Dermatology · Medicine
Epidermolysis bullosa acquisita
Also known as Epidermolysis bullosa acquisita (EBA) · EBA · Acquired epidermolysis bullosa · Pemphigoid gestationis-like EBA
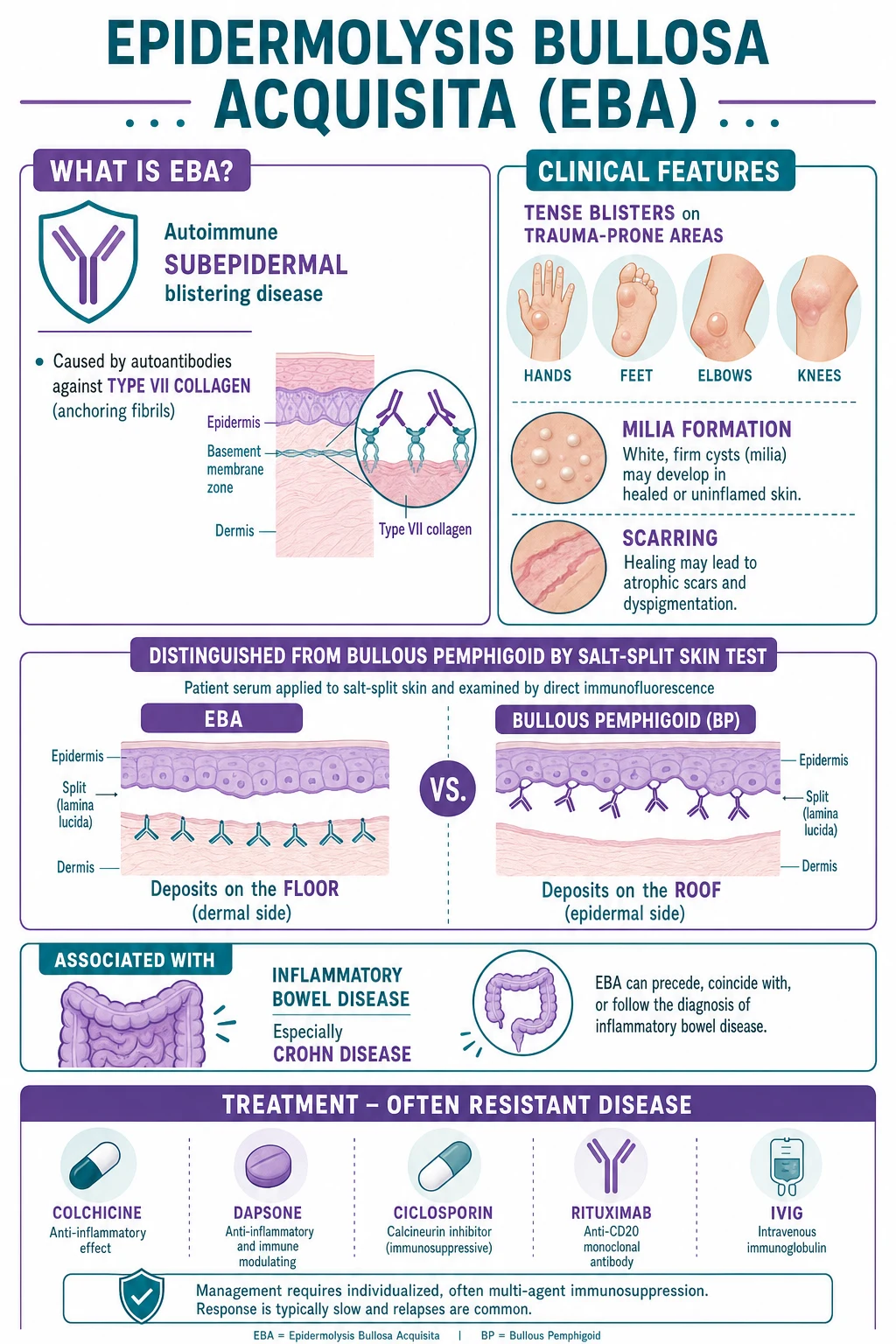
Epidermolysis bullosa acquisita (EBA) is an acquired autoimmune subepidermal blistering disease caused by IgG autoantibodies against type VII collagen (the major structural protein of anchoring fibrils at the dermoepidermal junction). It produces trauma-induced tense bullae on extensor surfaces that heal with milia and scarring, distinguishing it from bullous pemphigoid. The salt-split skin test is the key discriminator: EBA labels the FLOOR (dermal side) while bullous pemphigoid labels the ROOF (epidermal side). The classical variant mimics porphyria cutanea tarda; the mucous membrane variant mimics mucous membrane pemphigoid. Strong association with inflammatory bowel disease (Crohn's). Notoriously treatment-resistant.
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags
Overview & Definition
Epidermolysis bullosa acquisita (EBA) is a chronic, acquired, autoimmune subepidermal blistering disease caused by immunoglobulin G (IgG) autoantibodies directed against type VII collagen, the major structural protein of the anchoring fibrils that secure the lamina densa of the basement membrane zone to the underlying papillary dermis.[1][2] Because type VII collagen is the structural keystone of dermoepidermal adhesion, its autoimmune destruction produces a split deep in the basement membrane zone — below the lamina densa — with a clinical signature of trauma-induced blistering, milia, and scarring that distinguishes EBA from the more common bullous pemphigoid.[1]
The name "acquisita" (Latin: acquired) is deliberate and diagnostic. EBA is the acquired immunological twin of inherited dystrophic epidermolysis bullosa, in which germline COL7A1 mutations produce defective or absent type VII collagen.[2] The two share the same structural target and the same clinical consequence (fragile skin, scarring, milia, nail dystrophy), but EBA arises in adults with no family history through humoral autoimmunity rather than through inherited gene defects. This parallel is the conceptual hinge of the disease and the reason the salt-split skin test is so critical: it localises the autoimmune target to the deep basement membrane and separates EBA from every other pemphigoid.
Classification
EBA is heterogeneous. The same autoantibody produces four recognisable clinical variants, and the International Bullous Diseases Group (IBDG) further stratifies cases by diagnostic certainty into definite, probable, and possible EBA.[3]
Classical (mechanobullous)
- Trauma-induced tense bullae on extensor surfaces (hands, feet, elbows, knees)
- Heals with milia and scarring
- Nail dystrophy, scarring alopecia
- Mimics inherited dystrophic EB and porphyria cutanea tarda
- Most characteristic variant
Inflammatory (BP-like)
- Widespread tense bullae on erythematous, urticarial base
- Pruritus common
- Little or no scarring
- Histology: eosinophil-rich infiltrate
- Easily misdiagnosed as bullous pemphigoid
Mucous membrane (MMP-like)
- Predominant oral, oesophageal, laryngeal, genital, ocular involvement
- Heals with scarring and strictures
- Mimics mucous membrane pemphigoid
- Highest morbidity (blindness, airway stenosis)
- Requires aggressive combination therapy
IgA-EBA
- Linear IgA deposition on DIF
- Overlaps with linear IgA bullous dermatosis
- Often dapsone-responsive
- Rare variant
Epidemiology & Risk Factors
EBA is rare. The estimated incidence is 0.2 new cases per million population per year — roughly one-hundredth the incidence of bullous pemphigoid — and prevalence figures are correspondingly low.[1][2] The condition shows a bimodal age distribution: a large adult peak in the fourth to sixth decade and a smaller childhood form (discussed separately).[9] There is no consistent sex predilection and no recognised racial or ethnic predisposition.[1]
The most important disease associations cluster around autoimmunity and inflammation. The single strongest link is with inflammatory bowel disease (IBD), particularly Crohn's disease; both conditions share type VII collagen as an autoimmune target, and the colonic epithelium expresses type VII collagen at its basement membrane, providing a plausible mechanistic bridge between mucosal inflammation and cutaneous autoimmunity.[10] Other well-described associations include systemic lupus erythematosus, rheumatoid arthritis, autoimmune thyroiditis, diabetes mellitus, and haematological malignancy (lymphoma in particular).[1][2]
Crohn's disease (strongest)
- Shared type VII collagen autoimmunity
- Colonic basement membrane expresses type VII collagen
- May precede, accompany, or follow IBD diagnosis
- Treat the blistering independently
Other autoimmune
- Systemic lupus erythematosus
- Rheumatoid arthritis
- Autoimmune thyroiditis
- Diabetes mellitus
Malignancy
- Haematological: lymphoma, leukaemia
- Rare solid tumours
- Consider paraneoplastic workup in refractory disease
Drug-induced
- Penicillamine (classic trigger)
- Captopril
- Vancomycin
- Resolves on drug cessation
Drug-induced EBA deserves separate mention. D-penicillamine is the classic trigger — the same drug that causes drug-induced pemphigus — and captopril has also been implicated, presumably through sulfhydryl-group interference with basement membrane proteins.[12] A careful drug history is part of the workup in any new case; cessation of the culprit agent may bring resolution without long-term immunosuppression.
Pathophysiology
Understanding EBA begins at the dermoepidermal junction. Type VII collagen, encoded by COL7A1 and a 290 kDa homotrimer, is the exclusive component of anchoring fibrils — curvilinear structures that project from the underside of the lamina densa into the papillary dermis, where they entrap interstitial collagen fibrils and so rivet the epidermis to its underlying stroma.[6] The amino-terminal NC1 domain of type VII collagen carries the dominant EBA epitopes and is the target of the commercial ELISA.[5]
The autoimmune cascade proceeds in three linked steps. First, IgG autoantibodies (mostly IgG1 and IgG4 subclasses) bind the NC1 domain of type VII collagen on the anchoring fibrils.[6] Second, antibody binding activates complement — generating the potent neutrophil chemoattractant C5a, which recruits neutrophils to the basement membrane zone. Third, degranulating neutrophils release proteolytic enzymes, including neutrophil elastase and matrix metalloproteinase-9, that digest the anchoring fibrils themselves.[6] The resulting loss of mechanical adhesion between lamina densa and dermis produces a subepidermal split that, by electron microscopy, sits below the lamina densa — the anatomical signature of the disease.
The antibody subclass and the patient's Fc-receptor genotype together shape the clinical phenotype.[6] IgG1 fixes complement efficiently and drives the inflammatory picture, whereas IgG4 — which dominates in chronic disease — fixes complement poorly and tends to produce the mechanobullous phenotype through direct interference with the collagen-binding function of the anchoring fibril. Polymorphisms in activating Fc-gamma receptors (notably Fc-gamma-RIIIa on neutrophils and natural killer cells) modulate neutrophil recruitment and so tilt the disease toward or away from inflammation; the same biology explains why some patients respond to neutrophil-directed drugs (colchicine, dapsone) while others require B-cell depletion. Complement-independent pathways — direct steric hindrance of anchoring-fibril assembly, and antibody-dependent cellular cytotoxicity by Fc-receptor-bearing cells — account for blistering in biopsy specimens that show little or no inflammatory infiltrate.[6]
Two pathophysiological nuances explain the clinical variants. In the classical mechanobullous variant, antibody binding alone appears sufficient to disrupt adhesion: small conformational perturbations of the anchoring fibril impair its collagen-binding function even without heavy inflammatory infiltrate, so blisters appear on trauma-prone extensor surfaces where shearing forces exceed the weakened adhesion, and the deeper split heals with milia and scarring.[1] In the inflammatory variant, abundant complement activation and neutrophil recruitment dominate the histology, producing widespread tense blisters on an erythematous base that mimic bullous pemphigoid.[4] The split is always subepidermal but the clinical expression depends on the balance between mechanical (antibody-mediated) and inflammatory (complement- and neutrophil-mediated) injury.

The pathogenesis is not purely humoral. Type VII collagen-specific CD4+ T cells provide help to autoreactive B cells and sustain autoantibody production; this T-cell arm is the rationale for B-cell-depleting therapies such as rituximab (which removes CD20-positive B-cell precursors and so indirectly throttles the antigen-presenting and antibody-secreting compartment).[7] The disease has been faithfully reproduced in animal models by active immunisation with type VII collagen, confirming that the autoantibody is pathogenic, not merely a marker.[6]
Clinical Presentation
The clinical picture of EBA is shaped by which of the four variants dominates, and a single patient can shift between patterns over time. Recognising each variant is essential because the inflammatory and mucous membrane forms are easily mistaken for more common diseases, and the wrong diagnosis leads to ineffective treatment. [1]
Classical (mechanobullous) variant
The most characteristic presentation. Tense bullae and erosions arise on trauma-prone extensor surfaces — the dorsal hands, feet, elbows, knees, knuckles, and buttocks — at sites of minor friction or shearing.[1] Blisters heal slowly with milia (tiny firm white keratin cysts) and atrophic scarring, giving the skin a fragile, "torn" appearance reminiscent of inherited dystrophic epidermolysis bullosa. Nail dystrophy (onychogryphosis, pterygium, nail loss) and scarring alopecia of the scalp are common adjuncts. Pruritus is variable. The dorsal-hand distribution with milia and scarring can closely mimic porphyria cutanea tarda.[2]
Inflammatory (bullous pemphigoid-like) variant
Widespread large tense bullae arise on an erythematous, urticarial base across the trunk, flexures, and proximal limbs, often with prominent pruritus.[4] Milia and scarring are less prominent than in the classical variant because the split is more superficially inflamed and heals faster. Histology shows an eosinophil-rich infiltrate that further blurs the distinction from bullous pemphigoid — only the salt-split skin test reliably separates the two.
Mucous membrane (mucous membrane pemphigoid-like) variant
The most morbid form. Painful oral erosions and desquamative gingivitis dominate, but involvement extends to the oesophagus (strictures causing dysphagia and weight loss), the larynx (hoarseness, stridor, stenosis), the conjunctiva (symblepharon, synechiae, corneal scarring, blindness), and the anogenital mucosa (scarring, stenosis).[2] Healing is by scarring, so the damage is cumulative and often irreversible; this variant mimics mucous membrane pemphigoid and demands aggressive early combination immunosuppression to prevent permanent ocular and airway damage.
IgA-EBA and childhood EBA
A rare IgA-mediated variant overlaps with linear IgA bullous dermatosis and is often dapsone-responsive.[5] Childhood EBA typically presents with the inflammatory or IgA phenotype, may involve mucous membranes, and is distinguished from inherited dystrophic EB by adult-style onset in an otherwise well child with no family history and positive immunofluorescence.[9]
Differential Diagnosis
The differential of EBA is the entire family of subepidermal blistering diseases. The discriminators are clinical distribution, presence of scarring and milia, immunofluorescence pattern, and — above all — the salt-split skin localisation.[5]
Bullous pemphigoid
- Older patients (greater than 70 years typical)
- Widespread flexural tense bullae on erythematous base
- No scarring, no milia
- Antigen: BP180 and BP230 (hemidesmosome)
- DIF: linear IgG/C3 at BMZ
- Salt-split: ROOF (epidermal side)
- Steroid-responsive
Mucous membrane pemphigoid
- Mucosal-dominant, ocular and oral scarring
- Skin involvement limited
- Antigen: BP180 NC16A, beta-4 integrin, laminin 332
- Salt-split: ROOF pattern
- Requires dapsone + cyclophosphamide/rituximab
Linear IgA bullous dermatosis
- String-of-pearls annular tense bullae
- Drug-induced (vancomycin) in adults
- DIF: linear IgA at BMZ
- Salt-split: floor pattern (overlaps EBA)
- Dapsone-responsive
Porphyria cutanea tarda
- Sun-exposed dorsal hands, face
- Milia, scarring, hypertrichosis, hyperpigmentation
- Skin fragility and bullae
- Urine/plasma porphyrins elevated
- Normal immunofluorescence
- UROD deficiency; trigger: alcohol, hepatitis C, iron, oestrogens
Dermatitis herpetiformis
- Intensely pruritic extensor vesicles (knees, elbows, buttocks, scalp)
- DIF: granular IgA at dermal papillae
- Gluten-sensitive enteropathy (coeliac)
- Dapsone-responsive
Pemphigus vulgaris
- Flaccid suprabasal blisters, oral erosions
- Nikolsky positive
- DIF: intercellular (fishnet) IgG/C3
- Antigen: desmoglein 3 and 1
- Salt-split: not applicable (intraepidermal)
Inherited dystrophic EB
- Onset at birth or infancy
- Positive family history (autosomal recessive or dominant)
- COL7A1 germline mutation
- Same type VII collagen target but genetic, not autoimmune
The classical EBA variant is most often confused with porphyria cutanea tarda (dorsal hands, milia, scarring) — the discriminator is sun-exposed distribution plus elevated urine porphyrins plus normal immunofluorescence in PCT.[2] The inflammatory variant is confused with bullous pemphigoid — only the salt-split test and ELISA separate them reliably. The mucous membrane variant is confused with mucous membrane pemphigoid — but EBA tends to have more prominent skin involvement and a floor pattern on salt-split, whereas classical MMP has a roof pattern.[5]
Two further differentials deserve explicit treatment because they are classic examination stems. Pseudoporphyria (also called bullous dermatosis of haemodialysis) mimics porphyria cutanea tarda and EBA on the dorsal hands with fragility, tense bullae, milia, and scarring in a sun-exposed distribution, but it has normal urine, plasma, and faecal porphyrins.[2] It is most often drug-induced (non-steroidal anti-inflammatory drugs, especially naproxen and furosemide) or arises in patients on haemodialysis or peritoneal dialysis; immunofluorescence is normal (no linear basement-membrane deposition), which separates it cleanly from EBA. Inherited dystrophic epidermolysis bullosa shares the type VII collagen target and the mechanobullous phenotype — scarring, milia, nail dystrophy — but begins at or soon after birth, carries a positive family history (autosomal recessive severe or autosomal dominant forms), and results from germline COL7A1 mutations rather than autoimmunity.[2] The distinction is made on history (age of onset, family history), immunofluorescence (negative in inherited EB, linear IgG/C3 in EBA), and ELISA (no circulating anti-type VII collagen antibodies in the inherited form).
Clinical & Bedside Assessment
A focused examination determines the variant, screens for mucosal damage, and looks for associated systemic disease. Examine the distribution of blisters: extensor trauma-prone sites suggest the classical variant; widespread flexural blisters suggest the inflammatory variant; mucosal-dominant disease suggests the mucous membrane variant.[1] Note the morphology of individual lesions (tense bulla, erosion, crust), the presence of milia and scarring at healed sites (the hallmarks of EBA), and the nails (dystrophy, loss) and scalp (scarring alopecia).
The mucosal examination is non-negotiable. Inspect the oral cavity (erosions, desquamative gingivitis), ask about dysphagia and hoarseness (oesophageal and laryngeal involvement), examine the conjunctivae (symblepharon, sicca, keratopathy), and inspect the anogenital mucosa (scarring, stenosis).[2] Any ocular involvement mandates urgent ophthalmology referral because scarring progression can be rapid and irreversible.
Screen for associated disease in the history. Ask about abdominal pain, diarrhoea, rectal bleeding, and weight loss (Crohn's disease); arthralgia, morning stiffness, and malar rash (lupus and rheumatoid arthritis); and constitutional symptoms that might suggest lymphoma or other malignancy.[10] Take a thorough drug history, specifically enquiring about penicillamine, captopril, and vancomycin.[12] Confirm that blistering began in adulthood with no family history, which distinguishes EBA from inherited dystrophic EB.
The bedside reasoning runs along three axes. First, the tempo and distribution of blistering: trauma-induced extensor bullae with milia and scarring point to the classical mechanobullous variant; widespread flexural bullae on an urticarial base point to the inflammatory variant; mucosal-dominant scarring disease points to the mucous membrane variant. Second, the immunofluorescence pattern on perilesional biopsy: linear IgG and C3 along the basement membrane zone confirm a pemphigoid but cannot distinguish EBA from bullous pemphigoid or mucous membrane pemphigoid — that distinction rests on the salt-split test. Third, the salt-split localisation of patient serum: a floor (dermal) pattern is EBA; a roof (epidermal) pattern is bullous pemphigoid or mucous membrane pemphigoid.[3][5] Where the clinical picture and salt-split pattern are discordant, request the anti-type VII collagen NC1 ELISA and, if necessary, immunoelectron microscopy before committing to a diagnosis — because the treatment of EBA differs fundamentally from that of bullous pemphigoid, and mislabelling a steroid-resistant "BP" wastes months of ineffective corticosteroid therapy.
Investigations
EBA cannot be diagnosed on histology alone — the subepidermal blister is shared with several pemphigoids. Diagnosis requires a panel of investigations that together localise the autoimmune target to type VII collagen below the lamina densa.[3][5]

Histology (H&E)
A punch biopsy of the edge of a fresh blister (including adjacent perilesional skin) shows a subepidermal blister with a neutrophil-predominant or mixed inflammatory infiltrate; eosinophils may predominate in the inflammatory variant, mimicking BP.[2] Chronic lesions show fibrosis in the papillary dermis, consistent with the scarring phenotype. Histology alone cannot distinguish EBA from BP or linear IgA disease.
Direct immunofluorescence (DIF)
A perilesional biopsy incubated with fluorescein-tagged anti-human IgG, IgA, IgM, and C3 shows linear deposition of IgG and C3 along the basement membrane zone.[5] This pattern is identical to bullous pemphigoid and to mucous membrane pemphigoid, so DIF alone is never diagnostic of EBA — it only establishes that the disease is a basement-membrane pemphigoid and prompts the salt-split test.
Salt-split skin indirect immunofluorescence (IIF)
This is the critical discriminator. The patient's serum is incubated on normal human skin that has been split through the lamina lucida by incubation in 1 mol/L sodium chloride.[5] The artificial split places BP180 and BP230 (hemidesmosome antigens) on the roof and type VII collagen (anchoring fibrils) on the floor.
- EBA: IgG labels the FLOOR (dermal side).[1][3]
- Bullous pemphigoid: IgG labels the ROOF (epidermal side).[5]
The reason is anatomical: type VII collagen sits below the lamina densa and stays with the dermal floor; BP180 and BP230 sit in the hemidesmosome above the lamina lucida and stay with the epidermal roof. This single test converts "basement-membrane pemphigoid" into a specific diagnosis. [1]
A note on substrate and sensitivity is worth stating at the viva. Conventional IIF on monkey oesophagus or guinea-pig oesophagus detects circulating basement-membrane antibodies in roughly half of EBA sera — a lower sensitivity than for bullous pemphigoid — and cannot localise the antigen to floor or roof; it is therefore being superseded by salt-split human skin as the first-line IIF substrate.[5] The salt-split technique itself has a sensitivity of about 80 per cent and a specificity above 90 per cent for EBA when the floor pattern is observed, but it requires both a control serum (a known BP patient giving a roof pattern) and adequate split quality (the NaCl incubation must cleave cleanly through the lamina lucida).[3] A floor pattern on salt-split skin is not unique to EBA — linear IgA bullous dermatosis, anti-laminin gamma-1 pemphigoid, and anti-p200 pemphigoid can also label the floor — so a floor pattern mandates confirmation by the anti-type VII collagen NC1 ELISA before EBA is declared definite.[3][5]
Anti-type VII collagen ELISA
The commercially available ELISA uses the NC1 domain of type VII collagen as substrate. A positive result confirms circulating anti-type VII collagen antibodies and is both sensitive and specific for EBA.[5] Antibody titres correlate imperfectly with disease activity but are useful for diagnosis and for monitoring response to therapy. Anti-BP180 and anti-BP230 ELISAs should be requested in parallel to exclude bullous pemphigoid.
Immunoelectron microscopy (historical gold standard)
The definitive ultrastructural localisation: immune deposits lie below the lamina densa in the upper dermis, within the anchoring fibril zone.[1] Immunoelectron microscopy is technically demanding, not widely available, and has been substantially supplanted by salt-split IIF plus ELISA in routine practice, but it remains the arbiter in difficult cases and is written into the IBDG 2018 criteria.[3]
Additional workup
Request full blood count, urea and electrolytes, liver function tests, glucose, and inflammatory markers as a baseline and to screen for associated disease (anaemia of chronic inflammation, IBD, diabetes). Screen for IBD with a careful history, faecal calprotectin, and colonoscopy where indicated. Consider a malignancy screen (lymphoma, solid organ) in refractory disease or where paraneoplastic triggers are suspected. Check glucose-6-phosphate dehydrogenase (G6PD) activity before starting dapsone, and thiopurine methyltransferase (TPMT) before azathioprine.[1]
Management — Resuscitation
EBA is rarely a true dermatological emergency, but two scenarios demand urgent action. The first is mucosal airway compromise: laryngeal involvement may progress to stridor and airway stenosis, requiring urgent ear-nose-throat assessment and, rarely, airway protection.[2] The second is extensive skin loss with secondary infection or sepsis, particularly in the inflammatory variant or in immunosuppressed patients, where the principles of fluid balance, wound care, and antimicrobial therapy mirror those of severe burns or toxic epidermal necrolysis.
In every patient, the immediate priorities are gentle wound care with non-adherent dressings (silicone or paraffin-based), pain control (erosions are painful), infection surveillance and treatment (swabs, topical antiseptics, systemic antibiotics for cellulitis or sepsis), and nutritional support — particularly when oesophageal strictures impair intake.[1] An acute oesophageal stricture causing dysphagia and weight loss needs nutritional supplementation (oral supplements, nasogastric, or, rarely, parenteral feeding) and urgent gastroenterology referral for endoscopic dilatation.[2] Any ocular involvement mandates same-day ophthalmology referral, topical lubricants and steroids, and division of forming synechiae to prevent irreversible symblepharon and corneal blindness.
Management — Definitive & Stepwise
EBA is notoriously difficult to treat and responds poorly to the corticosteroid regimens that control bullous pemphigoid.[1][2] The reasons are partly mechanistic — type VII collagen sits in a mechanically loaded, deep basement-membrane location where antibody deposition is continuously replenished — and partly immunological: the humoral and cellular autoimmune response is sustained and aggressive. Treatment therefore tends to combine anti-neutrophilic agents (colchicine, dapsone) with disease-modifying immunosuppression (steroids, azathioprine, mycophenolate, ciclosporin), escalating to biologic and antibody-removal strategies in refractory disease.[4]

Stepwise treatment of EBA
First line — anti-neutrophilic monotherapy: colchicine 0.5 to 1.5 mg per day (inhibits neutrophil chemotaxis; monitor FBC and gastrointestinal tolerability) and/or dapsone 50 to 150 mg per day (check G6PD first; monitor for haemolysis and methaemoglobinaemia). Effective for the classical and IgA variants.
Inflammatory flare — corticosteroids: prednisolone 0.5 to 1 mg/kg per day for rapid control, almost always combined with a steroid-sparing agent because monotherapy is insufficient and toxicity accumulates.
Steroid-sparing immunosuppression: azathioprine 1 to 3 mg/kg per day (check TPMT genotype first), mycophenolate mofetil 2 g per day, methotrexate 10 to 25 mg weekly, or ciclosporin 3 to 5 mg/kg per day. Used as maintenance and as steroid-sparing agents.
Refractory disease — biologics and IVIG: rituximab 375 mg/m2 weekly for four weeks (anti-CD20 B-cell depletion; supported by the 2024 systematic review) with or without IVIG 2 g/kg per cycle every 4 weeks.
Severe, rapid, or antibody-driven disease — antibody removal: immunoadsorption on protein A or tryptophan columns, or plasmapheresis, to deplete circulating anti-type VII collagen antibodies (usually combined with rituximab to prevent rebound).
Supportive care at every stage: gentle non-adherent dressings, trauma avoidance, wound care, nutritional support, oesophageal dilatation for strictures, ocular lubricants and topical steroids, physical therapy to prevent contractures.
Anti-neutrophilic agents (first line)
Colchicine at 0.5 to 1.5 mg per day is widely used first-line for the classical and IgA variants; it inhibits neutrophil chemotaxis and microtubule-mediated degranulation, targeting the inflammatory limb of the pathogenic cascade.[1] Monitor for gastrointestinal intolerance (diarrhoea, nausea) and cytopenia. Dapsone at 50 to 150 mg per day is the other key anti-neutrophilic agent; the mandatory G6PD check precedes the first dose because G6PD-deficient patients develop severe haemolysis. During therapy, monitor full blood count, methaemoglobin, and liver function for haemolysis, methaemoglobinaemia, and the dapsone hypersensitivity syndrome.[4] The IgA variant of EBA is often dapsone-responsive, mirroring linear IgA bullous dermatosis.
Systemic corticosteroids
Prednisolone at 0.5 to 1 mg/kg per day is used for inflammatory flares and for the mucous membrane variant, but is rarely sufficient alone.[1] EBA's resistance to corticosteroids is the practical expression of its deep, mechanically loaded autoimmune target and its sustained T-cell-driven antibody production. Corticosteroids therefore serve as a bridge to a steroid-sparing agent rather than as definitive monotherapy. Pulse methylprednisolone is reserved for severe inflammatory or mucosal disease.
Steroid-sparing immunosuppressants
Azathioprine (1 to 3 mg/kg per day), mycophenolate mofetil (2 g per day), methotrexate (10 to 25 mg weekly), and ciclosporin (3 to 5 mg/kg per day) are the workhorses of maintenance therapy.[2] Azathioprine requires a pre-treatment TPMT assay to identify slow metabolisers at risk of severe myelosuppression; ciclosporin demands monitoring of renal function and blood pressure; methotrexate needs folate supplementation and monitoring of full blood count and liver function. These agents take weeks to months to work and are combined with corticosteroids during induction.
Intravenous immunoglobulin (IVIG)
IVIG at 2 g/kg per cycle, given over two to five days every four weeks, is used in refractory disease or in patients intolerant of conventional immunosuppression.[8] The 2023 systematic review of IVIG in autoimmune bullous disease found a substantial response rate in EBA, though the evidence is limited by the rarity of the disease and the absence of randomised trials.[8] Limitations include cost, intravenous access, infusion reactions, and a theoretical risk of thromboembolism.
Rituximab
Rituximab (anti-CD20) at 375 mg/m2 weekly for four weeks (lymphoma schedule) or 1 g on days 1 and 15 (rheumatology schedule) depletes CD20-positive B cells and indirectly throttles autoantibody production by removing the antigen-presenting and precursor compartment that feeds the plasma-cell pool.[7] Two independent systematic reviews (Kianfar 2024 and Xenopoulou 2025) report a high response rate in refractory EBA, including complete remission in a majority of treated patients, with an acceptable safety profile dominated by infusion reactions and opportunistic infection.[7] Rituximab is now the biologic of choice for severe, refractory, or rapidly progressive disease.
Immunoadsorption and plasmapheresis
For severe, antibody-driven disease — particularly the rapidly progressive mucous membrane variant — immunoadsorption on protein A or tryptophan columns (or plasmapheresis) physically removes circulating anti-type VII collagen antibodies.[4] Because the antibody rebounds within weeks, antibody-removal strategies are always combined with rituximab or cyclophosphamide to suppress resynthesis.
Supportive care
At every stage, supportive care is integral. Non-adherent silicone or paraffin dressings protect fragile skin; trauma and friction are minimised; secondary infection is treated promptly; nutritional support (oral supplements, enteral feeding) addresses weight loss from oesophageal strictures; endoscopic dilatation relieves oesophageal strictures; ocular lubricants and topical steroids protect the conjunctiva; and physical therapy prevents contractures in extensively scarred limbs.[1] Emerging therapies under investigation include type VII collagen-specific T-cell therapy, Janus kinase inhibitors (targeting the intracellular signalling downstream of type VII collagen-reactive T cells), and low-dose interleukin-2 to expand regulatory T cells.[4]
Therapeutic monitoring in EBA is demanding and runs in parallel with treatment. The clinical target is cessation of new blister formation, with healing of existing erosions and, in mucosal disease, arrest of scarring progression; a fall in the anti-type VII collagen ELISA titre provides objective evidence of response but lags behind the clinical picture by several weeks. Before azathioprine, assay thiopurine methyltransferase (TPMT) activity to avoid catastrophic myelosuppression in slow metabolisers; before dapsone, screen glucose-6-phosphate dehydrogenase (G6PD) and recheck full blood count at weeks 1, 2, 4, and 12 for haemolysis and methaemoglobinaemia; on ciclosporin, monitor renal function and blood pressure monthly; on methotrexate, supplement folic acid and check full blood count and liver function every two to three months.[1][4] For rituximab, screen for hepatitis B and C and tuberculosis beforehand (reactivation risk), give the first infusion slowly with pre-medication (paracetamol, antihistamine, hydrocortisone) to blunt infusion reactions, and track CD19 and CD20 B-cell counts and immunoglobulin levels — peripheral B cells are typically depleted within two weeks and remain undetectable for six to twelve months, during which live vaccines are contraindicated and inactivated vaccines (influenza, pneumococcus, COVID-19) should ideally be timed before depletion or after recovery.[7] Bone-protection (calcium, vitamin D, bisphosphonate where indicated), glycaemic and blood-pressure surveillance, and opportunistic-infection vigilance (oral candidiasis, herpes zoster, Pneumocystis prophylaxis on dual immunosuppression) complete the safety net.
Specific Subtypes & Scenarios
The treatment approach is tailored to the variant. The classical mechanobullous variant is managed with colchicine first-line, supplemented by dapsone or a steroid-sparing agent, with meticulous trauma avoidance and wound care; corticosteroids have a limited role because the inflammatory component is modest.[1] The inflammatory BP-like variant is managed much like bullous pemphigoid — corticosteroids for induction combined with a steroid-sparing agent — but is more refractory and more often escalates to rituximab.[4] The mucous membrane (MMP-like) variant demands the most aggressive approach: early combination immunosuppression (typically corticosteroid plus cyclophosphamide or rituximab) to prevent irreversible scarring, with parallel ophthalmology, gastroenterology, and ENT input, because the morbidity of untreated disease (blindness, stricture, airway stenosis) vastly exceeds the toxicity of aggressive therapy.[2]
Childhood EBA typically presents with the inflammatory or IgA phenotype. It is distinguished from inherited dystrophic EB by adult-style onset in an otherwise well child with no family history and positive immunofluorescence, and from linear IgA bullous dermatosis by the salt-split floor pattern and anti-type VII collagen ELISA.[9] Dapsone is often first-line in childhood EBA, with weight-based dosing and the same G6PD precautions as in adults. Drug-induced EBA (penicillamine, captopril, vancomycin) typically resolves on drug cessation with a short course of corticosteroids for bridging; a careful drug history is essential in any new case.[12] EBA associated with Crohn's disease is managed concurrently with the IBD; the shared type VII collagen target means that effective IBD control does not reliably control the blistering, which must be treated on its own merits.[10]
Complications & Pitfalls
The complications of EBA reflect both the disease and its treatment. Cutaneous complications — scarring, milia, nail dystrophy, nail loss, and scarring alopecia — are the visible legacy of the deep basement-membrane split and the reason the classical variant mimics inherited dystrophic EB.[1] Extensively scarred limbs may develop contractures that impair function. Mucosal complications are more dangerous: oral erosions impair nutrition and speech, oesophageal strictures cause dysphagia and weight loss, laryngeal stenosis threatens the airway, genital scarring impairs urinary and sexual function, and ocular scarring (symblepharon, synechiae, corneal opacity) leads to blindness if unchecked.[2] Systemic complications include secondary infection and sepsis (the leading acute threat in extensive disease), malnutrition from oesophageal strictures, and anaemia of chronic disease.
Cutaneous
- Scarring and atrophy
- Milia
- Nail dystrophy and nail loss
- Scarring alopecia
- Contractures
Mucosal
- Oral erosions and dysphagia
- Oesophageal strictures
- Laryngeal stenosis (airway)
- Genital scarring and stenosis
- Ocular symblepharon and blindness
Systemic
- Secondary infection and sepsis
- Malnutrition and weight loss
- Anaemia of chronic disease
- Reduced quality of life and depression
Treatment-related
- Corticosteroid toxicity (diabetes, osteoporosis, infection)
- Immunosuppression (opportunistic infection, malignancy)
- Dapsone haemolysis and methaemoglobinaemia
- Colchicine gastrointestinal toxicity
- Rituximab infusion reactions and late-onset neutropenia
- IVIG thromboembolism
The principal pitfall is misdiagnosis of EBA as bullous pemphigoid.[5] The two share a subepidermal blister and linear IgG/C3 on DIF; without a salt-split skin test, the patient is labelled as "treatment-resistant bullous pemphigoid" and exposed to escalating and ineffective corticosteroid regimens. The salt-split skin test, anti-type VII collagen ELISA, and immunoelectron microscopy are the safeguards. A second pitfall is missed ocular involvement: a patient may have minimal cutaneous disease but rapidly progressive conjunctival scarring, and the window for preventing symblepharon and blindness is narrow — every patient with mucous membrane EBA needs routine ophthalmology surveillance.[2] A third pitfall is under-treating the mucous membrane variant: conservative single-agent therapy allows irreversible scarring to accumulate, and the morbidity of untreated disease far outweighs the toxicity of aggressive combination immunosuppression.
Prognosis & Disposition
EBA is a chronic, relapsing, often treatment-resistant disease that may persist for years or decades. The prognosis is shaped by the clinical variant, the extent of mucosal involvement, the presence of associated disease, and the response to therapy.[1] The classical mechanobullous variant has the best skin prognosis but the most visible scarring; the inflammatory variant tends to relapse and remit; the mucous membrane variant has the worst prognosis, driven by irreversible ocular and airway damage. Mortality is significant and is largely attributable to complications of the disease (sepsis, malnutrition, airway compromise) and toxicity of treatment (opportunistic infection, corticosteroid and immunosuppressant side effects).[2] Spontaneous remission is described but unpredictable, and some patients enter durable remission only after years of immunosuppression or a successful biologic course.
Childhood EBA generally has a more favourable prognosis than adult disease, often responding well to dapsone and entering remission within a few years.[9] All patients require long-term dermatology follow-up; those with mucosal disease need parallel ophthalmology, gastroenterology, ENT, and (where relevant) gynaecology or urology input in a multidisciplinary clinic. Patients on rituximab or other immunosuppression require infection surveillance and vaccination optimisation (inactivated vaccines before rituximab, avoidance of live vaccines during and after B-cell depletion).
Special Populations
Children with EBA typically have the inflammatory or IgA phenotype; dapsone is often first-line, with weight-based dosing and the same G6PD precautions as in adults; the differential from inherited dystrophic EB and linear IgA disease rests on family history, immunofluorescence, and the salt-split pattern.[9] In pregnancy, drug selection is constrained: methotrexate, mycophenolate, and ciclosporin are teratogenic or contraindicated; dapsone is relatively safe in pregnancy (with folate supplementation) but the G6PD check remains essential; corticosteroids (prednisolone, which is largely placenta-inactivated) are the safest systemic agent; rituximab crosses the placenta and causes transient neonatal B-cell depletion, so it is avoided in the third trimester where possible.[4] In the elderly, comorbidity and frailty amplify the toxicity of corticosteroids and immunosuppression: malignancy screening (especially lymphoma and solid-organ tumours), bone protection (calcium, vitamin D, bisphosphonate), glycaemic and blood-pressure control, and infection surveillance are integral to safe management. The immunocompromised patient (post-transplant, on anti-TNF for IBD, HIV-positive) carries a heightened risk of opportunistic infection with rituximab and combination immunosuppression, and treatment must be co-ordinated with the relevant specialist teams.[10] In EBA associated with IBD, azathioprine is often already in use for the Crohn's disease; anti-TNF biologics (infliximab, adalimumab) may help both conditions but do not reliably control the blistering, which must be treated on its own merits.[10]
Evidence, Guidelines & Regional Differences
EBA is rare and the evidence base is correspondingly thin. No randomised controlled trial of any therapy for EBA has been completed, and treatment recommendations rest on case series, systematic reviews of observational data, and expert consensus.[4] The two most influential recent documents are the International Bullous Diseases Group 2018 consensus on diagnostic criteria, which standardised the definition of definite, probable, and possible EBA and remains the global reference for diagnosis;[3] and the Japanese Dermatologic Association 2019 guidelines for the management of pemphigoid (which explicitly includes EBA), providing the most detailed national treatment algorithm.[11] The 2024 systematic review by Kianfar and colleagues of rituximab in EBA reported a high response rate and an acceptable safety profile, establishing rituximab as the biologic of choice in refractory disease;[7] the 2023 systematic review of IVIG by the same group confirmed a substantial but more modest role for IVIG.[8] The 2024 state-of-the-art diagnosis review by van Beek and colleagues codified the contemporary diagnostic algorithm (DIF plus salt-split IIF plus anti-type VII collagen ELISA), with immunoelectron microscopy reserved for difficult cases.[5]
Regional guideline differences are modest but real. European (EADF/EDF) and Indian (IADVL) practice tends to favour colchicine and dapsone first-line for the classical and IgA variants, reflecting both efficacy and access. The Japanese guidelines recommend a corticosteroid-combination approach earlier and reserve rituximab for refractory disease. In the United States, where no EBA-specific guideline exists, rituximab is often deployed earlier in severe or refractory cases, driven by the systematic-review evidence and specialist preference.[7] The disease itself is global, but the drugs and the thresholds are regional — and the clinician should name the guideline being followed.[3][11]
The IBDG 2018 consensus diagnostic criteria apply worldwide and are the reference standard for diagnosis. Treatment choice is then guided by regional availability and specialist preference.
Australian and New Zealand practice follows EADF principles, with rituximab access through specialist immunology or dermatology centres for refractory disease.
Exam Pearls
FLOOR
Exam application bank (NEET-PG / INICET)
One-line answer
Epidermolysis bullosa acquisita (EBA) is an acquired autoimmune subepidermal blistering disease caused by IgG autoantibodies against type VII collagen (the major structural protein of anchoring fibrils at the dermoepidermal junction). It produces trauma-induced tense bullae on extensor surfaces that heal with milia and scarring, distinguishing it from bullous pemphigoid. The salt-split skin test is the key discriminator: EBA labels the FLOOR (dermal side) while bullous pemphigoid labels the ROOF (epidermal side). The classical variant mimics porphyria cutanea tarda; the mucous membrane variant mimics mucous membrane pemphigoid. Strong association with inflammatory bowel disease (Crohn's). Notoriously treatment-resistant.
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard. [1]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes. [1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change. [1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each. [1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory. [1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Epidermolysis bullosa acquisita.
[1]Salt-split skin: EBA deposits on which side?
The FLOOR (dermal side). Type VII collagen sits below the lamina densa and stays with the dermal floor when skin is split through the lamina lucida with 1 mol/L sodium chloride. Bullous pemphigoid deposits on the ROOF (epidermal side) because BP180 and BP230 sit in the hemidesmosome above the lamina lucida.
Which clinical variant of EBA is the most morbid, and why?
The mucous membrane (mucous membrane pemphigoid-like) variant. It causes cumulative, irreversible scarring of the ocular, oral, oesophageal, laryngeal, and anogenital mucosa — leading to blindness, dysphagia, airway stenosis, and genital scarring. It demands early aggressive combination immunosuppression to prevent permanent damage.
References
- [1]Kim JH, Kim SC. Epidermolysis bullosa acquisita J Eur Acad Dermatol Venereol, 2013.PMID 23368767
- [2]Koga H, Prost-Squarcioni C, Iwata H. Epidermolysis Bullosa Acquisita: The 2019 Update Front Med (Lausanne), 2018.PMID 30687710
- [3]Prost-Squarcioni C, Caux F, Schmidt E, et al. International Bullous Diseases Group: consensus on diagnostic criteria for epidermolysis bullosa acquisita Br J Dermatol, 2018.PMID 29165796
- [4]Miyamoto D, Gordilho JO, Santi CG. Epidermolysis bullosa acquisita An Bras Dermatol, 2022.PMID 35701269
- [5]van Beek N, Holtsche MM, Atefi I, et al. State-of-the-art diagnosis of autoimmune blistering diseases Front Immunol, 2024.PMID 38903493
- [6]Woodley DT, Remington J, Chen M. Autoimmunity to type VII collagen: epidermolysis bullosa acquisita Clin Rev Allergy Immunol, 2007.PMID 18058258
- [7]Kianfar N, Dasdar S, Marashi A, et al. Rituximab in the Treatment of Epidermolysis Bullosa Acquisita: A Systematic Review J Clin Aesthet Dermatol, 2024.PMID 39006807
- [8]Kianfar N, Dasdar S, Daneshpazhooh M, et al. A systematic review on efficacy, safety and treatment durability of intravenous immunoglobulin in autoimmune bullous dermatoses: Special focus on indication and combination therapy Exp Dermatol, 2023.PMID 37150538
- [9]Hignett E, Sami N. Pediatric epidermolysis bullosa acquisita: A review Pediatr Dermatol, 2021.PMID 34339066
- [10]Antonelli E, Bassotti G, Tramontana M, et al. Dermatological Manifestations in Inflammatory Bowel Diseases J Clin Med, 2021.PMID 33477990
- [11]Ujiie H, Iwata H, Yamagami J. Japanese guidelines for the management of pemphigoid (including epidermolysis bullosa acquisita) J Dermatol, 2019.PMID 31646663
- [12]Bialy-Golan A, Brenner S. Penicillamine-induced bullous dermatoses J Am Acad Dermatol, 1996.PMID 8912569