cardiology · cardiology
Restrictive Cardiomyopathy
Also known as Restrictive cardiomyopathy · RCM · Infiltrative cardiomyopathy · Stiff ventricle syndrome
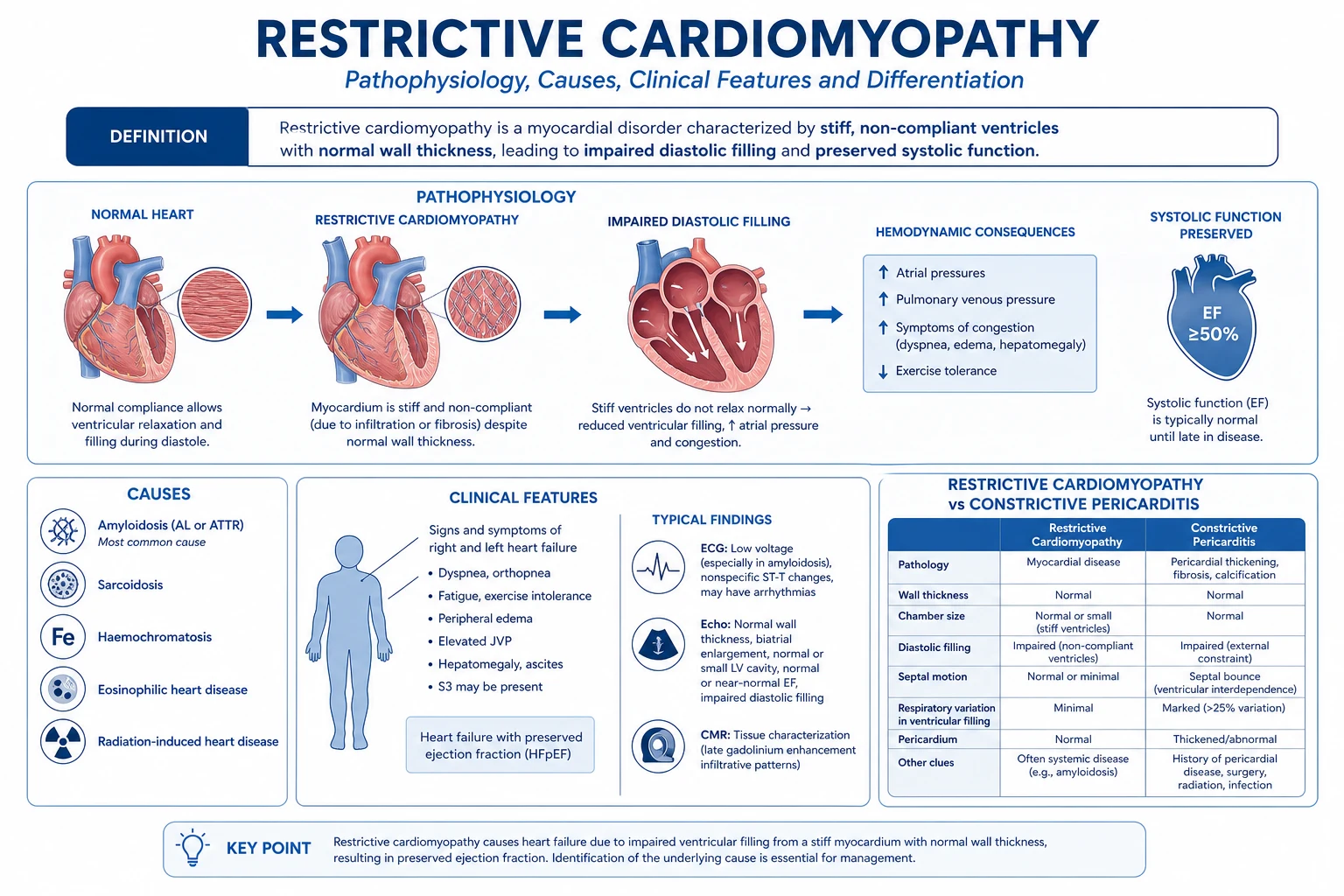
Restrictive cardiomyopathy (RCM) is the rarest of the three WHO cardiomyopathies, defined by non-compliant, stiff ventricles that resist filling in diastole, with reduced diastolic volume and preserved (or near-normal) systolic function, producing biventricular diastolic heart failure with bi-atrial enlargement. Leading cause in the developed world is cardiac amyloidosis (AL light-chain, and ATTR — wild-type/senile and hereditary variant); other causes are sarcoidosis, haemochromatosis, endomyocardial fibrosis (EMF) / Loffler endocarditis, radiation, carcinoid heart disease, glycogen storage diseases (Fabry, Pompe, Danon) and scleroderma. Presents with right-heart-failure signs (raised JVP, Kussmaul's sign, hepatomegaly, ascites, oedema), dyspnoea, fatigue and atrial fibrillation, in a patient with preserved EF. Differentiate from constrictive pericarditis by BNP (high in RCM, low in constriction), pericardial calcification (constriction only), septal bounce (constriction) and the square-root sign in both. Investigate with ECG (low voltages with thick walls = amyloid), echo (bi-atrial enlargement, granular myocardium, restrictive mitral inflow, low tissue e'), cardiac MRI (diffuse subendocardial LGE, T1/ECV mapping), serum free light chains for AL, bone-tracer scintigraphy for ATTR, and endomyocardial biopsy (gold standard). Treat the underlying cause; tafamidis 61 mg daily for ATTR-CM (ATTR-ACT trial); CyBorD/daratumumab for AL; steroids for sarcoid; phlebotomy/chelation for haemochromatosis. Cautious diuretics; avoid digoxin and calcium-channel blockers in amyloid. Heart transplant for end-stage.
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags
Overview & Definition
Restrictive cardiomyopathy (RCM) is a myocardial disorder defined by a functional phenotype: non-compliant, stiff ventricles that resist filling in diastole, with reduced ventricular diastolic volume, near-normal (or mildly reduced) wall thickness initially but thickened in infiltrative forms, and normal or near-normal systolic function, producing biventricular diastolic heart failure with bi-atrial enlargement.[5]
The defining physiology is a stiff ventricle with preserved pump — systolic ejection is normal early; the problem is the ventricle cannot accept the blood returning to it. Filling pressures rise steeply, the atria hypertrophy and dilate, and the patient develops systemic and pulmonary venous congestion with a low cardiac output once filling is critically impaired. [1]
RCM is the least common and least understood of the three WHO cardiomyopathies (hypertrophic most common in many populations, then dilated, then restrictive). The clinical skill is not the echocardiographic label — it is (a) recognising that a stiff ventricle is the cause of HFpEF-like symptoms, (b) finding the underlying infiltrative/storage disorder, and (c) distinguishing RCM from constrictive pericarditis — the single most important clinical decision, because pericardiectomy cures constriction but is futile (or harmful) in RCM.[5][6]
Classification
By the WHO/ESC functional framework — RCM is one of five cardiomyopathy phenotypes (DCM, HCM, RCM, ARVC, unclassified). Two sub-classifications matter clinically:[5]
-
By anatomical level of disease:
- Myocardial RCM — infiltration or fibrosis of the myocardium (amyloid, sarcoid, radiation, scleroderma, idiopathic).
- Endomyocardial RCM — disease of the endocardium with overlying thrombus and fibrosis (Loffler endocarditis, tropical endomyocardial fibrosis, carcinoid). [1]
-
By aetiology (the classification that drives treatment): [1]
| Mechanism | Examples |
|---|---|
| Infiltrative | Amyloidosis (AL, ATTRwt, ATTRv), sarcoidosis (rarely truly 'infiltrative' but mimics) |
| Storage | Haemochromatosis, Fabry disease, Pompe disease, Danon disease, glycogen storage diseases |
| Endomyocardial | Loffler endocarditis (hypereosinophilic syndrome), tropical endomyocardial fibrosis, carcinoid heart disease |
| Iatrogenic / toxic | Radiation (mantle), anthracyclines, serotonin/ergotamine, chronic serotonin excess |
| Connective tissue | Systemic sclerosis, mixed connective tissue disease |
| Idiopathic / familial | Troponin I, desmin, myosin-binding protein C mutations |
Cardiac amyloidosis (commonest in West)
- AL (primary, light-chain) — plasma-cell dyscrasia; most aggressive; needs urgent chemotherapy
- ATTRwt (senile/wild-type) — elderly men, carpal tunnel, 'HFpEF'; tafamidis-responsive
- ATTRv (hereditary variant) — TTR V122I (African descent, cardiac), T60M (Portuguese, neuropathy), V30M (FAP)
- Diffuse subendocardial LGE on MRI; voltage-mass discordance (low ECG, thick walls)
- 99mTc-PYP/DPD bone scintigraphy grade 2-3 — non-invasive ATTR diagnosis
Cardiac sarcoidosis
- Non-caseating granulomas; African-American, Scandinavian, Japanese, young adults
- High-grade AV block and VT in a young patient is the giveaway
- Patchy multifocal basal/subepicardial LGE; FDG-PET uptake (inflammation)
- Steroid-responsive if caught early; high sudden-death risk — ICD often
Endomyocardial disease (EMF / Loffler)
- Tropical equatorial belt (Uganda, Nigeria, Mozambique, Kerala coast) — EMF
- Loffler = hypereosinophilic syndrome in temperate zones with eosinophilia and endocardial thrombus
- Apical obliteration of one or both ventricles on echo; AV-valve regurgitation
- Steroids and cytotoxics for Loffler; endocardiectomy for advanced EMF
Storage diseases
- Hereditary haemochromatosis (HFE C282Y) — iron deposition, bronze diabetes, low T2* under 20 ms
- Fabry (X-linked, alpha-galactosidase A deficiency) — LVH + proteinuria + angiokeratomas
- Pompe, Danon — glycogen storage, paeds, with conduction and skeletal muscle disease
- Treat: phlebotomy/chelation (iron); enzyme replacement (Fabry/Pompe)

Epidemiology & Risk Factors
RCM is the rarest cardiomyopathy in adult cardiology practice. Its epidemiology is dominated by cardiac amyloidosis, which is increasingly recognised as a major cause of "HFpEF" in the elderly, especially elderly men.[1][9]
- AL amyloidosis — incidence 8 to 12 per million per year; cardiac involvement in around half at diagnosis; median age over 60; slight male predominance; arises from a plasma-cell dyscrasia (often lambda light chain).
- ATTRwt (senile) — underdiagnosed; autopsy prevalence 25% in those over 80; clinically presents in men over 65 with HFpEF, bilateral carpal tunnel syndrome, and often spinal stenosis or biceps tendon rupture years before the cardiac diagnosis.[9]
- ATTRv V122I variant — carried by 3 to 4% of African Americans; penetrant cardiac amyloid in late life; commonly mislabelled "hypertensive heart disease" or "HFpEF".
- Cardiac sarcoidosis — clinical cardiac involvement in 2 to 5% of systemic sarcoid, but subclinical in 20 to 25% on autopsy/PET; over-represented in African Americans, Scandinavians, Japanese; peak in young and middle-aged adults.[11]
- Endomyocardial fibrosis (EMF) — tropical equatorial belt (Uganda, Nigeria, Mozambique, Kerala coast of India, parts of South America); leading cause of heart failure in those regions in young people; eosinophilia and poverty/eosinophilic parasitic load are risk factors.[7]
- Hereditary haemochromatosis — HFE C282Y homozygosity in 1 in 200 of Northern Europeans; cardiac deposition in around one-third of untreated adults.[8]
- Radiation-induced RCM — typically 5 to 30 years after mediastinal radiotherapy (Hodgkin lymphoma, breast cancer); dose-dependent; combined pericardial, myocardial, valvular and coronary damage.
Risk-factor lens: "Stiff ventricle" should be suspected whenever a patient presents with right-heart failure and a preserved EF — and the cause is found by asking about age, ancestry (African), occupation/region (tropical), past radiotherapy, chronic inflammatory disease, carpal tunnel or orthopaedic signs, and family history. [1]
Pathophysiology
The defining abnormality is increased ventricular diastolic stiffness from infiltration, fibrosis, or endocardial scarring. Three structural problems converge on the same haemodynamic phenotype:[1][4]
- Reduced compliance — amyloid fibrils, granulomas, iron, or fibrous tissue displace contractile myocardium, so the passive pressure-volume curve shifts up and to the left. A small increment in ventricular volume produces a large rise in filling pressure.
- Restrictive (early, rapid) filling — because the ventricle cannot expand further, the early diastolic (E) inflow is rapid and abrupt, then stops suddenly as the stiff ventricle hits its limit. Atrial contraction (A wave) contributes little.
- Atrial barotrauma — the atria must pump against a non-compliant ventricle; they hypertrophy, dilate, and finally fail or fibrillate, producing the bi-atrial enlargement that is the radiological and echocardiographic hallmark. [1]
The haemodynamic signature is the dip-and-plateau (square-root) sign: in early diastole the ventricle fills briefly and rapidly (deep dip in pressure), then pressure plateaus abruptly as the stiff ventricle reaches its limit. LVEDP and RVEDP are both elevated; in RCM the LVEDP exceeds RVEDP by more than 5 mmHg at end-expiration, distinguishing it from constriction (where they equalise within 5 mmHg).[6]
Why is systolic function preserved early? The contractile machinery is initially intact — infiltration is interstitial, so myocyte shortening is preserved. As amyloid or iron load rises, myocyte toxicity, microvascular ischaemia and replacement fibrosis eventually impair systolic function late in the disease (this is sometimes called the "burnt-out" phase, and predicts poor outcome). [1]
Molecular basis of amyloid deposition. Amyloidosis is extracellular deposition of misfolded proteins in beta-pleated sheet conformation that stain with Congo red and show apple-green birefringence under polarised light. The cardiac-relevant precursor proteins are:
- Immunoglobulin light chains (AL) — monoclonal lambda more often than kappa, from a plasma-cell clone. Light chains are directly toxic to cardiomyocytes (oxidative stress, p38 MAPK, apoptosis), explaining the aggressive course of AL amyloid (median survival 6 months untreated from heart-failure onset) and its frequent troponin elevation.[3]
- Transthyretin (TTR) — a tetrameric thyroxine/retinol-binding protein made in the liver and choroid plexus. Wild-type (senile) TTR destabilises with age; hereditary variants (V122I in African descent, T60M Portuguese, V30M familial amyloid polyneuropathy) destabilise earlier. The TTR monomer misfolds, aggregates and deposits in the myocardium; far less directly toxic than AL, hence better prognosis.[3]
Conduction and arrhythmia. Amyloid infiltration of the conduction system causes fascicular and AV block (sarcoid is particularly arrhythmogenic with non-caseating granulomas in the basal septum). Atrial standstill and sick sinus syndrome are described in amyloid, and atrial fibrillation is common once the atria dilate. Intracardiac thrombus forms easily in the dilated, non-contractile atria — stroke risk is high. [1]
Clinical Presentation
The dominant picture is biventricular diastolic heart failure with preserved EF. Symptoms and signs cluster around venous congestion, low cardiac output, and arrhythmia:[4][5]
Symptoms:
- Dyspnoea on exertion, then at rest; orthopnoea and paroxysmal nocturnal dyspnoea (pulmonary venous congestion).
- Fatigue, lethargy, anorexia from low cardiac output.
- Abdominal swelling, right-upper-quadrant discomfort (hepatic congestion, ascites).
- Syncope or pre-syncope — conduction disease or arrhythmia.
- Palpitations — atrial fibrillation, ventricular ectopy.
- Lower-limb swelling. [1]
Signs at the bedside:
- Raised JVP — often markedly elevated; Kussmaul's sign (JVP rises paradoxically on inspiration because the stiff right ventricle cannot accept the augmented venous return). The y descent is rapid and deep (Friedreich's sign) — early diastolic filling is rapid, then arrested.
- Bi-atrial gallop, hypotension, narrow pulse pressure, cool peripheries (low stroke volume).
- Hepatomegaly, ascites, dependent oedema, occasionally anassarca.
- Murmurs of functional mitral or tricuspid regurgitation (papillary-muscle infiltration or annular dilatation).
- Signs of low cardiac output — weak pulse, prolonged capillary refill, occasionally peripheral cyanosis. [1]
Cause-specific clues (high-yield, examined):
- AL amyloidosis — macroglossia (large, scalloped tongue from teeth imprints), periorbital purpura (racoon-sign, classically after proctoscopy or prolonged Valsalva — the Valsalva-purpura sign), spontaneous biceps-tendon rupture ("Popeye sign"), bilateral carpal tunnel syndrome, autonomic orthostatic hypotension, nephrotic-range proteinuria, easy bruising, weight loss.
- ATTRwt — elderly man (over 65), bilateral carpal tunnel syndrome (often years earlier), lumbar spinal stenosis, biceps tendon rupture, "hypertensive heart failure" mislabel.
- ATTRv V122I — African-descent patient, "hypertensive heart failure" with increased wall thickness out of proportion, often a family history of "heart failure".
- Cardiac sarcoidosis — young/middle-aged patient, high-grade AV block (often complete heart block), sustained ventricular tachycardia, bilateral hilar lymphadenopathy, erythema nodosum, uveitis, hypercalcaemia.
- Haemochromatosis — bronze diabetes (skin pigmentation, insulin-dependent diabetes), hypogonadism, arthropathy (second/third metacarpophalangeal), hepatomegaly, cirrhosis, chondrocalcinosis.
- Loffler endocarditis — marked peripheral eosinophilia, systemic thromboembolism, restrictive filling with apical obliteration by thrombus; temperate-zone hypereosinophilic syndrome (often FIP1L1-PDGFRA).
- Endomyocardial fibrosis — young patient in tropical belt; mimics rheumatic mitral disease; apical murmur; ascites out of proportion to peripheral oedema.
- Radiation-induced — history of mantle radiotherapy 5-30 years before; combined pericardial constriction, myocardial restriction, valvular (aortic/mitral) regurgitation, premature coronary disease.
- Carcinoid heart disease — flushing, diarrhoea, bronchospasm, tricuspid and pulmonary regurgitation, right-sided plaques. [1]
Atypical presentations — elderly patients may present with falls, anorexia, weight loss, "failure to thrive" or new AF with heart failure, and are commonly misdiagnosed as HFpEF, diastolic dysfunction of ageing, or deconditioning. African-descent patients with V122I ATTR are often misdiagnosed as hypertensive heart disease. A low threshold to send serum free light chains and a bone-tracer scan is essential in any patient with unexplained HFpEF + increased wall thickness. [1]
Differential Diagnosis
The crucial differential is constrictive pericarditis — the bedside signs overlap (raised JVP, Kussmaul, hepatomegaly, ascites) but the management is opposite (pericardiectomy is curative for constriction, futile for RCM). The other mimics are Hypertrophic cardiomyopathy with restrictive physiology, severe tricuspid regurgitation, right ventricular infarction, and HFpEF.[5][6]
| Differential | Distinguishing features from RCM |
|---|---|
| Constrictive pericarditis | Pericardial knock (not S3); pericardial calcification on CXR/CT (RCM never calcifies the pericardium); septal bounce on echo; BNP normal or only mildly raised (RCM markedly raised); annulus paradoxus (septal e' preserved over 8 cm/s in constriction, both e' reduced in RCM); equalised LVEDP and RVEDP within 5 mmHg (RCM LVEDP exceeds RVEDP by over 5 mmHg); RVSP under 50 mmHg and RVEDP over one-third of RVSP (RCM: RVSP over 50 mmHg, RVEDP under one-third RVSP). |
| Hypertrophic cardiomyopathy | Asymmetric septal hypertrophy, dynamic LVOT obstruction, systolic anterior motion of mitral valve, small LV cavity with hyperdynamic EF; family history and genetic testing. |
| Severe tricuspid regurgitation | Systolic waves in JVP, pulsatile liver, RV heave, prominent v waves; echo shows flail/tethered tricuspid leaflets. |
| Right ventricular infarction | Acute inferior MI on ECG, right-sided ST elevation V4R, haemodynamic improvement with fluid loading; no chronic infiltration. |
| Severe aortic stenosis with diastolic dysfunction | Ejection systolic murmur, slow-rising carotid pulse, echo shows calcified bicuspid/tricuspid valve, high transvalvular gradient. |
| Elderly HFpEF | Modest LVH with normal atrial size for age, no granular myocardium, normal ECV; absence of an infiltrative cause. |
The single best discriminator between RCM and constriction is BNP/NT-proBNP — markedly raised in RCM (the myocardium is diseased and stretched), normal or low in constriction (the myocardium is normal, only encased).[6]
Clinical & Bedside Assessment
Vital signs and haemodynamics often show narrow pulse pressure, low cardiac output (cool peripheries, weak pulse), and hypotension in advanced disease. Atrial fibrillation with rapid ventricular response may precipitate decompensation. [1]
JVP — the most important bedside sign. Classify the waveform: in RCM the y descent is rapid and deep (Friedreich's sign) because early diastolic filling is rapid then arrested; the x descent is also present. Kussmaul's sign (rise in JVP on inspiration) is present in both RCM and constrictive pericarditis but absent in tamponade — useful for the three-way differential.[6]
Heart sounds:
- S3 (early diastolic) is heard in RCM (rapid deceleration of early filling). A pericardial knock is the equivalent of constriction — higher-pitched, earlier than S3, occurring as filling is abruptly arrested by the pericardium.
- S4 is common when the atrium is still contracting (sinus rhythm).
- Murmurs of MR/TR if papillary muscles or annuli are involved. [1]
Abdomen — hepatomegaly, ascites, pulsatile liver if TR; splenomegaly in advanced congestion. Lower limbs — pitting oedema, occasionally chronic venous stasis changes. [1]
Always look for systemic clues to the cause:
- Tongue (macroglossia — AL), eyelids (periorbital purpura — AL), skin (bronze pigmentation, angiokeratomas — haemochromatosis / Fabry), wrists and shoulders (carpal tunnel, biceps rupture — ATTR), joints (MCP arthritis — haemochromatosis), lymph nodes and skin (erythema nodosum, uveitis — sarcoid), abdomen (hepatosplenomegaly — sarcoid, glycogen storage). [1]
Investigations
The diagnostic strategy is staged: (1) confirm the restrictive phenotype (echo, cardiac MRI), (2) distinguish RCM from constriction, and (3) identify the underlying cause.[1][5]
First-line tests
- ECG — characteristically low voltage in amyloid (especially limb leads, under 0.5 mV QRS in standard limb leads), pseudoinfarct pattern (Q waves in anterior/inferior leads despite no infarct), AF, first-degree AV block, bundle-branch block, RV pacing for AV block. Sarcoid shows AV block, bundle-branch block, and scar-related VT. Voltage-mass discordance — low ECG voltages with increased LV wall thickness on echo — is the classic clue for cardiac amyloidosis.[1]
- Chest X-ray — bi-atrial enlargement (double-density right heart border, splaying of carina), pulmonary venous congestion, small-to-moderate pericardial effusion. Pericardial calcification = constriction, not RCM.
- Echocardiogram (the key imaging test) —
- Bi-atrial enlargement (massive).
- Normal or reduced LV cavity, normal or mildly reduced EF, increased wall thickness in infiltrative causes (LV wall over 12 mm) with granular/sparkling myocardial texture (amyloid).
- Restrictive mitral inflow on pulse-wave Doppler: E-dominant, E/A ratio over 2, deceleration time under 150 ms, short isovolumic relaxation time.
- Tissue Doppler — e' (mitral annular early diastolic velocity) reduced, under 5 cm/s at both septal and lateral annulus; E/e' ratio elevated (over 15) reflecting high LV filling pressure. Pericardial effusion small.
- Strain imaging (high yield) — apical sparing pattern on longitudinal strain ("cherry on top" — apical longitudinal strain preserved, basal and mid reduced) is highly suggestive of cardiac amyloidosis.[1]
- Bloods — NT-proBNP/BNP (markedly elevated — single best discriminator from constriction), troponin (elevated in AL — prognostic, included in Mayo staging), renal and liver function, full blood count and film (eosinophilia in Loffler), iron studies (ferritin, transferrin saturation in haemochromatosis), fasting glucose/HbA1c.
Confirmatory and subtype-specific tests
- Cardiac MRI (the gold standard for tissue characterisation) —
- Diffuse subendocardial late gadolinium enhancement (LGE) with difficulty nulling the myocardium (the blood pool and myocardium null together) is virtually diagnostic of amyloidosis.[1]
- Native T1 mapping elevated (over 1040 ms at 1.5 T) and extracellular volume (ECV) over 40% confirm the infiltrative amyloid phenotype.
- Patchy, multifocal basal/subepicardial LGE suggests sarcoidosis; focal FDG uptake on PET indicates active inflammation.
- T2-star (T2) under 20 ms* indicates iron overload (haemochromatosis).[8]
- 99mTc-pyrophosphate (PYP), 99mTc-DPD, or 99mTc-HMDP bone scintigraphy — Perugini grade 2 or 3 cardiac uptake diagnoses ATTR amyloidosis non-invasively (sensitivity and specificity over 99%) — provided monoclonal protein has been excluded by serum/urine electrophoresis and serum free light chains.[1][12]
- Serum free light chain assay with kappa/lambda ratio, serum and urine protein electrophoresis with immunofixation — diagnose the AL clone. Bone marrow biopsy confirms plasma-cell dyscrasia.[3]
- Endomyocardial biopsy — gold standard for tissue diagnosis; Congo red staining with apple-green birefringence under polarised light, with mass spectrometry typing to define the precursor protein (AL vs ATTR vs AA). Biopsy is now reserved for cases where non-invasive work-up is inconclusive.
- CT or MRI pericardial thickness — over 3 to 4 mm favours constriction rather than RCM.
- Cardiac catheterisation (when constriction is still suspected) — simultaneous LV and RV pressure tracings: dip-and-plateau (square-root) sign in both; in RCM, LVEDP exceeds RVEDP by more than 5 mmHg at end-expiration; in constriction, the two diastolic pressures equalise within 5 mmHg. RV systolic pressure over 50 mmHg and RVEDP less than one-third of RVSP favour RCM; the opposite favours constriction.[6]
Reproduced: key imaging thresholds and named signs
| Test | Sign / threshold | Implication |
|---|---|---|
| ECG | Low limb-lead voltage with thick LV on echo | Voltage-mass discordance — amyloid until excluded |
| Echo tissue Doppler | Both septal and lateral e' under 5 cm/s | Restrictive physiology |
| Echo tissue Doppler | Septal e' preserved (over 8 cm/s), lateral reduced | Annulus paradoxus — constriction |
| Echo strain | Apical sparing (cherry-on-top) | Cardiac amyloidosis |
| CMR | Diffuse subendocardial LGE, ECV over 40% | Cardiac amyloidosis |
| CMR | T2* under 20 ms | Iron overload (haemochromatosis) |
| Bone scintigraphy | Perugini grade 2-3 uptake | ATTR amyloidosis (after light chains excluded) |
| Cath | LVEDP - RVEDP over 5 mmHg end-expiration | RCM (vs constriction) |
| BNP/NT-proBNP | Markedly elevated | RCM (low or normal in constriction) |
RCM — key numbers
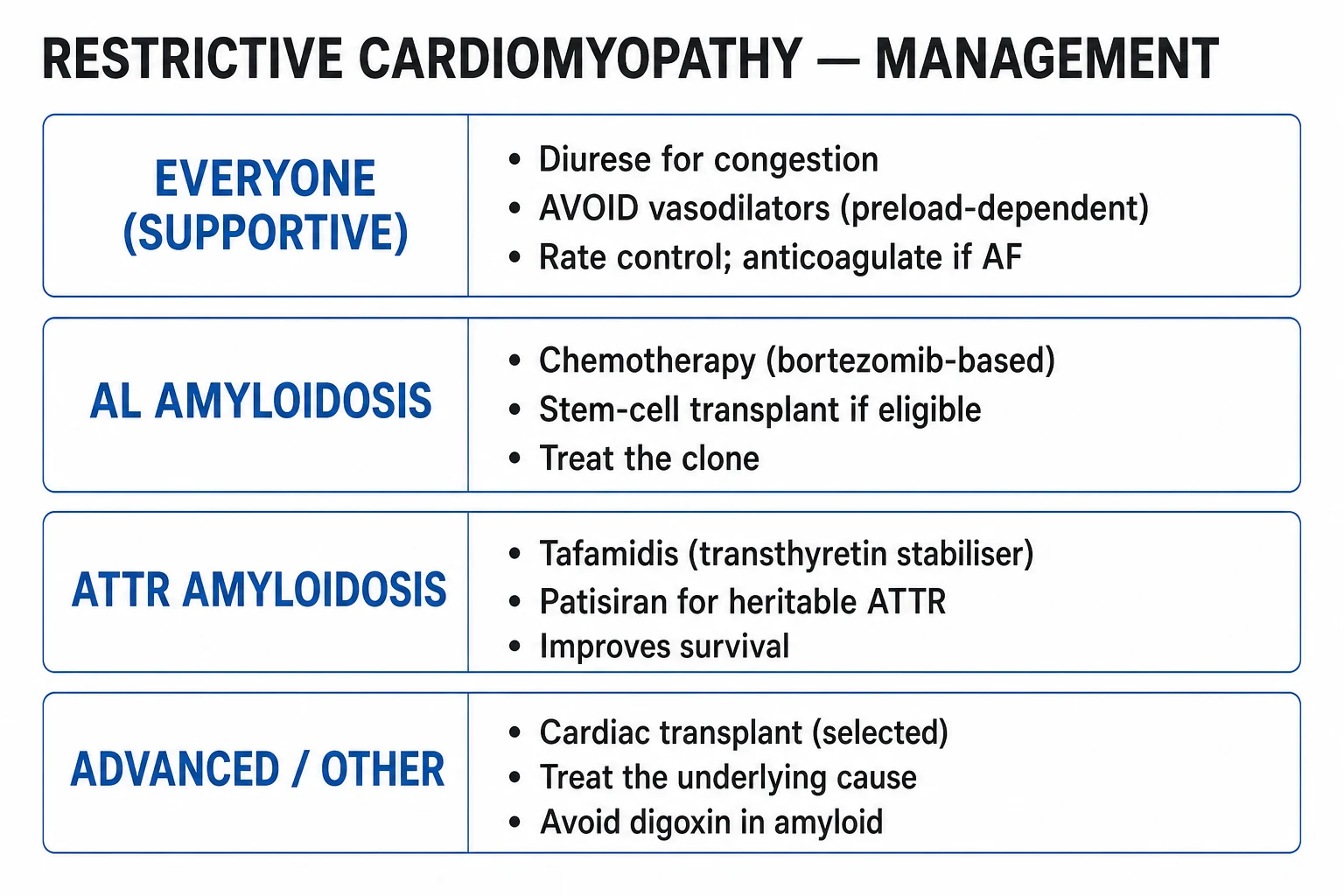
Management — Resuscitation
ABCDE assessment. Acute decompensated RCM typically presents with marked congestion, hypotension, and renal dysfunction — a difficult combination.[10]
- Cautious IV loop diuretic — furosemide 20 to 40 mg IV (or bumetanide 1 mg IV), titrated to urine output and renal function. The ventricles are preload-dependent; over-diuresis precipitates cardiogenic shock. Spironolactone 12.5 to 25 mg orally adds sodium retention control and is generally better tolerated than high-dose loop.
- Avoid nitrates, ACE inhibitors, ARBs, and hydralazine in decompensated disease — small falls in preload collapse cardiac output. Avoid beta-blockers in decompensated RCM — the tachycardia is compensatory for a small fixed stroke volume; slowing the rate drops cardiac output.
- Atrial fibrillation with rapid ventricular response — rate control with small doses of beta-blocker (metoprolol 25 to 50 mg orally BD, or IV metoprolol 2.5 to 5 mg cautiously). Avoid digoxin (binds amyloid fibrils, dangerous toxicity) and avoid non-dihydropyridine calcium-channel blockers (verapamil/diltiazem — negative inotrope, worsens HF, binds amyloid fibrils). Amiodarone 200 mg orally TDS for one week, then 200 mg OD for rhythm control in unstable AF.
- Anticoagulation for AF, intracardiac thrombus, or severe atrial standstill (e.g. apixaban 5 mg orally BD or warfarin with INR 2 to 3 — note AL amyloid patients bleed easily).
- Temporary pacing for symptomatic high-grade AV block while planning permanent device.
- Search for and treat reversible precipitants — anaemia, infection, thyroid disease, AF, NSAIDs (sodium retention). [1]
Management — Definitive & Stepwise
The overarching principle is treat the underlying cause — modern therapy has transformed prognosis for AL and ATTR amyloidosis, making early, accurate diagnosis critical.[1][3]
Disease-specific therapy
- AL amyloidosis — urgent plasma-cell-directed therapy to halt light-chain production:
- CyBorD — cyclophosphamide, bortezomib (a proteasome inhibitor), dexamethasone — first-line in many centres; bortezomib subcutaneously at standard doses (e.g. 1.3 mg/m squared on days 1, 4, 8, 11 of a 21-day cycle) with dexamethasone 40 mg weekly and cyclophosphamide 300 mg/m squared weekly.
- Daratumumab (anti-CD38 monoclonal antibody, subcutaneous) added for higher-risk disease.
- Autologous stem cell transplant in selected fit patients with limited cardiac involvement.
- Tafamidis is NOT useful in AL amyloid (it stabilises TTR, not light chains).
- ATTR amyloidosis —
- Tafamidis 61 mg orally once daily — the ATTR-ACT trial showed 30% reduction in all-cause mortality and 32% reduction in CV hospitalisations over 30 months in ATTR-CM (both wild-type and hereditary).[2]
- Diflunisal (off-label NSAID that stabilises TTR tetramer) — use is limited by renal/GI toxicity.
- Patisiran (RNA interference) and inotersen/vutrisiran (antisense/RNA interference) — primarily for hereditary ATTR with polyneuropathy; emerging cardiac data.
- Acoramidis (next-generation stabiliser) — recent positive phase-3 trial.
- Cardiac sarcoidosis — corticosteroids: prednisolone 0.5 to 1 mg/kg/day orally (typically 30 to 40 mg daily), tapering over 6 to 12 months guided by symptoms, ECG and imaging. Steroid-sparing agents — methotrexate 7.5 to 15 mg orally weekly, mycophenolate mofetil 1 g orally BD, or azathioprine 1 to 2 mg/kg/day — for refractory disease or steroid toxicity. ICD for sustained VT or primary prevention in high-risk scar; pacemaker for high-grade AV block.[11]
- Hereditary haemochromatosis — therapeutic phlebotomy of one unit (450 to 500 mL, around 200 to 250 mg iron) weekly until serum ferritin under 50 microgram/L, then maintenance phlebotomy every 1 to 3 months. Iron chelation — deferoxamine (subcutaneous infusion 20 to 40 mg/kg/day over 8 to 12 hours), or oral deferasirox 14 to 28 mg/kg/day — for those who cannot tolerate phlebotomy (anaemia, severe disease).[8]
- Loffler endocarditis (hypereosinophilic syndrome) — corticosteroids (prednisolone 1 mg/kg/day) ± cytotoxic therapy (hydroxyurea, or imatinib if FIP1L1-PDGFRA positive), anticoagulation, and surgical endocardiectomy with AV-valve repair in advanced disease.
- Tropical endomyocardial fibrosis — medical therapy for heart failure; endocardiectomy with valve repair/replacement offers symptomatic relief in selected surgical candidates.[7]
- Radiation-induced RCM — supportive; pericardiectomy if constriction coexists; valve replacement and CABG as indicated.
- Carcinoid heart disease — somatostatin analogue — octreotide LAR 20 to 30 mg IM every 4 weeks, or lanreotide autogel 120 mg deep SC every 4 weeks; tricuspid/pulmonary valve replacement (preferably biological) for symptomatic right-heart failure.
- Fabry disease — enzyme replacement (agalsidase alfa 0.2 mg/kg IV every 2 weeks, or agalsidase beta 1 mg/kg IV every 2 weeks) or migalastat (oral chaperone) for amenable mutations.
Symptomatic heart-failure therapy (universal principles)
- Loop diuretic — furosemide 20 to 40 mg orally (BD as needed), titrated to euvolaemia.
- Aldosterone antagonist — spironolactone 12.5 to 25 mg orally daily — modest benefit and potassium sparing.
- Cautious ACE inhibitor / ARB — only if blood pressure tolerates; avoid in AL amyloid with nephrotic syndrome (hypotension, AKI).
- Avoid — digoxin (binds amyloid fibrils, toxicity), calcium-channel blockers (negative inotropy, bind amyloid), high-dose beta-blockers (collapse cardiac output).
- Anticoagulation — for AF, intracardiac thrombus, or severe atrial dysfunction. [1]
Devices and advanced therapy
- Pacemaker for symptomatic bradycardia or high-grade AV block.
- ICD for sustained VT/VF, or primary prevention in selected high-risk sarcoid or HCM phenocopies (benefit uncertain in end-stage amyloid; consider life expectancy and shock burden).
- Cardiac resynchronisation therapy only if there is a wide QRS and dyssynchrony with systolic dysfunction.
- Heart transplantation — for end-stage RCM with a treatable or absent extra-cardiac cause; 5-year survival 70 to 80% in selected patients. Combined heart-liver transplant for ATTRv (liver is the source of mutant TTR); combined heart-kidney transplant for AL with renal failure.
- Mechanical circulatory support — bridge to transplant in selected. [1]
Escalation triggers
- Worsening heart failure despite optimal medical therapy — refer to a specialist amyloid/cardiomyopathy centre for disease-modifying treatment.
- New high-grade AV block — permanent pacemaker.
- Sustained VT — ICD.
- Symptomatic severe valvular disease — surgical/transcatheter intervention in a high-volume centre.
- End-stage HF — transplant assessment. [1]
Specific Subtypes & Scenarios
Cardiac amyloidosis (AL) — most aggressive; presents with heart failure + autonomic neuropathy + nephrotic proteinuria + macroglossia/periorbital purpura. Troponin elevation is universal; ECG low voltage with thick LV on echo. Diagnosis: serum free light chains, immunofixation, bone marrow biopsy; CMR shows diffuse subendocardial LGE. Mayo 2004 biomarker staging (cTnT, NT-proBNP) and revised 2013 (cTnT, NT-proBNP, difference between involved and uninvolved free light chains) stage prognosis. Treatment: CyBorD +/- daratumumab; transplant in selected.[3]
Cardiac amyloidosis (ATTRwt) — elderly men, HFpEF + bilateral carpal tunnel + spinal stenosis + biceps tendon rupture. Diagnosis: 99mTc-PYP/DPD grade 2-3 uptake (after light chains excluded) avoids biopsy. Treatment: tafamidis 61 mg daily.[2][9]
Cardiac amyloidosis (ATTRv V122I) — African-descent patient with hypertensive heart disease phenotype (LVH out of proportion, family history of "heart failure"). Screen with genetic testing in suspected patients and first-degree relatives. Tafamidis responsive. [1]
Cardiac sarcoidosis — basal/subepicardial LGE on CMR, FDG uptake on PET. HRS diagnostic criteria (2014) require histological diagnosis from cardiac or extracardiac tissue, OR a combination of clinical/imaging features with exclusion of other causes. Steroid-responsive if caught early; high sudden cardiac death risk.[11]
Loffler endocarditis — hypereosinophilic syndrome (eosinophil count over 1500 per microlitre for over 6 months), endocardial fibrosis with overlying thrombus, apical obliteration, systemic thromboembolism. Treat the eosinophilia (steroids, hydroxyurea, imatinib if FIP1L1-PDGFRA). [1]
Tropical endomyocardial fibrosis (EMF) — leading cause of heart failure in young people in the tropical belt; apical fibrosis of one or both ventricles with AV-valve tethering; ascites out of proportion to peripheral oedema. Endocardiectomy for advanced disease.[7]
Radiation-induced RCM — 5 to 30 years after mediastinal radiotherapy; mixed constriction + restriction + valvular (aortic/mitral) disease + premature coronary disease. Pericardiectomy risk is high; valve surgery carries high operative mortality. [1]
Carcinoid heart disease — serotonin and tachykinins cause right-sided endocardial plaques, tricuspid and pulmonary regurgitation, with restrictive filling. Treat with somatostatin analogue and valve surgery. [1]
Glycogen storage diseases — Fabry (X-linked alpha-galactosidase A deficiency; LVH + angiokeratomas + anhidrosis + proteinuria + corneal verticillata), Pompe (acid alpha-glucosidase, infantile hypotonia + HCM), Danon (LAMP2, young males, HCM + WPW + skeletal muscle + intellectual disability). [1]
Scleroderma cardiac disease — patchy myocardial fibrosis, microvascular disease, pericardial effusion; restrictive physiology with Raynaud, skin changes, anti-Scl-70 antibodies. [1]
Complications & Pitfalls
Cardiac complications:
- Progressive biventricular heart failure — the natural history.
- Arrhythmias — AF (atrial standstill in advanced amyloid), high-grade AV block (especially sarcoid), sustained VT, sudden cardiac death.
- Thromboembolism — atrial stasis, intracardiac thrombus (especially in EMF and amyloid); systemic embolisation may be the presenting event.
- Hypotension and cardiogenic shock from over-diuresis or vasodilator use.
- Renal failure from low cardiac output, congestion, and (in AL) nephrotic syndrome.
- Hepatic congestion progressing to cardiac cirrhosis. [1]
Treatment complications:
- Digoxin toxicity in amyloidosis — binds amyloid fibrils, dangerous arrhythmias; digoxin is contraindicated.
- Calcium-channel-blocker toxicity — worsens heart failure in amyloid; avoid.
- Over-diuresis precipitates cardiogenic shock.
- Bleeding on anticoagulation in AL amyloid (factor X depletion, vascular fragility from amyloid). [1]
Diagnostic pitfalls:
- Misdiagnosing as constrictive pericarditis and subjecting the patient to futile pericardiectomy — avert by checking BNP, pericardial calcification, septal bounce, and annulus paradoxus.
- Misdiagnosing as HFpEF in the elderly — missing disease-modifying therapy for ATTRwt or AL.
- Misdiagnosing as hypertensive heart disease in African-descent patients with ATTRv V122I.
- Missing the underlying cause — sending only an echo and not checking serum free light chains, immunofixation, and a bone-tracer scan. [1]
Prognosis & Disposition
Prognosis is cause-dependent and has been transformed by modern therapy: [1]
- AL amyloidosis — median survival under 6 months untreated from heart-failure onset; with CyBorD +/- daratumumab, median survival now exceeds 3 to 5 years. Mayo 2013 stage (NT-proBNP, cTnT, dFLC) stratifies: stage I 5-year survival over 90%; stage IV under 25%.
- ATTRwt — median survival 3 to 5 years untreated; with tafamidis, all-cause mortality reduced 30% over 30 months.[2]
- ATTRv V122I — median 2 to 3 years untreated; tafamidis-responsive.
- Cardiac sarcoidosis — variable; high sudden-death risk; responds to steroids if caught early before scar establishes.
- Radiation-induced — progressive over decades; median survival under 2 years once cardiac symptoms appear.
- Tropical EMF — high early mortality (around 25% in 1 year); survivors stabilise with chronic heart failure.
- Hereditary haemochromatosis — good prognosis with phlechotomy if caught before cirrhosis and diabetes; cardiac function often reverses with iron removal.
- Heart transplant — 5-year survival 70 to 80% in selected cases; recurrence of amyloid in the graft is described (especially AL if clone persists), and combined heart-liver transplant prevents recurrence in ATTRv.
Poor-outcome indicators: advanced NYHA class, troponin elevation, severe RV dysfunction, low cardiac output, advanced Mayo stage, sustained VT, severe autonomic involvement. [1]
Special Populations
- Elderly — ATTRwt is the dominant cause; suspect in any man over 65 with "HFpEF" and bilateral carpal tunnel or biceps rupture. Lower threshold to send bone-tracer scintigraphy and serum free light chains.
- African descent — screen TTR V122I in patients presenting with hypertensive heart failure and LVH out of proportion; 3 to 4% carrier rate.
- Tropical regions — EMF is the dominant RCM cause; constrictive pericarditis from TB is the dominant mimic — both common.
- Pregnancy — rare; avoid ACE/ARB/MRA; diurese cautiously (furosemide acceptable, spironolactone crosses placenta); deliver in a cardiac centre; consider early anaesthesia for labour.
- Paediatrics — glycogen storage diseases (Pompe, Danon), infantile sarcoma; different natural history and management.
- Renal impairment — AL often has nephrotic syndrome; kidney biopsy may give the diagnosis non-invasively; ACE inhibitor for proteinuria is poorly tolerated; dialysis + heart transplant may be combined.
- Family screening — all first-degree relatives of ATTRv patients should be offered genetic testing; cascade screening identifies pre-symptomatic carriers.
- Hereditary haemochromatosis — screen siblings with genetic testing (HFE C282Y) and iron studies. [1]
Evidence, Guidelines & Regional Differences
- 2021 ESC Position Statement on Cardiac Amyloidosis (Garcia-Pavia et al., EJHF) — comprehensive diagnostic algorithm: red-flag screening, non-invasive bone-tracer scintigraphy for ATTR, serum free light chains for AL, and disease-specific therapy (tafamidis, chemotherapy).[1]
- 2021 ESC Monitoring Consensus — surveillance echocardiography, cardiac biomarkers, and clinical review intervals for ATTR-CM on tafamidis.[12]
- 2021 ESC HF Guideline and 2016 ESC HF Guideline (Ponikowski) — RCM included in HFpEF/Heart Failure with mildly reduced EF; advocate early referral for cardiomyopathy assessment.[10]
- ATTR-ACT trial (Maurer 2018, NEJM) — tafamidis 61 mg or 80 mg daily vs placebo over 30 months in ATTR-CM: all-cause mortality 29.5% vs 42.9% (hazard ratio 0.70), fewer cardiovascular hospitalisations, slower decline in 6-minute walk and Kansas City Cardiomyopathy Questionnaire.[2]
- Mayo 2004 and 2013 AL amyloid staging systems — biomarker-based (troponin, NT-proBNP, free light-chain difference) staging driving transplant decisions and prognosis.[3]
- HRS 2014 Expert Consensus on Cardiac Sarcoidosis (Birnie) — diagnostic criteria and arrhythmia management; established the Wonderology criteria for non-invasive diagnosis and the role of steroids/immunosuppression.[11]
Regional differences:
- Tropical Africa, Kerala coast, South America — endomyocardial fibrosis is the dominant RCM cause; rheumatic and tuberculous pericarditis dominate the constriction differential.[7]
- Indian subcontinent — TB-endemic; constrictive pericarditis from tuberculosis is the most frequent RCM mimic; amyloidosis (especially AL with leprosy/chronic inflammation, AA amyloid from TB) is also common.
- African populations — high TTR V122I carrier rate; ATTRv cardiac amyloid under-recognised.
- UK/NHS — tafamidis commissioned via the National Amyloidosis Centre with NYHA-based stop criteria.
- US — Medicare covers PYP scintigraphy for suspected ATTR-CM; tafamidis FDA-approved.
Controversies — tafamidis cost-effectiveness (UK NICE rejected, then negotiated; high annual cost); role of patisiran/inotersen in isolated cardiomyopathy; timing of combined heart-liver transplant in ATTRv; ICD benefit in amyloid given competing risks. [1]
Exam Pearls
RCM causes — AMISH
AMISH
AL, ATTRwt (senile), ATTRv (V122I, T60M, V30M) — commonest in the West
Fabry, Pompe, Danon, glycogen storage diseases
HFE C282Y; bronze diabetes; T2* under 20 ms
Granulomas; African-American; AV block and VT in the young
Loffler endocarditis, tropical EMF, radiation-induced RCM
- RCM triad — stiff ventricles, restrictive diastolic filling, preserved EF.
- Rarest of the three cardiomyopathies; commonest cause in the West is amyloidosis, in the tropics endomyocardial fibrosis.
- Kussmaul's sign — raised JVP on inspiration — present in RCM and constriction, absent in tamponade.
- Square-root (dip-and-plateau) sign — in both RCM and constriction.
- Voltage-mass discordance — low ECG voltages with thick LV walls on echo = amyloidosis until excluded.
- Pericardial calcification on CXR/CT = constrictive pericarditis, NOT RCM.
- BNP — markedly raised in RCM, normal or low in constrictive pericarditis — the single best discriminator.
- Annulus paradoxus — septal e' preserved in constriction, both e' reduced in RCM.
- Apical sparing on strain ("cherry on top") — cardiac amyloidosis.
- Diffuse subendocardial LGE on CMR — amyloidosis.
- Perugini grade 2-3 bone scintigraphy — ATTR amyloidosis (after light chains excluded).
- Macroglossia, periorbital purpura, carpal tunnel, biceps rupture — amyloid clues.
- Avoid digoxin (binds amyloid fibrils, toxicity) and calcium-channel blockers (negative inotropy, bind amyloid) in amyloidosis.
- Tafamidis 61 mg daily — ATTR-CM (NOT AL); ATTR-ACT trial.
- AL amyloid — serum free light chains + immunofixation; treat with CyBorD +/- daratumumab.
- Cardiac sarcoid — high-grade AV block in a young patient; treat with steroids, ICD for sustained VT.
- Haemochromatosis — bronze diabetes, weekly phlebotomy to ferritin under 50 microgram/L.
- EMF — tropical, apical obliteration, ascites out of proportion to peripheral oedema.
- Congo red stain with apple-green birefringence — gold standard for amyloid.
- Heart transplant 5-year survival 70-80%; combined heart-liver transplant for ATTRv. [1]
Exam application bank (NEET-PG / INICET)
One-line answer
Restrictive cardiomyopathy (RCM) is the rarest of the three WHO cardiomyopathies, defined by non-compliant, stiff ventricles that resist filling in diastole, with reduced diastolic volume and preserved (or near-normal) systolic function, producing biventricular diastolic heart failure with bi-atrial enlargement. Leading cause in the developed world is cardiac amyloidosis (AL light-chain, and ATTR — wild-type/senile and hereditary variant); other causes are sarcoidosis, haemochromatosis, endomyocardial fibrosis (EMF) / Loffler endocarditis, radiation, carcinoid heart disease, glycogen storage diseases (Fabry, Pompe, Danon) and scleroderma. Presents with right-heart-failure signs (raised JVP, Kussmaul's sign, hepatomegaly, ascites, oedema), dyspnoea, fatigue and atrial fibrillation, in a patient with preserved EF. Differentiate from constrictive pericarditis by BNP (high in RCM, low in con
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard. [1]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes. [1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change. [1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each. [1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory. [1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Restrictive Cardiomyopathy.
References
- [1]Garcia-Pavia P, Bengel F, Cuddy S, et al. Diagnosis and treatment of cardiac amyloidosis. A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases Eur J Heart Fail, 2021.PMID 33826207
- [2]Maurer MS, Elliott P, Comenzo R, et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy N Engl J Med, 2018.PMID 30145929
- [3]Muchtar E, Buadi FK, Dispensieri A, Gertz MA. Systemic amyloidosis from A (AA) to T (ATTR): a review J Intern Med, 2021.PMID 32929754
- [4]Falk RH. Diagnosis and management of the cardiac amyloidoses Circulation, 2005.PMID 16186440
- [5]Rapezzi C, Arbustini E, Caforio AL, et al. Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases Eur Heart J, 2013.PMID 23211230
- [6]Hatle LK, Appleton CP, Popp RL. Differentiation of constrictive pericarditis and restrictive cardiomyopathy by Doppler echocardiography Circulation, 1989.PMID 2914352
- [7]Mocumbi AO. Endomyocardial fibrosis: recent advances and future therapeutic targets Nat Rev Cardiol, 2025.PMID 40011660
- [8]Murphy CJ, Oudit GY. Iron-overload cardiomyopathy: pathophysiology, diagnosis, and treatment J Card Fail, 2010.PMID 21055653
- [9]Grogan M, Scott CG, Kyle RA, et al. Natural History of Wild-Type Transthyretin Cardiac Amyloidosis and Risk Stratification Using a Novel Staging System J Am Coll Cardiol, 2016.PMID 27585505
- [10]Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure Rev Esp Cardiol (Engl Ed), 2016.PMID 27894487
- [11]Birnie DH, Sauer WH, Bogun F, et al. HRS expert consensus statement on the diagnosis and management of arrhythmias associated with cardiac sarcoidosis Heart Rhythm, 2014.PMID 24819193
- [12]Garcia-Pavia P, Rapezzi C, Adler Y, et al. Expert consensus on the monitoring of transthyretin amyloid cardiomyopathy Eur J Heart Fail, 2021.PMID 33915002