Dermatology · Medicine
Graft-versus-host disease (GVHD) — cutaneous manifestations
Also known as Graft-versus-host disease (GVHD) · Acute GVHD · Chronic GVHD · Cutaneous GVHD · Allogeneic stem cell rejection
Graft-versus-host disease (GVHD) is a multisystem complication of allogeneic haematopoietic stem cell transplantation (HSCT) in which donor T-lymphocytes attack host HLA-mismatched tissues — primarily the skin, gastrointestinal tract, and liver. Acute GVHD (classically ≤100 days post-transplant) presents with an acral maculopapular rash (Stage I less than 25% BSA → Stage IV bullae/desquamation, TEN-like), GI symptoms (secretory/bloody diarrhoea), and cholestatic hepatitis. Chronic GVHD (classically 100 days, NIH-defined by clinical features) presents with lichenoid (lichen planus-like) or sclerodermoid (morphea/SSc-like) cutaneous changes, oral mucositis, sicca syndrome, bronchiolitis obliterans, and fasciitis with joint contractures. The skin is the most commonly affected organ and is often the first site of clinically apparent disease. Grading uses the Glucksberg/IBMTR system (acute) and the NIH 2025 consensus (chronic). First-line treatment: systemic corticosteroids (methylprednisolone 1–2 mg/kg/day); steroid-refractory chronic GVHD treated with three FDA-approved targeted agents — ruxolitinib 10 mg twice daily (JAK1/2 inhibitor; REACH-3), belumosudil 200 mg once daily (ROCK2 inhibitor; ROCKstar), and ibrutinib 420 mg once daily (BTK inhibitor; PCYC-1129) — plus rituximab and extracorporeal photopheresis. Acute steroid-refractory disease: ruxolitinib (REACH-2). Prophylaxis: calcineurin inhibitor + methotrexate or post-transplant cyclophosphamide (haploidentical).
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags
Definition and overview
Graft-versus-host disease (GVHD) is a multisystem immunological complication of allogeneic haematopoietic stem cell transplantation (HSCT) in which immunocompetent donor T-lymphocytes engrafted in the recipient recognise host tissues as foreign and mount an inflammatory attack. The skin is the most commonly affected organ (up to 80% of patients) and is often the first site of clinically apparent disease. GVHD is the leading cause of non-relapse mortality after allogeneic HSCT and the single most important determinant of long-term quality of life in transplant survivors. The traditional classification divides GVHD by timing — acute (classically ≤100 days post-transplant) and chronic (>100 days) — but the modern NIH consensus (2005, updated 2014 and 2025) defines the two forms by clinical features, not by timing, allowing recognition of late-onset acute, persistent acute and overlap syndromes.[1][4][7]

Pathophysiology
Billingham criteria
GVHD develops only when three conditions are simultaneously met: [1]
- The graft contains immunocompetent cells — donor T-lymphocytes capable of mounting an immune response.
- The host is immunocompromised — incapable of rejecting the graft (the fundamental difference from solid-organ transplant rejection).
- Histoincompatibility — HLA mismatch and/or disparity at minor histocompatibility antigens between donor and host.[5]
Three-phase model of acute GVHD
Three phases of acute GVHD — 'D-C-E'
DCE
Conditioning regimen (chemotherapy ± total body irradiation) damages host tissues → 'danger' signals (DAMPs, LPS translocation from gut) → cytokine release (TNF-α, IL-1, IL-6) → host APC activation. Tissue injury primes the immune response and explains why intensity of conditioning correlates with GVHD severity.
Donor T-cells encounter host antigens presented by host/donor APCs in spleen and lymph nodes → CD4+ Th1 and CD8+ cytotoxic T-cell activation, clonal expansion, trafficking to skin / gut / liver — the organs of tropism. Minor histocompatibility antigens (HA-1, HA-2, HY) trigger reactions even in HLA-matched transplants.
Activated cytotoxic T-cells (perforin, granzyme, Fas-FasL) plus inflammatory cytokines (TNF-α, IFN-γ, IL-1, IL-2, IL-6, IL-17) → keratinocyte apoptosis (skin basal layer), GI crypt cell death (mucosal denudation, diarrhoea), and bile duct epithelial injury (cholestasis). This is the 'cytokine storm' that drives clinical acute GVHD.
Acute GVHD is dominated by CD8+ cytotoxic T-cells and a Th1 cytokine milieu (TNF-α, IFN-γ, IL-1, IL-2) producing direct cytotoxic damage to the rapidly dividing epithelial cells of the skin, GI tract, and biliary epithelium. Chronic GVHD is a fundamentally different process — a dysregulated immune response combining autoimmunity and alloimmunity, with Th2 cytokines, B-cell activation, alloantibody formation, germinal-centre dysregulation and TGF-β-driven fibrosis. The result is lichenoid inflammation, sclerodermoid dermal sclerosis, sicca glandular destruction and ectopic lymphoid neogenesis.[5]
Organ tropism (skin, gut, liver) is explained by: [1]
- Shared minor histocompatibility antigens (HA-1, HA-2, HA-3, HA-8, HY) preferentially expressed on epithelial cells.
- Conditioning-induced damage that preferentially injures rapidly dividing epithelial cells (skin basal keratinocytes, GI crypts, biliary epithelium).
- Microbial translocation from conditioning-damaged gut mucosa amplifies systemic inflammation. [1]
The balance with graft-versus-leukaemia (GVL) is clinically important: donor T-cells that mediate GVL also mediate GVHD, and complete abrogation of GVHD (e.g. aggressive T-cell depletion) increases relapse risk. Regulatory T-cells (Tregs) are depleted by conditioning and their reconstitution correlates with GVHD control. [1]
Acute GVHD (cutaneous)
Clinical features
The cutaneous manifestations of acute GVHD follow a graded spectrum of severity:[3][4]
| Stage | Cutaneous involvement | Description |
|---|---|---|
| I | Maculopapular rash less than 25% BSA | Mild erythematous macules and papules, often beginning on the palms, soles, ears, and upper trunk (acral onset is characteristic) |
| II | Rash 25–50% BSA | Generalised maculopapular rash; may be pruritic or tender |
| III | Generalised erythroderma (>50% BSA) | Confluent red, hot, oedematous skin |
| IV | Bullae and desquamation | Toxic epidermal necrolysis (TEN)-like picture with full-thickness epidermal necrosis; life-threatening |
Key features: [1]
- Acral onset is the hallmark of cutaneous acute GVHD — rash often begins on the palms, soles, and ears — a distinguishing feature from drug eruptions (which tend to be truncal and symmetric).
- Associated organ involvement: GI (anorexia, nausea, vomiting, watery or bloody diarrhoea, abdominal pain, ileus) and liver (cholestatic hepatitis with rising bilirubin and alkaline phosphatase).
- Timing: typically 2–6 weeks post-transplant; engraftment syndrome overlaps at 7–14 days.
- Onset before day 14 may indicate hyperacute GVHD — a fulminant, treatment-refractory form with very poor prognosis.[3]
- A positive Nikolsky sign distinguishes Stage IV from early stages and predicts epidermal detachment akin to TEN.

Chronic GVHD (cutaneous and multisystem)
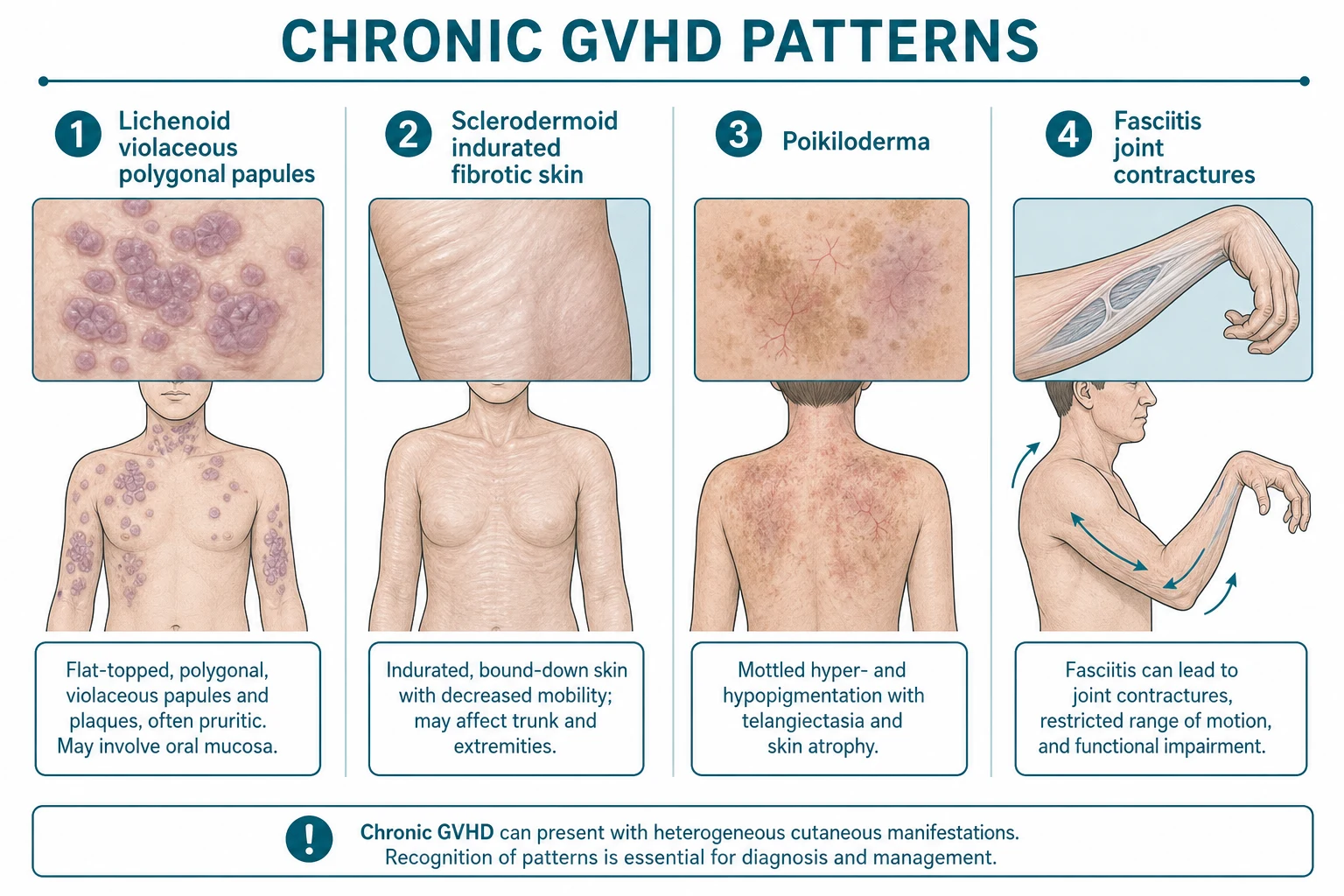
Chronic GVHD affects 30–50% of long-term HSCT survivors and is the leading cause of late non-relapse mortality. Cutaneous involvement is the most common manifestation and presents in several morphological patterns:[1][6]
1. Lichenoid chronic GVHD
- Violaceous, polygonal, flat-topped papules and plaques identical to lichen planus — especially on the wrists, forearms, trunk, and oral mucosa (Wickham striae-like white reticulate patches).
- May progress to hyperpigmentation and atrophy with prolonged disease.
- Oral involvement: lichenoid lesions on the buccal mucosa, tongue, gingivae; oral sicca (dry mouth from salivary gland involvement); mucosal sensitivity (pain to spicy/acidic foods); pseudo-membranous and erosive forms occur. [1]
2. Sclerodermoid chronic GVHD
- Indurated, bound-down, fibrotic plaques resembling morphea or systemic sclerosis — may be localised (morphea-like, often unilateral) or generalised.
- Fasciitis: deep fibrosis involving the fascia → restricted joint mobility and joint contractures (peau d'orange skin, cable-like fibrosis, "groove sign" of superficial veins).
- Poikiloderma: atrophy, telangiectasia, and pigmentary change in a reticulated pattern.
- Nail dystrophy: pterygium, longitudinal ridging, onycholysis, anonychia.
- Scalp alopecia: scarring and non-scarring forms.
- Risk of squamous cell carcinoma (SCC) in chronic GVHD-affected skin and mucosa (especially oral) — long-term surveillance essential.[6]
3. Other chronic GVHD manifestations
- Sicca syndrome: dry eyes (keratoconjunctivitis sicca) and dry mouth (xerostomia) from lymphocytic infiltration of lacrimal and salivary glands.
- Mucosal involvement: oral lichenoid lesions, genital erosions, vaginal stenosis, oesophageal webs/strictures.
- Hair: premature grey hair (leukoderma-like scalp), scarring alopecia.
- Systemic: bronchiolitis obliterans (pulmonary), myasthenia gravis, autoimmune cytopenias, polymyositis, peripheral neuropathy. [1]

Histopathology
- Acute GVHD: basal vacuolar degeneration (apoptotic keratinocytes with eosinophilic bodies — "dyskeratotic cells" / Civatte bodies), lymphocytic satellitosis (lymphocytes adjacent to apoptotic keratinocytes), spongiosis; in severe cases, full-thickness epidermal necrosis with subepidermal blistering (TEN-like).
- Chronic GVHD: lichenoid pattern (similar to lichen planus with band-like lymphocytic infiltrate obscuring the dermo-epidermal junction) or sclerodermoid pattern (dermal sclerosis with loss of adnexal structures, thickened collagen bundles, resembling morphea/systemic sclerosis). Eosinophilic fasciitis-like involvement of subcutis is seen in deep biopsies of fasciitis.[7]
Grading
Overall acute GVHD grading (Glucksberg/IBMTR system)
| Overall Grade | Skin | Liver (bilirubin) | GI (stool volume/day) |
|---|---|---|---|
| I | Stage 1–2 | None | None |
| II | Stage 3 | Stage 1 | Stage 1 |
| III | — | Stage 2–3 | Stage 2–4 |
| IV | Stage 4 | Stage 4 | Stage 4 |
(Grade IV = any organ with Stage 4 severity — life-threatening.)[3]
Chronic GVHD NIH severity scoring (2025 update)
Classified as mild, moderate, or severe based on the number and severity of organ involvements. The NIH 0–3 organ scoring covers skin, mouth, eyes, GI, liver, lungs, joints/fascia, and genital tract.[9]
- Mild: ≤2 organs involved, none with score >1, no lung involvement.
- Moderate: ≥3 organs involved OR any single organ with score 2 OR lung score 1.
- Severe: any organ with score 3 OR lung score 2–3. [1]
Management
Acute GVHD
- First-line: Systemic corticosteroids — methylprednisolone 1–2 mg/kg/day IV (or prednisolone equivalent). Responds in ~50% of patients.[3]
- Steroid-refractory (progression after 5–7 days or no improvement after 14 days):
- Ruxolitinib (JAK1/2 inhibitor) — FDA-approved for steroid-refractory acute GVHD; the preferred second-line agent.[3]
- Extracorporeal photopheresis (ECP).
- Anti-TNF (infliximab), mesenchymal stromal cells, tocilizumab, basiliximab, vedolizumab.
- Topical: for skin-limited disease — topical corticosteroids, calcineurin inhibitors (tacrolimus/pimecrolimus).[4]
Chronic GVHD
- First-line: Systemic corticosteroids (prednisolone 1 mg/kg/day) ± a calcineurin inhibitor (tacrolimus or cyclosporine); continued for at least 4–8 weeks before considering steroid-refractory designation.[2]
- Second-line / steroid-refractory agents — see detailed drug section below for mechanism, dose, and trial evidence.[2][8][9]
- Topical/supportive: topical corticosteroids and calcineurin inhibitors for cutaneous and oral disease; phototherapy (narrowband UVB, PUVA) for skin-limited disease; moisturisers, sun protection, and squamous cell carcinoma surveillance.[6]
Chronic GVHD second-line agents — detailed pharmacology
Approximately 50–60% of chronic GVHD patients become steroid-refractory or steroid-dependent and require second-line therapy. Three targeted agents have transformed this space and are now the preferred options after failure of first-line corticosteroids plus calcineurin inhibitor: [1]
- Ruxolitinib — oral JAK1/2 inhibitor, 10 mg twice daily (start 5 mg BD in cytopenic patients; titrate up). FDA-approved for steroid-refractory acute GVHD (REACH-2 trial, NEJM 2020) and chronic GVHD (REACH-3 trial, NEJM 2021). In REACH-3, ruxolitinib significantly improved overall response rate versus best available therapy (49.7% vs 25.6%) at week 24, with superior failure-free survival; benefit was consistent across skin, oral, ocular, and lung involvement. Dose adjustments required for renal/hepatic impairment and drug interactions (CYP3A4). Key adverse effects: cytopenias (thrombocytopenia, anaemia, neutropenia — dose-limiting), infection risk (CMV reactivation, PML), and rare lipid abnormalities. Monitor CBC weekly for the first 8 weeks.[10][13]
- Belumosudil — oral ROCK2 (rho-associated coiled-coil kinase 2) inhibitor, 200 mg once daily. FDA-approved in 2021 for chronic GVHD after failure of at least two prior lines of systemic therapy (ROCKstar trial, Blood 2021). ROCK2 inhibition rebalances Th17/Treg differentiation and reduces pro-fibrotic cytokine signalling (TGF-β), making it particularly useful for sclerodermoid disease and fasciitis where conventional immunosuppression often fails. Overall response rate approximately 74% with median duration of response ~14 months; corticosteroid dose reduction achieved in two-thirds of patients. The 200 mg once-daily dose is non-inferior to higher doses with better tolerability. Common adverse effects: fatigue, nausea, diarrhoea, hypertension, transaminitis; manageable. Fewer cytopenias than ruxolitinib.[12]
- Ibrutinib — oral Bruton tyrosine kinase (BTK) and IL-2-inducible T-cell kinase (ITK) inhibitor, 420 mg once daily. FDA-approved in 2017 for chronic GVHD after failure of one or more lines of systemic therapy (PCYC-1129 study, Blood 2017). Overall response rate ~67%, with responses across involved organs including skin (lichenoid), mouth, GI, and lungs. Particularly effective for autoantibody-driven and sclerodermoid chronic GVHD because BTK blockade impairs B-cell receptor signalling and alloantibody production. Key adverse effects: afibrinogenaemia and bleeding (impaired platelet function), atrial fibrillation (5–10% incidence), cytopenias, infection; drug interactions via CYP3A4. Reduced dose (140–280 mg) in hepatic impairment or with strong CYP3A4 inhibitors.[11]
- Rituximab — anti-CD20 monoclonal antibody, 375 mg/m² weekly × 4 doses. Effective for autoantibody-mediated chronic GVHD (immune cytopenias, myasthenia, sclerodermoid, oral lichenoid); B-cell depletion reverses alloantibody-driven tissue injury.
- Extracorporeal photopheresis (ECP) — apheresis with 8-methoxypsoralen + UVA, given on 2 consecutive days every 1–2 weeks. Particularly effective for sclerodermoid chronic GVHD, oral lichenoid disease and steroid-refractory acute cutaneous GVHD; steroid-sparing; minimal infection risk.[2]
- Mycophenolate mofetil (MMF) — 1–1.5 g twice daily; well-tolerated oral agent often added to calcineurin inhibitor in steroid-refractory disease.
- Methotrexate — low-dose (5–15 mg weekly) — useful in joint/fascia involvement.[8]
Ruxolitinib
- Class: oral JAK1/2 inhibitor
- Dose: 10 mg twice daily (5 mg BD in cytopenia)
- Approval: steroid-refractory acute GVHD (REACH-2, NEJM 2020) and chronic GVHD (REACH-3, NEJM 2021) — FDA-approved for both
- Mechanism: blocks IFN-γ, IL-2, IL-6, IL-15 signalling — broad T-cell and dendritic-cell suppression
- Best for: inflammatory acute GVHD and any chronic GVHD subtype with active inflammation
- Key toxicities: cytopenias (thrombocytopenia, anaemia, neutropenia), CMV reactivation, PML risk
- Trial endpoint: REACH-3 — 49.7% ORR vs 25.6% BAT at week 24; superior failure-free survival
Belumosudil
- Class: oral ROCK2 inhibitor
- Dose: 200 mg once daily
- Approval: chronic GVHD after ≥2 prior lines of systemic therapy (ROCKstar, Blood 2021) — FDA-approved
- Mechanism: ROCK2 inhibition rebalances Th17/Treg; downregulates pro-fibrotic TGF-β signalling
- Best for: sclerodermoid chronic GVHD and fasciitis where fibrosis dominates
- Key toxicities: fatigue, nausea, diarrhoea, hypertension, transaminitis — fewer cytopenias
- Trial endpoint: ROCKstar — 74% ORR; corticosteroid taper achieved in two-thirds of patients
Ibrutinib
- Class: oral BTK + ITK inhibitor
- Dose: 420 mg once daily (140–280 mg in hepatic impairment)
- Approval: chronic GVHD after ≥1 prior line (PCYC-1129, Blood 2017) — first FDA-approved targeted chronic GVHD drug
- Mechanism: BTK blockade impairs B-cell receptor signalling and alloantibody production; ITK blockade modulates Th2 responses
- Best for: autoantibody-driven and sclerodermoid chronic GVHD; skin, mouth, GI, lung involvement
- Key toxicities: bleeding (impaired platelet function), atrial fibrillation (5–10%), cytopenias
- Trial endpoint: PCYC-1129 — 67% ORR; durable responses across involved organs
Organ-specific therapy for chronic GVHD [1]
Systemic therapy alone rarely controls established chronic GVHD; organ-specific (topical, intralesional, intraluminal) therapy is essential for symptom control and steroid sparing, and is delivered in parallel with the systemic regimen. The NIH 2025 consensus and HSCT-Core-Curriculum update emphasise early escalation to combined systemic-plus-local therapy in symptomatic moderate-severe organ disease.[2][9]
- Ocular GVHD (keratoconjunctivitis sicca, cicatrising conjunctivitis, dry eye) — aggressively managed by an ophthalmologist familiar with HSCT ocular disease. First-line topical: preservative-free lubricant drops, topical cyclosporine 0.05% ophthalmic emulsion 1 drop BD-QID, topical tacrolimus 0.03% ophthalmic ointment, topical corticosteroid (fluorometholone 0.1% or prednisolone acetate 1%) for short courses; punctal plugs (collagen or silicone) for refractory dryness; autologous serum tears 20–50% for severe epitheliopathy; moisture goggles, humidified environments, and oral doxycycline 100 mg daily or omega-3 fatty acids for meibomian gland dysfunction. For moderate-severe ocular cGvHD (NIH score 2–3): escalate systemic therapy with ruxolitinib 10 mg BD (the only agent with proven ocular response in REACH-3) or ibrutinib 420 mg OD; consider scleral contact lenses (PROSE) for corneal protection; scleral thinning or corneal melt requires urgent ophthalmology referral.[2][10]
- Oral GVHD (lichenoid mucositis, xerostomia, ulceration) — cornerstone is topical super-potent corticosteroids plus systemic therapy: clobetasol 0.05% solution or paste (apply BD-TDS to affected sites, expectorate, do not swallow), dexamethasone 0.5 mg/5 mL mouthwash swish-and-spit TDS, tacrolimus 0.1% mouthwash (compounded; 2 mg/5 mL TDS, effective for refractory ulceration), or betamethasone 0.5 mg/5 mL oral rinse; intralesional triamcinolone acetonide 0.1–0.3 mL of 10 mg/mL for isolated erosions; phototherapy (narrow-band UVB, PUVA, or extracorporeal photopheresis) for generalised lichenoid disease; saliva substitutes, sugar-free gum, pilocarpine 5 mg TDS or cevimeline 30 mg TDS for residual salivary function in xerostomia; scrupulous oral hygiene and annual dental review (SCC surveillance). Systemic agents with proven oral response: ruxolitinib, belumosudil, ibrutinib, ECP, rituximab.[2][6]
- Pulmonary GVHD (bronchiolitis obliterans syndrome, BOS) — the leading non-cutaneous cause of chronic GVHD mortality; early recognition and aggressive immunosuppression are essential. Inhaled FAM regimen (Fluticasone 250 µg-metered-dose inhaler, 2 puffs BD via spacer + Azithromycin 250 mg orally 3 times per week + Montelukast 10 mg orally once daily) slows FEV1 decline and is now standard for NIH lung score 1–2. Add systemic corticosteroids (prednisolone 1 mg/kg/day, then taper) and a calcineurin inhibitor; escalate to ruxolitinib 10 mg BD or ibrutinib 420 mg OD for refractory or progressive disease; extracorporeal photopheresis (ECP) is effective steroid-sparing therapy for BOS and may stabilise FEV1 in 30–40% of treated patients. End-stage disease (NIH lung score 3, FEV1 less than 40% predicted) requires timely referral for lung transplantation — the only definitive therapy. Avoidance of inhalational irritants, pulmonary rehabilitation, pneumococcal / annual influenza vaccination, and PJP prophylaxis are mandatory supportive measures.[2][9][11]
- Hepatic GVHD (cholestatic hepatitis) — overlap of acute and chronic patterns often coexisting. Ursodeoxycholic acid (UDCA) 13–15 mg/kg/day orally in 2–4 divided doses is standard both for prophylaxis and treatment of cholestatic cGvHD; it improves LFTs and may reduce steroid dependence. Continue systemic immunosuppression with prednisolone ± calcineurin inhibitor; escalate to ruxolitinib 10 mg BD for refractory cholestasis; ECP reduces bilirubin and pruritus in ~50% of hepatic cGvHD patients; rituximab 375 mg/m² weekly × 4 is effective for steroid-dependent cholestatic disease. Screen for and treat concurrent hepatitis B / C / E viral reactivation (entecavir or tenofovir prophylaxis for HBsAg-positive recipients); avoid hepatotoxic drugs and alcohol.[2][9]
- Genital GVHD (vulvovaginal, penile lichen sclerosus-like and sclerotic disease) — frequently unrecognised but affects up to one-third of women post-HSCT and a smaller proportion of men; symptoms include dyspareunia, vaginal dryness, synechiae, vaginal stenosis, labial sclerosis, and in men urethral meatal stenosis, phimosis, and lichen sclerosus-like changes. First-line: topical ultrapotent corticosteroid (clobetasol propionate 0.05% ointment TDS initially, taper to twice weekly) applied to affected vulvovaginal or penile mucosa; topical tacrolimus 0.1% ointment BD for steroid-sparing maintenance and refractory disease; oestrogen vaginal cream for postmenopausal atrophy; vaginal dilator therapy initiated early and continued 10–15 minutes daily to prevent synechiae and stenosis; intralesional triamcinolone for focal sclerotic plaques. For moderate-severe NIH genital score 2–3, escalate systemic therapy (ruxolitinib 10 mg BD, ibrutinib 420 mg OD, or belumosudil 200 mg OD); surgical release (vulvovaginal adhesiolysis, perineoplasty, or circumcision) for established stenosis unresponsive to medical therapy; sexual dysfunction counselling and psychosexual support are integral.[2][9][12]
Prophylaxis
- Calcineurin inhibitor + methotrexate: tacrolimus or cyclosporine + short-course methotrexate — the standard prophylactic regimen.[1]
- Post-transplant cyclophosphamide (PTCy): increasingly used for haploidentical transplants; depletes alloreactive T-cells while sparing regulatory T-cells.
- T-cell depletion (ex vivo, e.g. CD34+ selection or alemtuzumab): reduces GVHD risk but increases relapse and infection.
Differential diagnosis
- Drug eruption: morbilliform rash from antibiotics, conditioning regimen drugs; timing and temporal relationship help distinguish; acral onset favours GVHD.
- Viral exanthem: CMV, adenovirus, HHV-6 reactivation post-transplant.
- Morphea/systemic sclerosis: identical to sclerodermoid chronic GVHD — transplant history and distribution help. [1]
Acute vs chronic GVHD at a glance
Chronic GVHD — second-line drug doses for the ward
Acute GVHD
- Timing: classically ≤100 days post-HSCT (NIH: clinical features, not just timing)
- Dominant effector cells: CD8+ cytotoxic T-cells + Th1 cytokines (TNF-α, IFN-γ, IL-1, IL-2)
- Skin: maculopapular rash, acral onset, Stage I less than 25% BSA → Stage IV bullae/desquamation
- Gut: secretory/bloody diarrhoea, abdominal pain, ileus (endoscopy with biopsy)
- Liver: cholestatic pattern (rising bilirubin, ALP) — distinct from VOD/SOS
- Histology: basal vacuolar degeneration, apoptotic keratinocytes (Civatte bodies), lymphocytic satellitosis
- First-line: methylprednisolone 1–2 mg/kg/day IV; steroid-refractory → ruxolitinib (JAK1/2)
- Prophylaxis: calcineurin inhibitor + methotrexate, or post-transplant cyclophosphamide (PTCy)
Chronic GVHD
- Timing: classically >100 days post-HSCT (NIH: lichenoid or sclerodermoid features)
- Dominant effector cells: Th2 cytokines, B-cell activation, alloantibodies, TGF-β fibrosis
- Skin: lichenoid (lichen planus-like) and/or sclerodermoid (morphea/SSc-like); fasciitis; poikiloderma
- Multisystem: oral lichenoid, sicca syndrome (eye/mouth), bronchiolitis obliterans, vaginal sclerosis, oesophageal web
- Liver: cholestasis; musculoskeletal: joint contractures, fasciitis, myositis
- Histology: lichenoid (band-like infiltrate) OR sclerodermoid (dermal sclerosis, adnexal loss)
- First-line: prednisolone 1 mg/kg/day ± calcineurin inhibitor; steroid-refractory → ruxolitinib, belumosudil, ibrutinib, ECP, rituximab
- Late effects: SCC in chronic skin/oral disease, BOS, joint contractures, end-stage non-relapse mortality
GVHD — key numbers for the ward
Complications & long-term sequelae
Chronic GVHD survivors face a wide spectrum of late effects that drive their long-term morbidity and mortality: [1]
- Cutaneous SCC — chronic cutaneous and oral lichenoid GVHD is a premalignant state; up to 5-fold increased risk of SCC, often multiple, with early metastases. Requires annual full-skin and oral examination; photoprotection and prompt biopsy of any non-healing ulcer or persistent leukoplakia.
- Bronchiolitis obliterans syndrome (BOS) — progressive obstructive airway disease (FEV1 decline ≥10% over 6 months, FEV1/FVC less than 0.7, air trapping on CT). The leading non-cutaneous cause of chronic GVHD mortality. The FAM regimen (fluticasone/azithromycin/montelukast) slows progression; lung transplantation is the only definitive therapy.
- Joint contractures and fasciitis — sclerodermoid deep fibrosis around joints (especially wrists, ankles, shoulders); physiotherapy and ruxolitinib/belumosudil escalation are first-line.
- Endocrine: hypothyroidism (radiation/immune), gonadal failure, growth failure in children, metabolic syndrome and osteoporosis from chronic steroids.
- Infectious: CMV, EBV (PTLD risk), VZV, PJP, invasive aspergillosis, atypical mycobacteria — antimicrobial prophylaxis (aciclovir, PJP prophylaxis, antifungals) is mandatory. [1]
Outcomes & prognosis
- Grade I acute GVHD — minimal impact on survival; full recovery expected.
- Grade II — 5-year survival approximately 70–80% with appropriate therapy.
- Grade III–IV acute GVHD — non-relapse mortality 60–80%; survival depends on steroid responsiveness.
- Steroid-refractory acute GVHD — less than 20% 1-year survival historically, modestly improved with ruxolitinib (REACH-2 trial).
- Chronic GVHD by NIH 2025 severity: mild (5-year OS >90%), moderate (60–80%), severe (less than 50%). [1]
Special situations
- Hyperacute GVHD (onset less than 14 days) — fulminant; poor prognosis; consider ATG.
- Engraftment syndrome — fever, rash, weight gain, pulmonary oedema at neutrophil recovery (day 7–14); responds to brief corticosteroids.
- Overlap syndrome — simultaneous acute and chronic features (per NIH criteria).
- Donor lymphocyte infusion (DLI)-induced GVHD — typical timing 2–6 weeks after DLI; managed similarly to post-HSCT GVHD.
- Transfusion-associated GVHD (TA-GVHD) — in immunocompetent recipients (e.g. cardiac surgery) transfused with cellular blood products from HLA-homozygous donors; >90% mortality. Prevention: irradiated blood products for immunocompromised patients and directed donations from relatives.
- Maternal-fetal GVHD — in severe combined immunodeficiency (SCID) / Omenn syndrome; engrafted maternal T-cells cause erythroderma, hepatosplenomegaly, lymphadenopathy. [1]
Prevention & patient education
- Transplant-modifying strategies: HLA-matched donors (high-resolution typing at HLA-A, -B, -C, -DRB1, -DQB1), umbilical cord blood, T-cell depletion, post-transplant cyclophosphamide (PTCy) for haploidentical HSCT.
- Gut decontamination and dietary modification (low-microbial diet) reduce bacterial translocation and acute GI GVHD.
- Prophylactic ECP in high-risk regimens.
- Patient counselling: photoprotection, emollients, surveillance for SCC, compliance with immunosuppression, prompt reporting of rash, diarrhoea or joint stiffness.
- Vaccination: killed vaccines from 6 months post-HSCT; live vaccines deferred ≥24 months and only off immunosuppression. [1]
Red flags
Exam application bank (NEET-PG / INICET)
One-line answer
Graft-versus-host disease (GVHD) is a multisystem complication of allogeneic haematopoietic stem cell transplantation (HSCT) in which donor T-lymphocytes attack host HLA-mismatched tissues — primarily the skin, gastrointestinal tract, and liver. Acute GVHD (classically ≤100 days post-transplant) presents with an acral maculopapular rash (Stage I less than 25% BSA → Stage IV bullae/desquamation, TEN-like), GI symptoms (secretory/bloody diarrhoea), and cholestatic hepatitis. Chronic GVHD (classically >100 days, NIH-defined by clinical features) presents with lichenoid (lichen planus-like) or sclerodermoid (morphea/SSc-like) cutaneous changes, oral mucositis, sicca syndrome, bronchiolitis obliterans, and fasciitis with joint contractures. The skin is the most commonly affected organ and is often the first site of clinically apparent disease. Grading uses the Glucksberg/IBMTR system (acute)
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard. [1]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes. [1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change. [1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each. [1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory. [1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Graft-versus-host disease (GVHD) — cutaneous manifestations.
Expanded exam teaching (depth pass)
Clinical reasoning
For Graft-versus-host disease (GVHD) — cutaneous manifestations, examiners test whether you can prioritise life threats, choose the right first test, and give specific therapy (agent, dose, route, timing). Generic phrases without numbers score poorly.
Mechanism → feature map
Build a short chain: cause → pathophysiologic intermediate → clinical feature → complication. Every major symptom in the classic vignette should sit on that chain.
Investigation strategy
- Bedside/first-line tests that change immediate management
- Confirmatory or staging tests
- What a normal result does not exclude
- When not to delay treatment for imaging (unstable patient)
Management ladder
- Resuscitation / ABC / sepsis or haemorrhage bundle as relevant
- Specific antidote / procedure / antimicrobial / reperfusion / surgery
- Supportive care and monitoring targets
- Definitive long-term therapy and secondary prevention
- Disposition and safety-net advice
Special populations
Always prepare one line each for children, pregnancy, elderly, renal/hepatic impairment, and immunocompromised patients when the topic allows.
Pitfalls that fail candidates
- Treating the number not the patient
- Missing pregnancy status when relevant
- Imaging before stabilisation
- Wrong empiric cover or wrong antidote timing
- Incomplete counselling on recurrence, adherence, or red-flag return
Graft-versus-host disease (GVHD) is a multisystem complication of allogeneic haematopoietic stem cell transplantation (HSCT) in which donor T-lymphocytes attack host HLA-mismatched tissues — primarily the skin, gastrointestinal tract, and liver. Acute GVHD (classically ≤100 days post-transplant) presents with an acral maculopapular rash (Stage I less than 25% BSA → Stage IV bullae/desquamation, TEN-like), GI symptoms (secretory/bloody diarrhoea), and cholestatic hepatitis. Chronic GVHD (classica [1]
[1]References
- [1]Hymes SR, Farmer KL, Lewis AT, et al. Chronic graft-versus-host disease. Part I: Epidemiology, pathogenesis, and clinical manifestations J Am Acad Dermatol, 2024.PMID 36572065
- [2]Hymes SR, Farmer KL, Lewis AT, et al. Chronic graft-versus-host disease. Part II: Disease activity grading and therapeutic management J Am Acad Dermatol, 2024.PMID 36572064
- [3]Eshaghian A, Ziakas PD, Mylonakis E. Acute Graft-versus-Host Disease: An Update on New Treatment Options Drugs, 2023.PMID 37247105
- [4]Inoue R, Fuji S, Kato J, et al. Understanding and treatment of cutaneous graft-versus-host-disease Bone Marrow Transplant, 2023.PMID 37730800
- [5]Zeiser R, Blazar BR. Pathophysiology of Chronic Graft-versus-Host Disease and Therapeutic Targets N Engl J Med, 2017.PMID 29281578
- [6]Hsieh MM, Routt D, Jagasia M, et al. Cutaneous Chronic Graft-Versus-Host Disease: Clinical Manifestations, Diagnosis, Management, and Supportive Care Transplant Cell Ther, 2024.PMID 39370234
- [7]Hymes SR, Farmer ER, Lewis AT, et al. Cutaneous graft-versus-host disease Arch Dermatol, 1998.PMID 9606330
- [8]Vermeren S, Pidala J. Updates in chronic graft-versus-host disease management Am J Hematol, 2023.PMID 37483142
- [9]Lee SJ, Flower ME, Carter SL, et al. NIH Chronic Graft-Versus-Host Disease Consensus Conference 2025 Update Transplant Cell Ther, 2025.PMID 40409691
- [10]Zeiser R, Polverelli N, Ram R, et al. Ruxolitinib for Glucocorticoid-Refractory Chronic Graft-versus-Host Disease N Engl J Med, 2021.PMID 34260836
- [11]Miklos D, Cutler CS, Arora M, et al. Ibrutinib for chronic graft-versus-host disease after failure of prior therapy Blood, 2017.PMID 28924018
- [12]Cutler C, Lee SJ, Arai S, et al. Belumosudil for chronic graft-versus-host disease after 2 or more prior lines of therapy: the ROCKstar Study Blood, 2021.PMID 34265047
- [13]Zeiser R, von Bubnoff N, Butler J, et al. Ruxolitinib for Glucocorticoid-Refractory Acute Graft-versus-Host Disease N Engl J Med, 2020.PMID 32320566