7][8]
Disposition: [1]
- All CS patients belong in a critical-care environment (cardiac ICU / coronary care unit with MCS capability) with invasive monitoring and rapid access to PCI, surgery and MCS.
- Early transfer to a regional shock centre for the deteriorating patient or for MCS / transplant consideration.
- Survivors need lifelong cardiology follow-up — GDMT for HFrEF, device therapy (ICD/CRT) once stable, secondary prevention, cardiac rehabilitation, and palliative-care discussion for the non-recoverable, non-transplant candidate. [1]
Special Populations
- Elderly (over 75) — atypical presentation, higher mortality, SHOCK-trial benefit of early revascularisation did not extend to over-75 in the original analysis (decision-making with patient and family); nevertheless revascularisation is offered if meaningful survival is plausible.
- Pregnancy — peripartum cardiomyopathy (last month of pregnancy to 5 months postpartum) — avoid teratogenic drugs in pregnancy (ACEi/ARB/MRA); use hydralazine-nitrate, beta-blocker (metoprolol), furosemide; bromocriptine postpartum; anticoagulate if EF below 30 percent; high recovery rate. Massive peripartum PE, amniotic-fluid embolism are other causes of CS in pregnancy. Left-lateral tilt to relieve aortocaval compression. Multidisciplinary obstetric-cardiology-anaesthetic care.
- Children / paediatric CS — usually viral myocarditis, congenital heart disease (post-operative or uncorrected), cardiomyopathy, arrhythmia, sepsis. Weight-based dosing of all drugs; VA-ECMO the dominant MCS; heart transplantation for end-stage disease.
- Diabetic — silent MI, worse microvascular disease, higher risk of CS and worse outcome; manage glucose 7.8 to 10 mmol/L.
- Chronic kidney disease — adjust drug doses; contrast during PCI — weigh risk-benefit; renal replacement therapy for severe AKI; contrast-induced nephropathy prevention with isotonic saline.
- Anticoagulated patient on warfarin / DOAC — reverse before emergency PCI / surgery (vitamin K + PCC / FFP for warfarin; specific antidotes — andexanet for apixaban/rivaroxaban, idarucizumab for dabigatran; PCC for other DOACs).
- Post-cardiac-arrest — targeted temperature management (32 to 36 degrees C for 24 hours) if comatose after ROSC; CS in this setting carries very high mortality; early revascularisation if STEMI.
- Immunocompromised / transplant patient — transplant rejection, drug cardiotoxicity, opportunistic myocarditis; endomyocardial biopsy if rejection suspected. [1]
Evidence, Guidelines & Regional Differences
Key guidelines: 2023 ESC ACS Guidelines (Byrne et al.) for the acute MI-CS pathway (early revascularisation, culprit-lesion-only PCI); 2022 AHA/ACC/HFSA Heart Failure Guideline (Heidenreich et al.) for HF-CS; SCAI SHOCK classification (Baran 2019, Naidu 2022) for staging; National Cardiogenic Shock Initiative standardised protocol (Tehrani et al., JACC:HF 2020) for the shock funnel.[6][7][9][12][13]
Landmark trials and statements every exam candidate must know: [1]
- SHOCK trial (Hochman et al., NEJM 1999) — randomised early revascularisation (PCI or CABG within 48 hours) vs initial medical stabilisation in MI-CS. Early revascularisation did NOT significantly reduce 30-day all-cause mortality (46.7 percent vs 56.0 percent, P equal to 0.11) but DID reduce 6-month mortality (50.3 percent vs 63.1 percent, P equal to 0.027) — cite it for the 6-month and 1-year survival benefit, NOT for a 30-day mortality win.[1]
- SHOCK 1-year follow-up (Hochman et al., JAMA 2001) — 1-year survival 46.7 percent vs 33.6 percent (P under 0.03), with the benefit confined to age under 75. This trial established early revascularisation as the standard of care in MI-CS.[2]
- IABP-SHOCK II (Thiele et al., NEJM 2012) — routine IABP in MI-CS did NOT reduce 30-day mortality (39.7 percent vs 41.3 percent, P equal to 0.69) — the basis for demoting routine IABP from Class I to Class IIa/III. IABP retained a role in selected mechanical-complication cases.[3]
- CULPRIT-SHOCK (Thiele et al., NEJM 2017) — in MI-CS with multivessel disease, culprit-lesion-only PCI reduced the composite of death OR severe renal failure needing RRT (45.9 percent vs 55.4 percent; RR 0.83, P equal to 0.01). Death alone RR 0.84 (P equal to 0.03); RRT alone RR 0.71 (P equal to 0.07, not significant). Cite the composite. Multivessel PCI deferred to a staged procedure.[4]
- TRIUMPH (Alexander et al., JAMA 2007) — the NOS inhibitor tilarginine (L-NMMA) in MI-CS did NOT reduce mortality and trended toward harm; trial stopped early for futility. A landmark negative trial — targeted NO modulation in CS does not work.[5]
- SOAP-II (De Backer et al., NEJM 2010) — dopamine vs noradrenaline in shock: more arrhythmic events with dopamine and a trend to higher mortality in the cardiogenic subgroup — the basis for noradrenaline as preferred first-line vasopressor in CS (referenced in the SCAI update).
- SCAI 2019 (Baran et al.) and 2022 update (Naidu et al.) — the 5-stage classification (A to E) with the B-to-C boundary defined by the appearance of hypoperfusion; validated against in-hospital mortality across registries (Jentzer 2019, Naidu 2022).[6][7][8]
Regional deltas: [1]
- US (SCAI/AHA) — SCAI SHOCK staging drives triage; early revascularisation in MI-CS Class I; culprit-lesion-only PCI (CULPRIT-SHOCK adopted); Impella and VA-ECMO at tertiary shock centres; National Cardiogenic Shock Initiative standardised protocol.
- UK (NICE / NHS England) — developing national CS network with regional shock centres, ECMO retrieval; routine IABP use declined post-IABP-SHOCK II; early revascularisation via cardiac networks.
- Europe (ESC) — 2023 ESC ACS Guidelines endorse early revascularisation and culprit-only PCI; levosimendan used in some countries for refractory CS.
- India (NEET-PG / INICET context) — primary PCI not universally available within 90 minutes; fibrinolysis (streptokinase historically, now tenecteplase) remains common as first reperfusion; delayed presentation typical (mean 6 to 12 hours); resource limits constrain PA catheter, Impella and VA-ECMO availability — concentrated in tertiary centres; high prevalence of rheumatic valvular disease and cardiomyopathy as non-MI causes; cost-benefit discussions with family are central; dobutamine and noradrenaline widely available and affordable. [1]
Current controversies: (1) optimal MCS choice (Impella vs VA-ECMO vs ECPELLA — the RECOVER IV, DANGER SHOCK, ECLS-SHOCK trials); (2) routine use of steroids for vasoplegia; (3) superurgent complete revascularisation timing after CULPRIT-SHOCK; (4) microcirculatory monitoring; (5) outcome prediction with machine-learning models; (6) ethical allocation of MCS and transplant. [1]
Exam Pearls
- CS = end-organ hypoperfusion due to cardiac pump failure — SBP below 90 mmHg (or on inotropes) + CI below 2.2 L/min/m2 + signs of hypoperfusion + wedge over 18 mmHg. Perfusion fails before BP collapses — watch lactate, urine, mentation, skin.[6]
- Commonest cause: acute MI (80 percent), typically large anterior STEMI. Mechanical complications at day 3 to 6 post-MI — new murmur with collapse.[1]
- SCAI SHOCK stages A to E — mortality 4 to over 80 percent; the B-to-C boundary is the appearance of overt hypoperfusion.[6][7]
- Forrester subsets — cold and wet (PCWP over 18, CI below 2.2) is classic CS (Subset IV, mortality about 60 percent).[10]
- Cardiac Power Output (CPO) below 0.6 W is the most powerful single haemodynamic predictor of mortality.[7]
- The single biggest mortality-reducing intervention is early coronary revascularisation — SHOCK trial reduced 6-month and 1-year mortality (NOT 30-day; benefit under 75).[1][2]
- CULPRIT-SHOCK — culprit-lesion-only PCI reduced the composite of death OR RRT-needing renal failure (RR 0.83).[4]
- IABP-SHOCK II — routine IABP did NOT reduce mortality (demoted from Class I); TRIUMPH — tilarginine (NOS inhibitor) did NOT reduce mortality (trend to harm).[3][5]
- First-line inotrope: dobutamine 2 to 20 mcg/kg/min (or milrinone 0.125 to 0.75); first-line vasopressor: noradrenaline (dopamine has more arrhythmia per SOAP-II).[13]
- RV infarct = clear-lung-fields shock — fluid challenge, avoid nitrates/diuretics/beta-blockers, reperfusion.[9]
- Tamponade = Beck triad + pulsus paradoxus — emergency pericardiocentesis; massive PE = sudden syncope, RV strain — systemic thrombolysis (alteplase 100 mg over 2 h); tension pneumothorax — needle decompression then chest drain.
- MCS: IABP (modest, demoted), Impella (LV-dominant, 2.5 to 5 L/min), VA-ECMO (biventricular, hypoxaemia, arrest; watch limb ischaemia and LV distension), TandemHeart (LA to artery, complex).
- In-hospital mortality 40 to 50 percent despite modern care.[1]
Causes of cardiogenic shock — mnemonic
DROPS
Acute MI — large anterior STEMI (the commonest, 80 percent); fulminant myocarditis
VT, VF, severe bradycardia, complete heart block, AF with rapid rate in a failing ventricle
Cardiac tamponade, massive PE, tension pneumothorax (obstructive mimics — immediately reversible)
Acute severe MR/AR/MS, papillary muscle rupture or VSR (day 3 to 6 post-MI), end-stage cardiomyopathy, hypertrophic obstructive cardiomyopathy
Drug overdose (beta-blocker, calcium-channel blocker, digoxin); severe acidosis; hypothermia
Exam application bank (NEET-PG / INICET)
One-line answer
Cardiogenic shock (CS) is a state of end-organ hypoperfusion due to cardiac pump failure, defined for acute MI as systolic blood pressure below 90 mmHg for at least 30 minutes (or needing inotropes/vasopressors to keep it above this), cardiac index below 2.2 L/min/m2, and signs of hypoperfusion (cool peripheries, oliguria under 0.5 mL/kg/hour, altered mentation, raised lactate), with pulmonary capillary wedge pressure over 18 mmHg (confirming a cardiac cause). The commonest cause is acute MI with left ventricular failure (about 80 percent) — typically large anterior STEMI, but also right ventricular infarction and mechanical complications (papillary muscle rupture, ventricular septal rupture, free-wall rupture) at a mean of 3 to 6 days post-MI. Other causes: acute decompensated heart failure, fulminant myocarditis, end-stage cardiomyopathy, arrhythmia, valvular catastrophe (acute severe [1]
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard. [1]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes. [1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change. [1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each. [1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory. [1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Cardiogenic Shock.
References
- [1]Hochman JS, Sleeper LA, Webb JG, et al. Early revascularization in acute myocardial infarction complicated by cardiogenic shock. SHOCK Investigators. Should We Emergently Revascularize Occluded Coronaries for Cardiogenic Shock N Engl J Med, 1999.PMID 10460813
- [2]Hochman JS, Sleeper LA, White HD, et al. One-year survival following early revascularization for cardiogenic shock JAMA, 2001.PMID 11176812
- [3]Thiele H, Zeymer U, Neumann FJ, et al. Intraaortic balloon support for myocardial infarction with cardiogenic shock N Engl J Med, 2012.PMID 22920912
- [4]Thiele H, Akin I, Sandri M, et al. PCI Strategies in Patients with Acute Myocardial Infarction and Cardiogenic Shock N Engl J Med, 2017.PMID 29083953
- [5]Alexander JH, Reynolds HR, Stebbins AL, et al. Effect of tilarginine acetate in patients with acute myocardial infarction and cardiogenic shock: the TRIUMPH randomized controlled trial JAMA, 2007.PMID 17387132
- [6]Baran DA, Grines CL, Bailey S, et al. SCAI clinical expert consensus statement on the classification of cardiogenic shock: This document was endorsed by the American College of Cardiology (ACC), the American Heart Association (AHA), the Society of Critical Care Medicine (SCCM), and the Society of Thoracic Surgeons (STS) in April 2019 Catheter Cardiovasc Interv, 2019.PMID 31104355
- [7]Naidu SS, Baran DA, Baumgartner LA, et al. SCAI SHOCK Stage Classification Expert Consensus Update: A Review and Incorporation of Validation Studies: This statement was endorsed by the American College of Cardiology (ACC), American College of Emergency Physicians (ACEP), American Heart Association (AHA), European Society of Cardiology (ESC) Association for Acute Cardiovascular Care (ACVC), International Society for Heart and Lung Transplantation (ISHLT), Society of Critical Care Medicine (SCCM), and Society of Thoracic Surgeons (STS) in December 2021 J Am Coll Cardiol, 2022.PMID 35115207
- [8]Jentzer JC, Bennett C, Wiley BM, et al. Cardiogenic Shock Classification to Predict Mortality in the Cardiac Intensive Care Unit J Am Coll Cardiol, 2019.PMID 31548097
- [9]Byrne RA, Rossello X, Coughlan JJ, et al. 2023 ESC Guidelines for the management of acute coronary syndromes Eur Heart J, 2023.PMID 37622654
- [10]Forrester JS, Diamond G, Chatterjee K, et al. Medical therapy of acute myocardial infarction by application of hemodynamic subsets (first of two parts) N Engl J Med, 1976.PMID 790191
- [11]Forrester JS, Diamond GA, Chatterjee K, et al. Medical therapy of acute myocardial infarction by application of hemodynamic subsets (second of two parts) N Engl J Med, 1976.PMID 790194
- [12]Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines J Am Coll Cardiol, 2022.PMID 35379503
- [13]Tehrani BN, Truesdell AG, Sherwood MW, et al. A Standardized and Comprehensive Approach to the Management of Cardiogenic Shock JACC Heart Fail, 2020.PMID 33121700