Dermatology · Medicine
Bullous pemphigoid
Also known as Bullous pemphigoid · BP · Pemphigoid
Bullous pemphigoid (BP) is the commonest autoimmune blistering disease, predominantly affecting the elderly, caused by IgG (and IgE) autoantibodies against BP180 (collagen XVII) and BP230 (BPAG1) at the basement membrane zone, producing subepidermal tense bullae with intense pruritus. Diagnosis rests on histology (subepidermal blister with eosinophilic infiltrate), direct immunofluorescence (linear IgG/C3 at the BMZ — the gold standard), indirect immunofluorescence on salt-split skin (epidermal-side binding), and anti-BP180 NC16A ELISA (disease-activity marker). Management has shifted from systemic corticosteroids toward potent topical clobetasol combined with doxycycline ± nicotinamide (safer in the elderly); refractory disease is treated with rituximab, omalizumab or dupilumab. Fellowship-level assessment demands mastery of the DIF vs pemphigus (linear vs intercellular), salt-split skin interpretation (epidermal = BP, dermal = EBA), drug triggers (DPP-4 inhibitors, loop diuretics, checkpoint inhibitors), the association with neurological disease (Parkinson's, dementia, stroke), and the first-line topical-first paradigm that minimises steroid toxicity in elderly patients.
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags
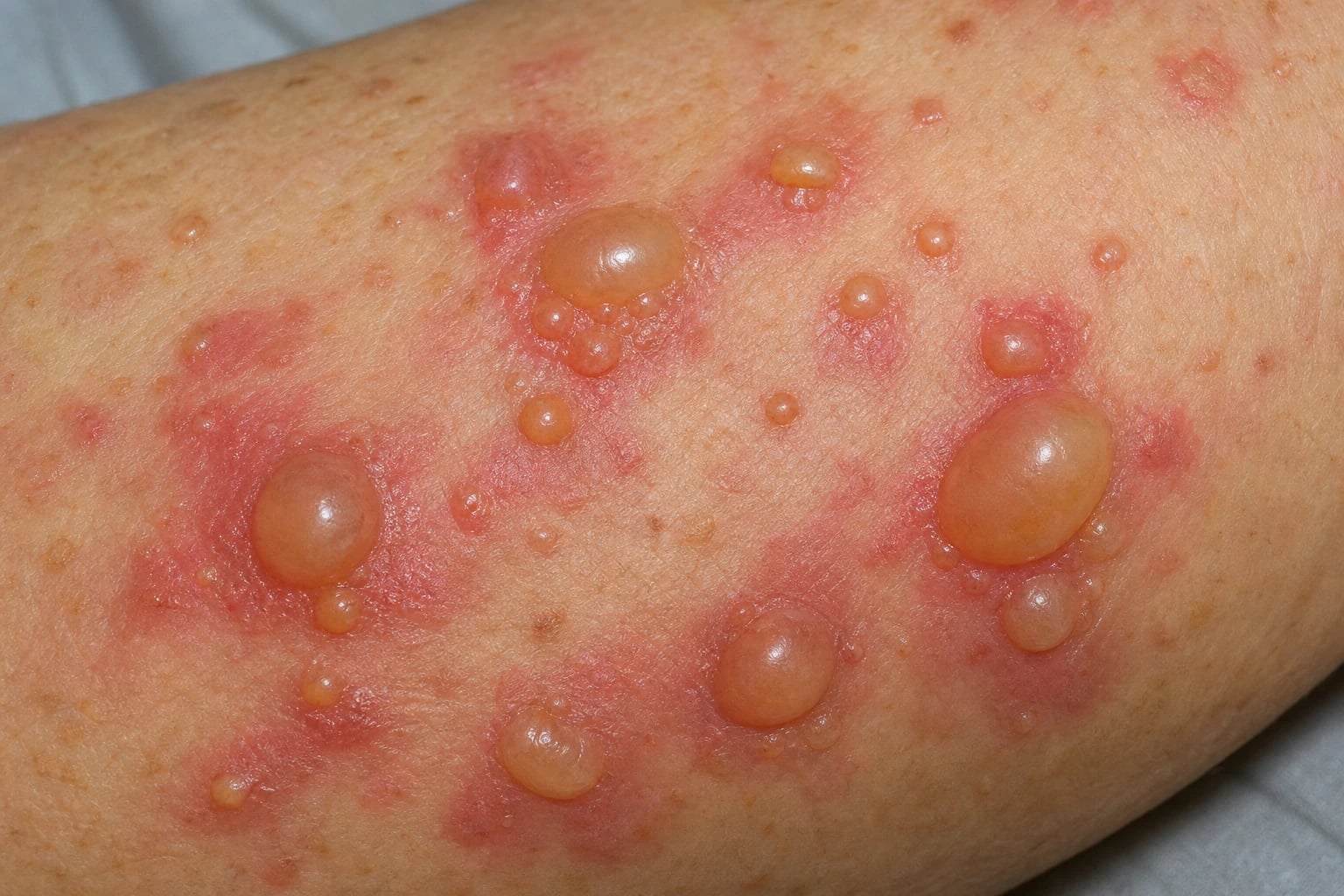
Definition & Classification
Bullous pemphigoid (BP) is the commonest autoimmune blistering disease, predominantly affecting the elderly, caused by autoantibodies against BP180 (collagen XVII) and BP230 (BPAG1) at the basement membrane zone (BMZ), producing a subepidermal blister.[1][3]
| Subtype | Antibody | Features |
|---|---|---|
| Classical BP | Anti-BP180, anti-BP230 | Tense bullae, elderly, pruritic |
| Mucous membrane pemphigoid (MMP) | Anti-BP180, anti-laminin 332, anti-integrin α6β4 | Predominantly mucosal, scarring (conjunctival, oral) |
| Pemphigoid gestationis | Anti-BP180 NC16A | Pregnancy (2nd-3rd trimester) or postpartum |
| Drug-induced pemphigoid | Anti-BP180/230 | DPP-4 inhibitors, loop diuretics, checkpoint inhibitors, PUVA, some antibiotics |
| Anti-p200 pemphigoid | Anti-laminin γ1 | Lower legs, often post-viral |
| IgA pemphigoid | IgA anti-BP180 | Vesicopustular; linear IgA on DIF |
| Pemphigoid nodularis | Anti-BP180/230 | Persistent prurigo-nodularis-like plaques with linear IgG at BMZ; often misdiagnosed as nodular prurigo for years |
| Pemphigoid vegetans | Anti-BP180/230 | Vegetating plaques in flexures (groin, axillae); resembles pemphigus vegetans clinically but DIF is linear IgG at BMZ |
| Erythrodermic BP | Anti-BP180/230 | Generalised erythroderma with secondary bullae; rare; mimics Sézary syndrome or erythrodermic CTCL |
| Paediatric BP | Anti-BP180 (often NC16A) | Rare in patients under 18 yr; often localised (genital, perioral); same immunopathology as adult BP |
Subtype notes (fellowship / examiner-relevant):[1][9][4][12]
- Mucous membrane pemphigoid (MMP / cicatricial pemphigoid) — a heterogeneous subset dominated by anti-BP180 (especially C-terminal domain), anti-laminin 332 (anti-epiligrin — paraneoplastic association), anti-laminin 331, and anti-integrin α6β4. Mucosal-dominant (oral, conjunctival, genital, laryngeal, oesophageal); scarring, symblepharon and eventual blindness if untreated — a key reason MMP is treated more aggressively than cutaneous BP.
- Pemphigoid gestationis (PG, "herpes gestationis") — pregnancy-specific BP; onset in 2nd-3rd trimester or postpartum; periumbilical urticarial plaques progressing to bullae; recurs in subsequent pregnancies, often earlier and more severe; managed with potent topical ± oral prednisolone.
- Drug-induced BP — DPP-4 inhibitors, PD-1/PD-L1 inhibitors, loop diuretics, spironolactone, penicillins; may have a latency of weeks-months; often resolves on drug withdrawal.
- Pemphigoid nodularis & pemphigoid vegetans — clinicopathological mimickers (nodular prurigo / pemphigus vegetans); linear IgG/C3 at BMZ on DIF is the discriminator.
- Erythrodermic BP — rare; generalised exfoliative erythroderma; secondary bullae formation; low threshold to biopsy.
- Paediatric BP — extremely rare; usually localised; reports of vaccination-associated BP in children. [1]
Epidemiology
- Mortality is 2-6× higher than age-matched controls at 1 year, driven by steroid toxicity (infection, osteoporosis, cardiovascular events), the underlying neurological comorbidity, and falls from pruritus — which is why the field has shifted decisively toward steroid-sparing regimens.
- Geographical variation: BP incidence is rising in industrialised countries (Germany: 3-4× in 20 yr), partly explained by ageing populations, increased DPP-4 inhibitor prescribing, and improved awareness/diagnosis. In pemphigus-endemic regions (Brazil, India, North Africa) BP is comparatively less common relative to pemphigus foliaceus and vulgaris.
- HLA associations: HLA-DQB103:01 and DRB104 are reproducibly associated with BP across multiple cohorts.
- Bidirectional neurological link: cohort studies demonstrate a higher prevalence of pre-existing or new-onset Parkinson's disease (~3×), Alzheimer's dementia, vascular dementia, stroke, epilepsy and multiple sclerosis in BP patients compared to age-matched controls — often preceding BP by years, suggesting a shared autoimmune mechanism against BP180 isoforms expressed in the nervous system.[6][8]
- Drug triggers: DPP-4 inhibitors (vildagliptin, linagliptin, sitagliptin), loop diuretics (furosemide), aldosterone antagonists (spironolactone), checkpoint inhibitors (PD-1/PD-L1), PUVA, some antibiotics (penicillins).[6][7]
Pathophysiology
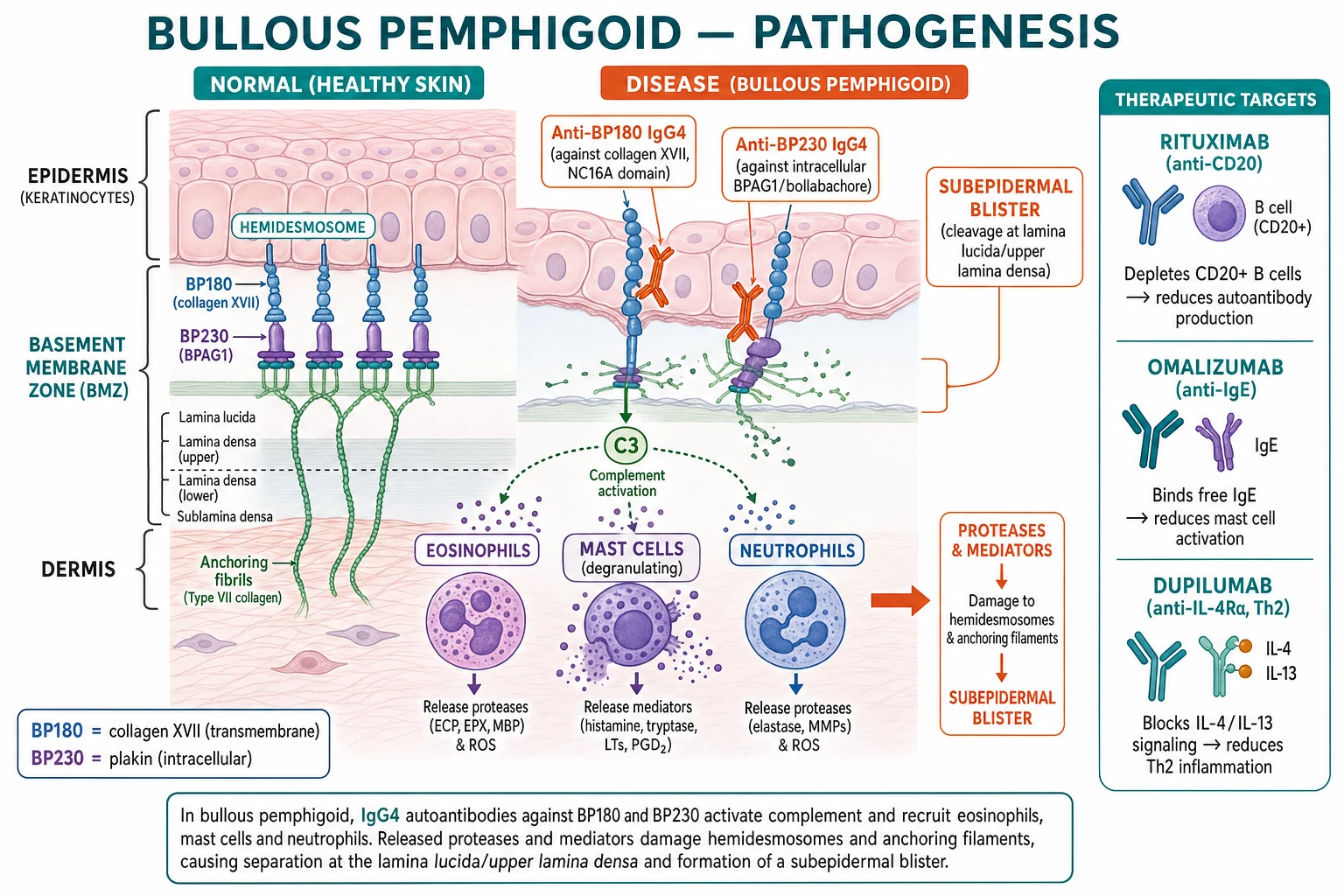
-
Autoantibodies: predominantly IgG4 and IgG1 against BP180 (collagen XVII), especially the NC16A extracellular domain (the immunodominant epitope), and BP230 (BPAG1), an intracellular plakin; IgE autoantibodies against BP180 are also important and drive pruritus.[1][3]
-
Mechanism: antibody binding activates complement (C3), recruits eosinophils and mast cells (degranulation), and neutrophils → release proteases (MMP-9, elastase) that cleave BP180 and damage the hemidesmosome–anchoring filament complex → separation at the lamina lucida → subepidermal blister.[1]
-
Th2-driven immune response with IL-4, IL-5, IL-13, IL-31 and high total IgE — explains the eosinophilia, pruritus, and the emerging role of dupilumab (anti-IL-4Rα).[5]
-
Disease-activity biomarker: circulating anti-BP180 NC16A ELISA titres correlate with disease activity, blister count and relapse risk — rising titres predict flare even before clinical relapse. Anti-BP230 antibodies are present in 50-70% and are more associated with pruritus and eosinophilia. Anti-BP180 NC16A is the most useful single biomarker for monitoring response to therapy and guiding taper.[1]
-
Genetic / molecular subtypes: classical IgG4-driven, IgE-driven (Th2-high, dupilumab-responsive), IgA-driven, drug-induced (DPP-4-inhibitor-associated often has anti-BP230 or anti-laminin reactivity), and BP with anti-BP230 alone (poorly immunoblot-validated — diagnostic caveat). Stratum-corneum and IgE deposition patterns may predict omalizumab responsiveness.[1][5]
Quick numbers for the examiner
Quick numbers for the examiner
TENSE BULLOUS — bullous pemphigoid features
Roof is thick; bullae don't rupture as easily as pemphigus
Commonest autoimmune blistering disease in the elderly
Lateral pressure does NOT cause epidermal slippage
Bulla is below the epidermis; eosinophil-rich infiltrate is characteristic
IgG against hemidesmosomal antigens; salt-split skin shows roof binding
Clinical Presentation [1]
- Prodromal phase: weeks to months of intense pruritus, eczematous or urticarial plaques (often misdiagnosed as eczema or scabies) BEFORE blisters appear.
- Established BP: tense bullae (>1 cm, clear or haemorrhagic fluid) on an erythematous or urticarial base — trunk, proximal limbs, flexures, lower abdomen, groin. Bullae do NOT rupture as easily as pemphigus (thick epidermal roof).[2][3]
- Nikolsky sign NEGATIVE (unlike pemphigus). Bullae may extend with pressure but the epidermis does not detach laterally.
- Mucosal involvement in ~20-30% (usually oral, mild; unlike pemphigus where mucosa is severe).
- Atypical presentations: localised BP (one area, often lower leg), prurigo nodularis-like, paediatric BP (rare), BP in pregnancy = pemphigoid gestationis.[9]
-
Body site distribution: most commonly the trunk, proximal limbs (upper inner arms, inner thighs), flexures (axillae, groin) and lower abdomen; palms/soles usually spared. Mucosal involvement (oral most common) occurs in 20-30% but is typically mild.[2][3]
-
Variant / atypical presentations (fellowship-relevant):[1][9]
- Pemphigoid nodularis — chronic prurigo-like nodules that, when biopsied (often after years of failed anti-pruritic therapy), show linear IgG/C3 at BMZ; anti-BP180/230 present.
- Pemphigoid vegetans — vegetating plaques in groin/axillae mimicking pemphigus vegetans clinically but linear (not intercellular) DIF.
- Erythrodermic BP — generalised exfoliative erythroderma with secondary bullae; differential includes erythrodermic CTCL/Sézary, drug reaction, psoriasis.
- Paediatric BP — typically localised (genital, perioral); post-vaccination in some cases.
- Pemphigoid gestationis (PG) — periumbilical onset in 2nd-3rd trimester; anti-BP180 NC16A; recurs in subsequent pregnancies.[10][11]
- Mucous-membrane pemphigoid (MMP) — predominantly oral/ocular/genital; scarring; ocular involvement is an emergency.
- Drug-induced BP — DPP-4 inhibitor, loop diuretic, checkpoint inhibitor — defined new entity; resolves with withdrawal.
- Non-bullous BP — chronic pruritic eczematous or urticarial plaques for months-years WITHOUT blister formation; positive DIF and/or ELISA; ~5-20% of BP diagnoses — important "forme fruste" for the examiner.
Differential Diagnosis
| Mimic | Distinguishing features |
|---|---|
| Pemphigus vulgaris | Flaccid blisters, severe oral involvement, Nikolsky +ve, DIF intercellular IgG4 |
| Dermatitis herpetiformis | Extensor grouped vesicles, coeliac, DIF granular IgA at papillae |
| Linear IgA disease | "String of pearls" sign; DIF linear IgA at BMZ |
| Epidermolysis bullosa acquisita | Mechanobullous, DIF linear IgG at BMZ; IIF binds dermal side of salt-split skin |
| Bullous drug eruption | Drug history, DIF negative, eosinophils |
| Bullous impetigo | Children, positive culture, superficial, DIF negative |
| Bullous lupus erythematosus | SLE, ANA+, sun-exposed, DIF granular BMZ |
| Pseudoporphyria | NSAID/dialysis, dorsal hands, histology similar, no antibodies |
Bedside mnemonic for the differential: ask yourself — is the bullae tense or flaccid (BP vs pemphigus vulgaris), is the DIF linear at BMZ or intercellular (BP or EBA vs pemphigus), is the antibody IgA or IgG (DH or linear IgA disease vs BP), is the DIF granular or linear (DH vs BP), is there a drug trigger (drug-induced BP or bullous drug eruption), is the bullae widespread or focal, is there scarring (MMP vs BP), and is there mucosal involvement (MMP or pemphigus vs BP)? Asking these 8 questions at the bedside correctly classifies >90% of bullous dermatoses.[1][9]
Diagnosis
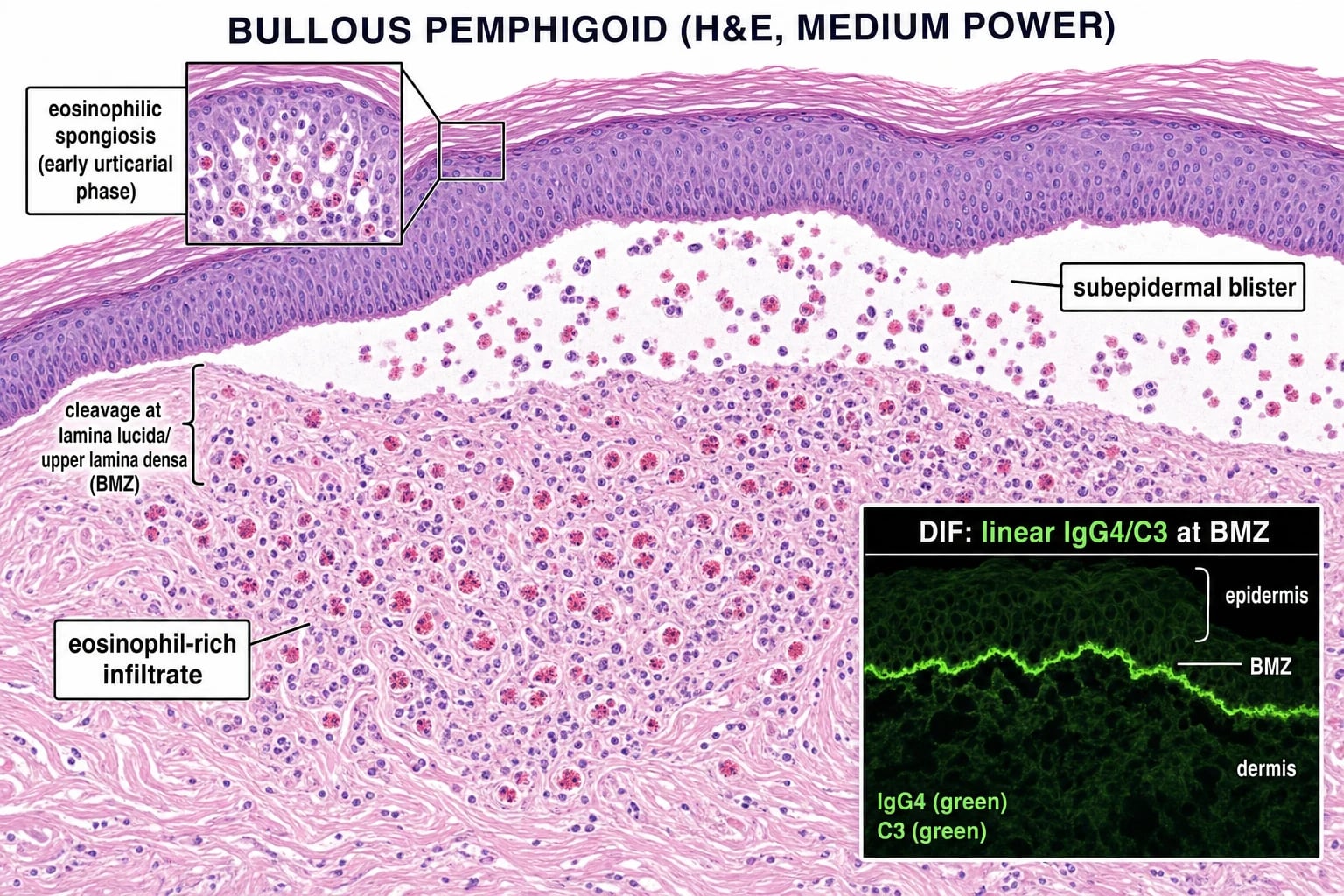
Diagnosis requires three investigations:[1][4]
- Histology (lesional biopsy): subepidermal blister with an eosinophil-rich infiltrate in the dermis and blister cavity. (Eosinophilic spongiosis in the prodromal phase.)
- Direct immunofluorescence (DIF) of perilesional skin — linear IgG4 (and C3) at the basement membrane zone (BMZ). This is the gold standard for diagnosis. (Contrast with pemphigus: intercellular/chicken-wire IgG4.)
- Serology:
- Indirect immunofluorescence (IIF) on salt-split skin — antibodies bind the epidermal side of the split (distinguishes BP from epidermolysis bullosa acquisita which binds the dermal side).
- Anti-BP180 NC16A ELISA — ~90% sensitivity; titres correlate with disease activity (monitoring biomarker).
- Anti-BP230 ELISA; total IgE often elevated (a Th2 marker). [1]
Salt-split skin — 'BED' rule
BED
Drug doses — BP cheat-sheet (fellowship numbers)
Investigations [1]
Bullous pemphigoid diagnosis requires three integrated investigations — never rely on serology alone (false positives common in elderly with background autoreactivity) or histology alone (cannot differentiate from EBA / DH):[1][4][2]
1. Skin biopsy for H&E (lesional biopsy) — 4 mm punch from a fresh, intact bulla (less than 24 h old). Show subepidermal blister with eosinophil-rich infiltrate in the dermis and within the blister cavity. Eosinophilic spongiosis (eosinophils tracking into the lower epidermis) is characteristic of the non-bullous prodromal phase and may be the only H&E finding in papular disease. [1]
2. Direct immunofluorescence (DIF) of perilesional skin — within 1-2 cm of an active lesion (NOT a bulla roof or floor — they produce false negatives). Perilesional skin is critical because lesional tissue shows consumption of immune deposits. Show linear IgG4 (with C3 in 70-80%) along the basement membrane zone — this is the gold-standard diagnostic test. Contrast with pemphigus: intercellular / chicken-wire IgG4. [1]
3. Serology (blood tests): [1]
- Indirect immunofluorescence (IIF) on 1 mol/L NaCl-split skin — substrate is healthy human skin split through the lamina lucida; BP antibodies bind the epidermal (roof) side; epidermolysis bullosa acquisita binds the dermal (floor) side. This is the BED rule.
- Anti-BP180 NC16A ELISA — sensitivity ~85-90% in active disease, specificity ~95%. Cut-off usually ≥20 U/mL is positive; titres correlate with disease activity, blister count and relapse risk and are the principal tool for monitoring response to therapy, guiding steroid taper and detecting early relapse.
- Anti-BP230 ELISA — positive in 50-70%; less disease-specific, but when isolated may indicate early or pre-bullous BP. Anti-BP230 IgE tracks pruritus.
- Total serum IgE and peripheral eosinophil count — usually elevated; high IgE predicts response to omalizumab; high eosinophil count tracks Th2 activity and dupilumab candidacy.
- Bedside Tzanck smear from a bulla floor — shows eosinophils (not acantholytic keratinocytes as in pemphigus); cheap but non-specific. [1]
4. Pre-treatment baseline work-up (essential before systemic therapy): full blood count, renal and liver function, HbA1c / fasting glucose, vitamin D, bone density (DEXA), chest radiograph, infection screening (TB, hepatitis B/C, HIV in those eligible for rituximab/dupilumab), G6PD activity (if dapsone planned), vaccination status (influenza, pneumococcal, COVID-19, shingles). [1]
Management
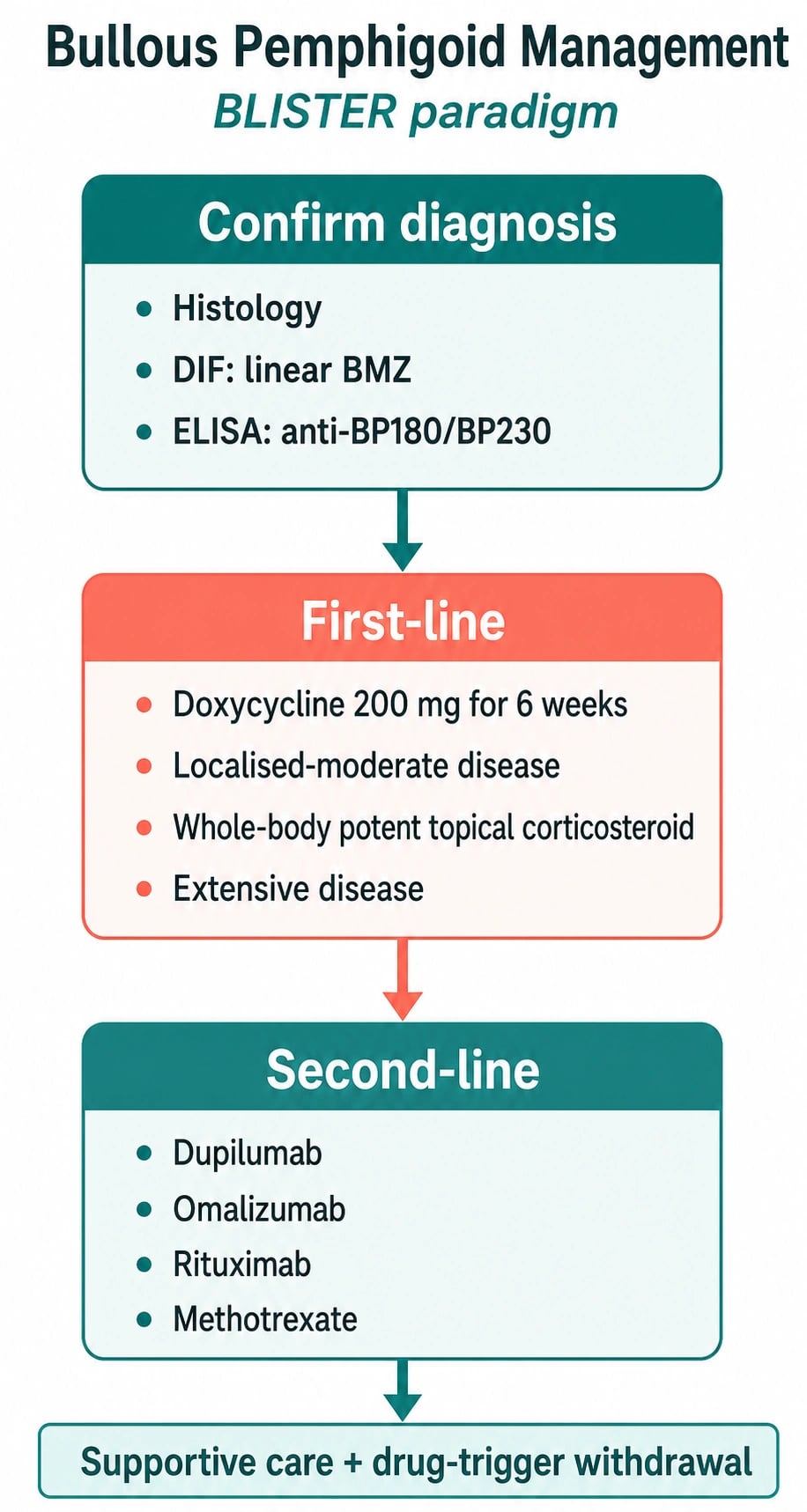
Management — Overview & First-line Therapy (Modern Paradigm)
- Potent topical corticosteroids (clobetasol propionate 0.05% cream/ointment) — apply to all affected AND clinically uninvolved skin at a typical dose of 30-40 g/day total-body application in adults, tapering over weeks-months. Surprisingly effective in moderate-severe BP; halves cumulative systemic-steroid exposure (the BLISTER trial showed doxycycline non-inferior to prednisolone for short-term control).[2]
- Doxycycline 200 mg orally daily (100 mg BD) ± nicotinamide 500 mg TDS — combination is steroid-sparing; doxycycline's anti-inflammatory and anti-protease (MMP-9 inhibition) effects at sub-antimicrobial doses; avoids infection, osteoporosis, diabetes and cardiovascular risks of systemic steroids in the elderly.[5]
- The modern paradigm prioritises topical clobetasol + doxycycline ± nicotinamide over systemic prednisolone wherever possible — the elderly are highly vulnerable to steroid complications.
- Antiseptic / emollient skin care, comorbidity management, bone protection (calcium 1 g + vitamin D 800-1000 IU daily; bisphosphonate if prolonged steroid course anticipated), and vaccination (influenza, pneumococcal, COVID-19, shingles) precede immunosuppression.
Management — Systemic Second-line Therapy
- Oral prednisolone 0.5-0.75 mg/kg/day (up to 1 mg/kg/day in extensive disease) — for moderate-to-severe BP failing topical clobetasol + doxycycline; taper once control achieved (typically to ≤10 mg/day by 8-12 weeks). Monitor BP, glucose, electrolytes, bone density, weight and infection at each visit.[3]
- Prednisolone taper protocol: reduce by 5-10 mg every 2 weeks until 20 mg/day, then by 2.5 mg every 2-4 weeks to 10 mg/day, then by 1 mg every 2-4 weeks; aim to discontinue within 6-9 months.
- Adjunctive steroid-sparing agents (see Steroid-Sparing & Refractory Therapy section below): doxycycline, dapsone, mycophenolate mofetil, azathioprine, methotrexate.
Drug triggers of bullous pemphigoid — 'GLIP-PS' rule
GLIP-PS
Steroid-Sparing & Refractory Therapy
For moderate-severe BP uncontrolled on topical clobetasol + doxycycline ± nicotinamide, or where prednisolone cannot be tapered, add or switch to a steroid-sparing agent or biologic:[3][5]
- Doxycycline 200 mg daily ± nicotinamide 500 mg TDS — first-line steroid-sparing; favoured over prolonged prednisolone (BLISTER trial evidence).[5]
- Dapsone 50-100 mg daily — particularly effective in neutrophil-rich BP variants and eosinophil-rich disease; G6PD must be checked first (haemolysis risk), and monitor methaemoglobin, reticulocytes and LFTs monthly.[3]
- Methotrexate 10-25 mg weekly (oral or subcutaneous) — useful in elderly with normal renal function; add folic acid 5 mg the day after; monitor FBC, LFTs every 4-8 weeks.
- Azathioprine 1-3 mg/kg/day — requires TPMT genotyping before initiation (avoid in homozygous deficiency); monitor FBC and LFTs.
- Mycophenolate mofetil 1-1.5 g BD (2-3 g/day) — preferred in younger patients with normal renal function; GI side effects common; monitor FBC.
- Rituximab 1 g IV × 2 doses 14 days apart (rheumatoid arthritis protocol; preferred over lymphoma 375 mg/m² × 4 due to better safety profile in BP) — anti-CD20 B-cell depletion with ~70% complete remission in largest systematic review; monitoring: hypogammaglobulinaemia, hepatitis B reactivation, progressive multifocal leukoencephalopathy.[5]
- Omalizumab 300-600 mg SC q4w — anti-IgE monoclonal; particularly effective when total serum IgE is elevated (>200 IU/mL) and in BP with prominent pruritus; works within 4-12 weeks.
- Dupilumab 600 mg SC loading dose, then 300 mg SC q2w — anti-IL-4Rα blocking IL-4/IL-13 Th2 axis (the LIBERTY-BP ADEPT trial met its primary and all key secondary endpoints in 2024, with patients on dupilumab five times more likely to achieve sustained disease remission and a marked steroid-sparing effect).[5]
- IVIG 2 g/kg/month divided over 3-5 days × 6 months — useful as adjunct in refractory or infection-prone patients; expensive.
- Emerging: JAK inhibitors (upadacitinib, baricitinib), anti-FcRn (efgartigimod) — promising case-series evidence; not yet first-line.
- Taper principle: once blistering is controlled on background therapy, taper oral prednisolone first (often to ≤10 mg/day within 8-12 weeks); allow steroid-sparing agent to take effect; monitor anti-BP180 NC16A titres every 3 months as a relapse biomarker.
Drug-Induced BP
A defined drug trigger is identifiable in roughly 15-25% of new BP diagnoses; withdrawal of the offending agent is mandatory and often results in resolution within weeks-months.[7]
- Stop the offending drug (DPP-4 inhibitor, loop diuretic, checkpoint inhibitor) immediately. Consider drug-induced causation in any new-onset BP — take a careful drug history including over-the-counter medications and recent vaccines.
- Time to onset: median 6-12 months after starting gliptin or loop diuretic (long latency may obscure attribution); checkpoint-inhibitor BP can develop within days to weeks of first infusion and tends to be severe, refractory, and may be accompanied by other immune-related adverse events.
- DPP-4 inhibitors (gliptins): vildagliptin, linagliptin, sitagliptin, saxagliptin, alogliptin — the most common drug trigger in elderly type 2 diabetics; BP arises via altered antigen presentation of BP180 by dysregulated e-cadherin. Risk is highest with vildagliptin and linagliptin.
- Loop diuretics and aldosterone antagonists: furosemide, bumetanide, spironolactone, eplerenone. Mechanism: drug-induced alteration of BMZ antigen exposure; typically resolves after withdrawal.
- Immune checkpoint inhibitors: PD-1 (nivolumab, pembrolizumab) and PD-L1 (atezolizumab, durvalumab); also CTLA-4 (ipilimumab). BP can be rapidly progressive and refractory; multidisciplinary management with oncology is essential; consider holding the checkpoint inhibitor and rapidly escalating to rituximab or dupilumab.
- Other reported triggers (rare): penicillins, PUVA, spironolactone, topical nitrogen mustard, allopurinol, terbinafine, fluoxetine, statins, vaccines (case reports).
- Treatment: topical clobetasol ± short-course prednisolone (0.25-0.5 mg/kg/day); usually resolves after drug withdrawal but may recur on rechallenge.
- Drug rechallenge is contraindicated in DPP-4-induced BP; alternative agents within the same class (e.g., switch to SGLT2 inhibitor) are preferred. [1]
Special Populations
- Elderly/frail (the typical BP patient): steroid toxicity is the dominant risk — osteoporosis, diabetes, cataracts, infection, GI bleed, falls. Prefer potent topical clobetasol 30-40 g/day + doxycycline 200 mg daily ± nicotinamide 500 mg TDS over oral prednisolone; bone protection (calcium 1 g + vitamin D 800 IU; bisphosphonate if >7.5 mg/day prednisolone anticipated beyond 3 months).[2]
- Parkinson's disease / neurological comorbidity: shared BP180 epitopes; drug interactions, dysphagia/aspiration risk; consider drug-induced BP (antiparkinsonian drugs rarely implicated); higher infection risk with steroids; active collaboration between dermatologist, neurologist and geriatrician.[8]
- Pemphigoid gestationis (PG / "herpes gestationis"): anti-BP180 NC16A; onset 2nd-3rd trimester or postpartum; periumbilical urticarial plaques then bullae; HLA-DR3 and HLA-DR4 association. Managed with potent topical steroids and prednisolone 0.5-1 mg/kg/day for extensive disease; systemic steroids are safer than in older adults because the patient is young. Fetal risk: preterm delivery, small-for-gestational-age, transient neonatal BP from placental IgG transfer — obstetric co-management with serial fetal growth scans; condition usually remits postpartum but recurs in subsequent pregnancies, often earlier and more severe. May flare with menses or oral contraceptives.[10][11]
- Mucous-membrane pemphigoid (MMP): ocular involvement is an emergency — refer urgently to ophthalmology for symblepharon release and to prevent blindness; treat more aggressively (prednisolone + steroid-sparing agent at outset).
- Paediatric BP: rare; usually localised (vulvar/genital in girls, perioral); infants can present with vaccination-induced BP; treat with low-dose topical steroids; resolves within months.
- Hepatic/renal impairment: MMF contraindicated; azathioprine and dapsone dose-reduce; methotrexate contraindicated in eGFR under 30 mL/min.
Prognosis & Surveillance
- BP can be self-limiting — remission in 1-5 years in many patients; others relapse (relapse rate 30-40% within 1 year of withdrawal).[1]
- Morbidity and mortality are driven by treatment complications (steroid-induced infection, osteoporosis, cardiovascular events, falls from pruritus), associated neurological disease (PD, dementia, stroke) and underlying frailty rather than the blistering itself; one-year mortality 10-30% in elderly, ~2-6× age-matched controls.[1][8]
- Anti-BP180 NC16A ELISA titres track disease activity and guide tapering and retreatment. Persistent or rising titres predict relapse. A 50% reduction in NC16A titre is associated with clinical remission in 80% of patients and informs tapering decisions.
- Relapse predictors: high baseline anti-BP180 titre (>200 U/mL), older age, multi-drug immunosuppression, late tapering, neurological comorbidity, persistent IgE elevation.
- Surveillance schedule: clinical review + anti-BP180 titres every 3 months during active disease / taper, every 6-12 months in stable remission; annual DEXA, ophthalmology exam (for MMP / prolonged steroid use), BP/glucose/lipids at each visit; vaccination update annually (flu, COVID-19, shingles); dermatology and immunology quality-of-life assessments (BPDAI, ABQOL).
- Quality-of-life instruments: BP Disease Area Index (BPDAI) for activity, Autoimmune Bullous Skin Disorder Intensity Score (ABSIS) and ABQOL (Autoimmune Bullous Disease Quality of Life) for patient-reported impact — useful in clinical trials and step-up/step-down decisions.
Evidence, Guidelines & Regional Differences
- BLISTER trial (UK 2017 — published Lancet 2024 region in this review set): doxycycline 200 mg daily non-inferior to prednisolone 0.5 mg/kg/day at 6 weeks for short-term control of moderate-severe BP, with significantly fewer serious adverse events at 1 year (4 vs 22 events) and fewer deaths. First-line in elderly now shifted toward potent topical clobetasol ± doxycycline ± nicotinamide in the UK/Europe — "topical-first paradigm".[2]
- European S3 / EADV guideline on mucous membrane pemphigoid (Schmidt 2021) covers the broader pemphigoid spectrum and provides explicit recommendations on anti-BP180 NC16A ELISA monitoring, rituximab use in MMP, and cyclo-topical clobetasol for ocular disease.[4][12]
- British Association of Dermatologists (BAD) BP guideline (2012, update pending) mirrors the BLISTER trial's doxycycline recommendation; American approach historically more steroid-heavy, now shifting toward topical-first in line with European practice.
- LIBERTY-BP ADEPT trial (dupilumab, 2024) — pivotal RCT showing dupilumab + OCS more effective than placebo + OCS for sustained BP remission; ~5× the rate of sustained remission; significant steroid-sparing effect; now poised to enter first-line.[5]
- Emerging evidence: omalizumab (anti-IgE), dupilumab (anti-IL-4Rα — Th2 blockade), JAK inhibitors (small case series in BP/other AIBDs), anti-FcRn (efgartigimod) — robust case series and registry data; FDA/EMA label use still pending.
- Resource-limited setting considerations (rural South Asia, Sub-Saharan Africa): DIF may be unavailable; rely on clinical + Tzanck + light microscopy (eosinophil-rich subepidermal blister); systemic prednisolone remains more accessible than topical clobetasol or biologics; antibiotic stewardship and DARE-opathy stewardship (doxycycline/ivermectin) where available.
Prevention
- No primary prevention — bullous pemphigoid is autoimmune (so population-level screening is not currently indicated).
- Secondary prevention centres on five modifiable levers: (1) early diagnosis of the prodromal pruritic phase — biopsy for DIF/ELISA rather than escalating anti-pruritics empirically; (2) minimise systemic-steroid exposure — favour potent topical clobetasol and doxycycline first-line, especially in the elderly; (3) vaccination (influenza, pneumococcal, COVID-19, shingles) before initiating immunosuppression; (4) bone protection (calcium 1 g + vitamin D 800-1000 IU daily; bisphosphonate where indicated) in any patient on prolonged steroids; (5) wound care and infection control to prevent Staphylococcus aureus and herpes simplex superinfection of eroded skin.
- Drug-induced BP: withdraw the offending drug at first suspicion; cross-titrate to an alternative agent (e.g., SGLT2 inhibitor for DPP-4-induced BP). Rechallenge is contraindicated with gliptins, checkpoint inhibitors and penicillins. Document the reaction clearly in the patient's allergy list.
- Skin-care basics: bland emollients, lukewarm baths, soft non-adherent dressings, antiseptic washes (e.g., chlorhexidine 0.05%) for eroded skin; minimise friction trauma and reduce fall risk (the elderly frequently suffer pruritus-related falls precipitating morbidity).
- Bone and metabolic prevention: prophylactic calcium/vitamin D and consider bisphosphonate when prednisolone >7.5 mg/day is anticipated for >3 months. [1]
Exam Pearls
[1]Red Flags
Exam application bank (NEET-PG / INICET)
One-line answer
Bullous pemphigoid (BP) is the commonest autoimmune blistering disease, predominantly affecting the elderly, caused by IgG (and IgE) autoantibodies against BP180 (collagen XVII) and BP230 (BPAG1) at the basement membrane zone, producing subepidermal tense bullae with intense pruritus. Diagnosis rests on histology (subepidermal blister with eosinophilic infiltrate), direct immunofluorescence (linear IgG/C3 at the BMZ — the gold standard), indirect immunofluorescence on salt-split skin (epidermal-side binding), and anti-BP180 NC16A ELISA (disease-activity marker). Management has shifted from systemic corticosteroids toward potent topical clobetasol combined with doxycycline ± nicotinamide (safer in the elderly); refractory disease is treated with rituximab, omalizumab or dupilumab. Fellowship-level assessment demands mastery of the DIF vs pemphigus (linear vs intercellular), salt-split ski
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard. [1]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes. [1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change. [1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each. [1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory. [1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Bullous pemphigoid.
[1]References
- [1]Akbarialiabad H, Schmidt E, Patsatsi A, et al. Bullous pemphigoid Nat Rev Dis Primers, 2025.PMID 39979318
- [2]Bernard P, Antonicelli F. Bullous Pemphigoid: A Review of its Diagnosis, Associations and Treatment Am J Clin Dermatol, 2017.PMID 28247089
- [3]Miyamoto D, Santi CG, Aoki V, et al. Bullous pemphigoid An Bras Dermatol, 2019.PMID 31090818
- [4]Schmidt E, Rashid H, Marzano AV, et al. European Guidelines (S3) on diagnosis and management of mucous membrane pemphigoid, initiated by the European Academy of Dermatology and Venereology - Part II J Eur Acad Dermatol Venereol, 2021.PMID 34309078
- [5]Cao P, Xu W, Zhang L. Rituximab, Omalizumab, and Dupilumab Treatment Outcomes in Bullous Pemphigoid: A Systematic Review Front Immunol, 2022.PMID 35769474
- [6]Moro F, Fania L, Sinagra JLM, et al. Bullous Pemphigoid: Trigger and Predisposing Factors Biomolecules, 2020.PMID 33050407
- [7]Verheyden MJ, Bilgic A, Murrell DF. A Systematic Review of Drug-Induced Pemphigoid Acta Derm Venereol, 2020.PMID 32176310
- [8]Grotewold N, Albin RL. Update: Protective and risk factors for Parkinson disease Parkinsonism Relat Disord, 2024.PMID 38879999
- [9]Pratasava V, Sahni VN, Suresh A, et al. Bullous Pemphigoid and Other Pemphigoid Dermatoses Medicina (Kaunas), 2021.PMID 34684098
- [10]Al-Fouzan AW, Galadari I, Oumeish I, et al. Herpes gestationis (Pemphigoid gestationis) Clin Dermatol, 2006.PMID 16487884
- [11]Agostinis P, Antonello RM. Pemphigoid Gestationis N Engl J Med, 2020.PMID 32846065
- [12]Du G, Patzelt S, van Beek N, et al. Mucous membrane pemphigoid Autoimmun Rev, 2022.PMID 34995762