Dermatology · Medicine
Incontinentia Pigmenti
Also known as Incontinentia pigmenti (IP) · Bloch-Sulzberger syndrome · Bloch-Siemens syndrome
Incontinentia pigmenti (IP; Bloch-Sulzberger syndrome) is an X-linked dominant genodermatosis caused by mutation in the IKBKG/NEMO gene (Xq28). It is lethal in most hemizygous males and almost exclusively affects females. The hallmark is a 4-stage Blaschkoid cutaneous eruption (vesicular, verrucous, hyperpigmented, atrophic/hypopigmented) with extracutaneous involvement of teeth, eyes, CNS, hair and nails.
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags

Overview and Definition
Incontinentia pigmenti (IP), also known as Bloch-Sulzberger syndrome or Bloch-Siemens syndrome, is a rare X-linked dominant multisystem genodermatosis. It is caused by mutation in the IKBKG gene (formerly called NEMO, NF-kappaB Essential Modulator), located at Xq28. The classic disease is characterised by a striking four-stage Blaschkoid cutaneous eruption and by ectodermal anomalies involving the teeth, eyes, central nervous system, hair and nails.[1][2]
IP is one of the best clinical examples of X-linked dominant inheritance with male lethality. Hemizygous affected males (XY) usually die in utero, so the overwhelming majority of live-born patients are female. The condition is therefore a cornerstone genetics topic in dermatology, paediatrics and medical genetics examinations: a rare disorder that illustrates lyonisation, functional mosaicism, Blaschkoid embryological cell migration and the developmental consequences of NF-kappaB signalling failure all at once. The severity of disease in females is highly variable, even within the same family, because of the randomness of X-inactivation and because different tissues may show different patterns of skewed X-inactivation. [1]
The name incontinentia pigmenti was chosen because, in the hyperpigmented stage, melanin appears to have lost continence and spilled from the epidermis into the dermis. The term highlights the central histological finding of melanin incontinence, which is seen in the third stage of skin disease and is one of the most distinctive pathological findings in dermatology. The condition was first described by Bloch and Sulzberger in the early twentieth century, and the clinical spectrum was later refined by the recognition of the four-stage eruption and the identification of the NEMO gene in 2000. [1]
IP is best understood as a developmental disorder of ectoderm. The ectoderm gives rise to the epidermis and its appendages, the lens and corneal epithelium, the retinal pigment epithelium and neural retina, the teeth enamel and oral mucosa, and the central nervous system. Failure of NF-kappaB signalling in these tissues explains why IP affects so many organ systems despite being caused by a single gene mutation. This multisystem involvement is what makes IP more than a simple dermatosis and why a coordinated multidisciplinary approach is essential. [1]
[1]Classification
IP is not formally divided into subtypes in the way cancers are staged, but several clinically important distinctions are made. [1]
- Classic IP — caused by loss-of-function mutations in IKBKG/NEMO, with the four-stage skin eruption and variable extracutaneous involvement. This is the form seen in examinations.
- IP with associated immunodeficiency — hypomorphic NEMO mutations can produce an overlapping phenotype with anhidrotic ectodermal dysplasia with immunodeficiency (EDA-ID). These patients have recurrent pyogenic infections, often due to Gram-positive bacteria, and may lack the full cutaneous IP phenotype.
- Affect males — extremely rare. Survival is usually explained by Klinefelter syndrome (47,XXY) or postzygotic somatic mosaicism. Such males may be underdiagnosed because the diagnosis is assumed to be female-limited. Males with mosaicism can have milder or patchy disease and may transmit the mutation if the germline is involved. Klinefelter males have a full second X chromosome, so lyonisation can produce a female-like mosaic pattern, but they also carry the long-term endocrine and fertility implications of Klinefelter syndrome.
- Familial IP versus de novo IP — de novo mutations account for a substantial proportion of cases, so a negative family history does not exclude the diagnosis. Paternal germline mosaicism can produce more than one affected child from an apparently unaffected father. [1]
Classic IP versus NEMO-related immunodeficiency overlap
| Feature | Classic IP | NEMO-related immunodeficiency (EDA-ID overlap) |
|---|---|---|
| Mutation type | Loss-of-function | Hypomorphic (partial function) |
| Skin stages | Usually all four | May be attenuated or absent |
| Infections | Not a dominant feature | Recurrent bacterial, mycobacterial, fungal |
| Immune work-up | Usually normal | Abnormal NF-kappaB response, low IgG subclasses |
| Management | Supportive + surveillance | Immunology input, IVIG, prophylactic antibiotics |
Epidemiology and Risk Factors
IP is rare, with an estimated incidence of approximately 1 in 40,000 live births, though ascertainment is incomplete because mildly affected adults may never be diagnosed. The condition is almost exclusively seen in females. The reported female-to-male ratio among recognised affected individuals is greater than 20:1, reflecting the high lethality of the fully mutant male conceptus.[2]
Because IP is X-linked dominant, an affected mother has a 50% chance of transmitting the mutation to each child. Daughters who inherit the mutation will be affected (with variable expressivity due to random X-inactivation). Sons who inherit the mutation usually die in utero, which is why many families have a history of recurrent male miscarriages. De novo mutations are common, and paternal gonadal mosaicism has been reported, so an unaffected father can still transmit the disease. The observed male miscarriage rate can make the family history more striking than the family itself, because affected males are rarely born. [1]
IP is not associated with ethnic or geographic predilection. Diagnosis is often delayed in populations with limited access to dermatology or genetics services. In such settings, the adult female may first present with dental problems, visual loss or neurological sequelae, while the infantile rash has long been forgotten or misdiagnosed as eczema or infection. [1]
Risk factors for severe extracutaneous disease are not well defined, but the following observations are clinically relevant: [1]
- Skewed X-inactivation that favours the mutant X chromosome in relevant tissues may worsen the phenotype.
- Early-onset, widespread or prolonged stage 1 skin disease may be associated with higher risk of neurological and ocular complications.
- Male sex is almost always lethal in the absence of Klinefelter syndrome or mosaicism. [1]
Epidemiology at a glance
Pathophysiology
The pathophysiology of IP is best understood at three levels: molecular, cellular and clinical-mosaic. [1]
Molecular level: IKBKG/NEMO and NF-kappaB
The IKBKG gene encodes the NEMO protein (NF-kappaB Essential Modulator), the regulatory subunit of the IKK (inhibitor of kappaB kinase) complex. NEMO is essential for activating the canonical NF-kappaB pathway after stimulation by tumour necrosis factor, interleukin-1, Toll-like receptors, antigen receptors and many other signals. Without functional NEMO, NF-kappaB cannot be released from its cytoplasmic inhibitor (IkappaB) and translocate to the nucleus. The result is failure of NF-kappaB-mediated transcription of survival genes, including anti-apoptotic genes such as BCL-xL and c-IAPs, and genes involved in immune and inflammatory responses. NF-kappaB also regulates genes that protect against tumour necrosis factor-induced apoptosis, so cells lacking NEMO are unusually vulnerable to TNF-mediated cell death.[1]
In IP, loss-of-function mutations (the classic deletion of exons 4-10 accounts for up to 80% of cases) abolish NEMO function. Cells that carry the mutant active X chromosome are therefore more susceptible to tumour necrosis factor-mediated apoptosis. Because many ectodermal structures depend on NF-kappaB for survival, migration and differentiation, the skin, teeth, retinal vasculature, brain and hair/nails are all vulnerable. The common exon 4-10 deletion removes the distal portion of the gene and usually produces a complete loss of function, whereas point mutations can create a milder or hypomorphic phenotype. [1]
Cellular level: X-inactivation and functional mosaicism
In females, one X chromosome is randomly inactivated in each somatic cell early in embryogenesis (lyonisation). In IP, this creates two populations of skin cells: cells in which the mutant X is active and cells in which the normal X is active. Mutant cells tend to die or are outcompeted by normal cells, producing the characteristic pattern of affected skin alternating with relatively normal skin along Blaschko's lines. These lines reflect the pathways of embryonic ectodermal cell migration, not nerves or blood vessels, which is why IP produces swirling, linear and whorled patterns rather than dermatomal patterns. Blaschko's lines are typically invisible in normal skin but become apparent when a mosaic genetic condition such as IP produces alternating populations of mutant and normal cells.[2]
The degree of skewing of X-inactivation varies between tissues and between individuals. In some tissues, the normal X may be preferentially active, producing milder disease; in others, the mutant X may dominate, producing more severe disease. This explains why two affected sisters can have very different phenotypes and why the same patient may have severe skin disease but normal neurology, or minimal skin findings but severe retinal disease. Understanding lyonisation is therefore essential for explaining variable expressivity to families and for approaching examination questions about phenotype prediction. [1]

Clinical pathophysiology of the four stages
The cutaneous stages reflect the evolving response to mutant-cell apoptosis and inflammation: [1]
- Stage 1 (vesicular) — intense inflammation and spongiosis with eosinophils; reflects the immune response to dying keratinocytes. Eosinophils are recruited because NF-kappaB deficiency alters the cytokine milieu, and the apoptotic keratinocytes release signals that attract inflammatory cells.
- Stage 2 (verrucous) — hyperkeratotic and papillomatous repair as the epidermis regenerates over areas of previous damage. The wart-like lesions represent a reactive epidermal proliferation rather than a true viral wart.
- Stage 3 (hyperpigmented) — melanin released from damaged basal keratinocytes is taken up by dermal melanophages; this is the melanin incontinence that gives the disease its name. The dermal melanin appears clinically as grey-brown or slate-blue pigmentation.
- Stage 4 (atrophic/hypopigmented) — reduced epidermal thickness and appendage loss in the affected clones produce pale, hairless, atrophic streaks. The atrophy is partly due to the loss of mutant cells and partly due to dermal scarring from the earlier inflammatory stages. [1]
Clinical Presentation
The clinical presentation of IP is dominated by the four-stage skin eruption and by the extracutaneous manifestations. A single patient may have all four stages simultaneously because they overlap in time and site. [1]

Stage 1: Vesicular/bullous (birth to approximately 4 months)
Stage 1 is the presentation that most often brings the newborn to medical attention. Infants develop linear or whorled inflammatory vesicles and bullae along Blaschko's lines, typically on the trunk, limbs and scalp. The lesions are often misdiagnosed as herpes simplex infection, bullous impetigo or epidermolysis bullosa until the characteristic distribution and eosinophilia are recognised. The eruption may be present at birth or appear within the first few weeks of life. There is often peripheral blood eosinophilia, and the skin biopsy shows eosinophilic spongiosis. Stage 1 usually resolves spontaneously within weeks to months.[4]
The vesicles may be filled with clear or haemorrhagic fluid, and the surrounding skin is often erythematous. Crusting and superficial erosions are common. The lesions are usually not painful but may be pruritic. Secondary bacterial infection can occur and should be treated. The scalp may be the first site affected, which can lead to confusion with scalp infection or epidermolysis bullosa simplex. [1]
Stage 2: Verrucous (approximately 4 to 6 months)
Stage 2 is characterised by wart-like (verrucous), hyperkeratotic papules and plaques arranged in linear streaks, especially on the limbs, hands, feet and digits. These lesions can be mistaken for epidermal naevi or common warts, but the history of preceding vesicles and the Blaschkoid distribution point to IP. Stage 2 also tends to resolve spontaneously, often leaving behind pigmented streaks. The verrucous stage can be the most persistent and can cause functional problems when lesions occur on the digits, flexures or around the nails. Localised treatment may be needed, but aggressive destructive therapy should be avoided because the lesions are self-limiting. [1]
Stage 3: Hyperpigmented (approximately 6 months to adolescence)
Stage 3 is the most recognisable stage. It consists of whorled, streaky, slate-grey to brown macules following Blaschko's lines, often described as a "marble-cake" pattern. The trunk, axillae and groin are commonly affected. This stage is produced by melanin incontinence: melanin drops from damaged basal keratinocytes into the upper dermis and is engulfed by melanophages. Stage 3 usually fades during adolescence, although pigmentary changes can persist in some areas. The pigment is often more prominent on the trunk and may be less obvious on the face and distal limbs. In darker skin phototypes, the hyperpigmented stage can be striking; in lighter skin, it may be subtle and overlooked. [1]
Stage 4: Atrophic/hypopigmented (adolescence to adulthood)
Stage 4 is the residual stage. It consists of pale, atrophic, hairless streaks along Blaschko's lines. In adult women, these streaks may be the only visible skin finding, and a careful history of neonatal blistering may be required to make the diagnosis. Vertex scarring alopecia is also common in this stage. The atrophic streaks are typically a few millimetres to a centimetre wide and can follow the classic S-shaped, V-shaped or whorled patterns of Blaschko's lines on the trunk and limbs. [1]
Extracutaneous features
- Dental (>90%) — the most consistent extracutaneous feature. Classic findings include peg-shaped or conical teeth, hypodontia or partial anodontia, delayed dentition, and malformed crowns. Dental anomalies can be the first clue in mild cases with subtle skin findings. The maxillary incisors are most commonly affected. Panoramic radiography often reveals the full extent of hypodontia and can show unerupted conical teeth. Early restorative care improves function, speech and appearance.[2]
- Ocular (~30-35%) — the most serious extracutaneous complication. Retinal vascular abnormalities are the core problem: the peripheral retina fails to vascularise normally, leading to avascular retina, neovascularisation, proliferative retinopathy, vitreous haemorrhage, traction and retinal detachment. Early laser photocoagulation or cryotherapy of the avascular retina can prevent blindness. Other ocular findings include strabismus, microphthalmia, cataracts, optic atrophy, blue sclerae and keratoconus. Retinal disease can be asymptomatic in the neonatal period, which is why screening must be performed before symptoms develop.[3]
- Neurological (~30%) — manifestations include seizures (infantile spasms, focal or generalised seizures), developmental delay, intellectual disability, motor delay, spastic hemiplegia or quadriplegia, and rarely stroke-like episodes from cerebral vasculopathy. Neurological injury is usually early and permanent. Infantile spasms can occur in the first year and may be mistaken for normal startle reflexes. Early recognition and treatment with appropriate anticonvulsants improves seizure control but does not reverse structural brain injury.[1]
- Hair and nails — patchy scarring alopecia at the vertex, coarse or woolly hair, and nail dystrophy (ridged, thickened, absent or dystrophic nails) may be seen.
- Breast and skeletal anomalies — supernumerary nipples, breast hypoplasia, skull asymmetry and delayed skeletal growth have been reported but are uncommon.
Atypical presentations
- Adult female with only stage 4 streaks — may be misdiagnosed as hypomelanosis of Ito or post-inflammatory change. A history of neonatal blistering, dental anomalies, ocular findings or family history clarifies the diagnosis.
- Affect male — should prompt consideration of Klinefelter syndrome (47,XXY) or postzygotic somatic mosaicism. Affected males may have a milder or patchy phenotype.
- Minimal skin disease, severe ocular/neurological disease — rare, but recognised; genetic testing is essential. [1]
Differential Diagnosis
The differential diagnosis of IP changes with the stage of skin disease. A careful history of the four-stage progression, the Blaschkoid distribution, eosinophilia and extracutaneous features usually distinguishes IP from mimics. It is important to remember that the same stage can occur in different anatomical areas at the same time, so an infant may have stage 1 vesicles on the trunk and stage 2 verrucous papules on the limbs simultaneously. [1]

Stage 1 vesicular mimics
- Herpes simplex infection — grouped vesicles on an erythematous base, may be disseminated in neonates. Positive PCR or direct fluorescent antibody from a vesicle, systemic illness, and lack of Blaschkoid distribution distinguish HSV. However, neonatal HSV must be excluded by testing in any febrile or unwell neonate with vesicles.
- Bullous impetigo — flaccid bullae, positive bacterial culture (Staphylococcus aureus), no eosinophilic spongiosis, no Blaschkoid pattern.
- Epidermolysis bullosa — mechanical or spontaneous blisters, often at sites of trauma; diagnosed by skin biopsy with immunofluorescence mapping or genetic testing of COL7A1, KRT5, KRT14, etc. No whorled hyperpigmentation.
- Linear IgA bullous dermatosis — "string of pearls" arrangement of vesicles, linear IgA deposition at the basement membrane on direct immunofluorescence, older age and drug triggers.
- Bullous pemphigoid — tense bullae, urticarial plaques, elderly patients, autoantibodies against BP180/BP230, subepidermal blister with eosinophils. Bullous pemphigoid is vanishingly rare in neonates but can occur. [1]
The most important clinical distinction is that Blaschkoid distribution + eosinophilia + eosinophilic spongiosis points to IP, whereas infection and autoimmune blistering disease have different distributions and histology. In practice, neonatal herpes simplex must be excluded in any unwell neonate with vesicles, even when IP is suspected, because untreated HSV is life-threatening. [1]
Stage 2 verrucous mimics
- Epidermal naevus — linear hyperkeratotic plaque present from birth or early childhood; does not progress through vesicular and hyperpigmented stages, no extracutaneous IP features.
- Verruca vulgaris — scattered hyperkeratotic papules, viral aetiology, no Blaschkoid systemic distribution.
- Nevus sebaceous — yellow-orange, waxy, hairless plaque on scalp, usually solitary, histology shows sebaceous glands without hair follicles. [1]
Stage 3 hyperpigmented mimics
- Linear and whorled nevoid hypermelanosis — streaky and whorled hyperpigmentation along Blaschko's lines, but no preceding vesicular or verrucous stages and no extracutaneous IP features.
- Dermatosis in a Blaschkoid distribution — lichen striatus, linear psoriasis, Blaschkoid lichen planus; these have characteristic histology and lack the four-stage progression. [1]
Stage 4 atrophic/hypopigmented mimics
- Hypomelanosis of Ito — streaky hypopigmentation along Blaschko's lines, often with neurological and musculoskeletal anomalies, but no preceding vesicular/verrucous stages and no IP-type dental anomalies. Chromosomal mosaicism is the underlying cause.
- Goltz syndrome (focal dermal hypoplasia) — X-linked dominant (male-lethal), linear atrophic lesions with fat herniation, osteopathia striata, papillomas, and dental anomalies. Differentiation is by morphology (fat herniation is pathognomonic) and genetic testing of PORCN. Goltz syndrome is caused by mutations in PORCN, a regulator of Wnt secretion, and shares X-linked dominant male lethality with IP, making it a particularly important differential in examinations.
- Atrophie lineaire — idiopathic linear atrophy; no other organ involvement and no preceding stages. [1]
Key discriminators for the examination
- Blaschkoid distribution + four-stage progression = IP
- Eosinophilic spongiosis + eosinophilia = stage 1 IP
- Peg-shaped teeth + retinal vasculopathy + Blaschkoid rash = IP
- Fat herniation in linear atrophic lesions = Goltz syndrome, not IP
Clinical and Bedside Assessment
The bedside examination should be systematic because IP is a multisystem disorder. Examiners may ask for a structured examination routine, and the following approach covers all essential domains. [1]
Skin
Map the distribution of lesions. Look for vesicles, bullae, erosions, verrucous papules, hyperpigmented whorls and atrophic streaks. Note whether lesions follow Blaschko's lines. Look for scarring alopecia at the vertex and nail dystrophy. Photographs are helpful for longitudinal comparison because the eruption evolves over months and years. [1]
Ocular examination
A red reflex examination should be performed in every newborn. Any infant with suspected IP requires urgent dilated indirect fundoscopy by an ophthalmologist experienced in retinopathy of prematurity or paediatric retinal disease. Look for avascular peripheral retina, neovascularisation, fibrovascular proliferation, retinal detachment, strabismus, microphthalmia and cataract. [1]
Dental examination
Inspect erupted teeth for peg-shaped or conical crowns, spacing from hypodontia, delayed eruption and malformed enamel. A panoramic radiograph is useful to count unerupted teeth and plan orthodontic or restorative treatment. [1]
Neurological examination
Assess gestational age-corrected developmental milestones, head circumference, tone, reflexes and vision. Ask specifically about seizures, infantile spasms, abnormal movements, feeding difficulty and motor asymmetry. [1]
Family history
Ask about recurrent male miscarriages, females with Blaschkoid rashes, females with dental anomalies, blindness, seizures, and any diagnosis of IP in the family. A three-generation pedigree is useful for genetic counselling. [1]
Investigations
The diagnosis of IP is usually clinical, supported by skin biopsy and confirmed by genetic testing. In the examination setting, questions often ask which investigation is most useful at each stage, or which combination of findings confirms the diagnosis. [1]
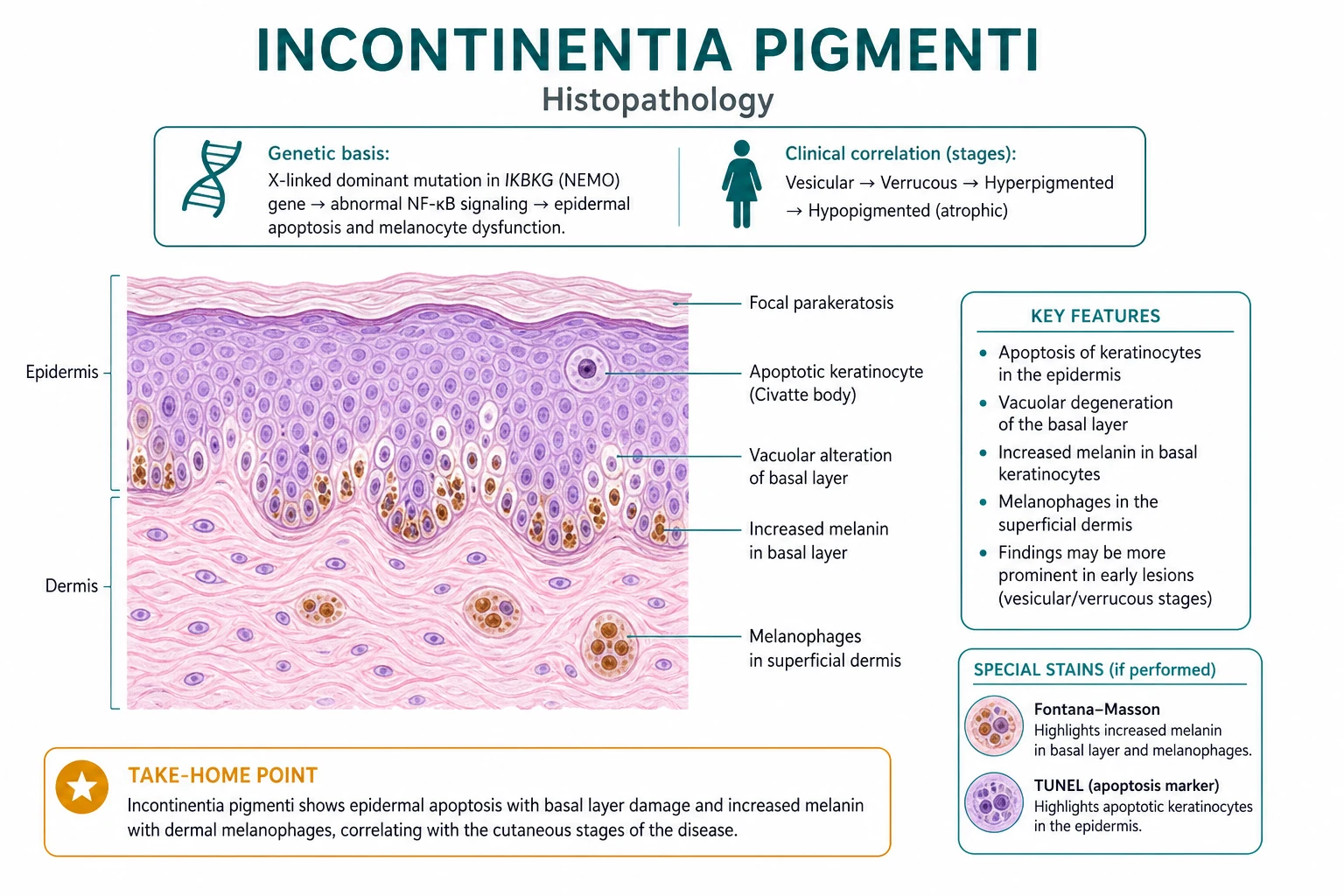
Skin biopsy with histopathology
Histology is stage-dependent:[4]
- Stage 1 (vesicular) — eosinophilic spongiosis: intraepidermal spongiosis with numerous eosinophils, dyskeratotic keratinocytes, and intraepidermal vesicle formation. This is the most pathognomonic finding. Inflammatory infiltrate may also include neutrophils and lymphocytes. The presence of eosinophils in the epidermis, rather than just the dermis, is the key distinguishing feature.
- Stage 2 (verrucous) — hyperkeratosis, papillomatosis, acanthosis and focal dyskeratosis. The histology resembles a viral wart or epidermal naevus, so the clinical history and Blaschkoid distribution are essential for diagnosis.
- Stage 3 (hyperpigmented) — melanin incontinence: melanophages in the upper dermis containing phagocytosed melanin, with decreased epidermal melanin. This is the lesion that gives the disease its name. Basal layer vacuolar degeneration may be seen, but the predominant finding is dermal pigment.
- Stage 4 (atrophic/hypopigmented) — epidermal atrophy, decreased or absent skin appendages, residual melanophages, and dermal fibrosis. The biopsy may be subtle, and the diagnosis at this stage often relies on clinical history and genetic testing. [1]
Laboratory tests
- Peripheral blood eosinophilia is common in stage 1 and may be marked; tissue eosinophilia is seen on biopsy.
- Complete blood count to document eosinophilia and screen for anaemia or infection. Eosinophil counts can be strikingly elevated, sometimes exceeding 5 x 10^9/L, and may correlate with the severity of the vesicular eruption.
- Serum immunoglobulins and lymphocyte subsets if recurrent infections suggest NEMO-related immunodeficiency overlap. [1]
Genetic testing
Genetic testing confirms the diagnosis and allows family counselling. The most common mutation is a deletion of exons 4-10 of IKBKG (the NEMO gene). Testing should include: [1]
- Sequence analysis of IKBKG for point mutations and small insertions/deletions. This detects mutations that are not large deletions.
- Deletion/duplication analysis for the common exon 4-10 deletion. This is the largest single mutation group and is the classic recurrent deletion caused by non-allelic homologous recombination between inverted repeats flanking the gene.
- Prenatal diagnosis for a known familial mutation when requested.
- X-inactivation studies are sometimes used in research but are not usually required for clinical diagnosis. They can be helpful in understanding variable expressivity in atypical families. [1]
Ophthalmic investigations
- Dilated indirect fundoscopy from birth, repeated regularly.
- Fluorescein angiography to define peripheral retinal avascular zones and neovascularisation.
- Optical coherence tomography (OCT) for macular or retinal structural detail.
- B-scan ultrasonography if media opacity prevents direct visualisation. [1]
Neurological investigations
- Brain MRI if there are seizures, developmental delay, focal neurological deficits, stroke-like episodes or abnormal head circumference. Findings may include cerebral atrophy, white-matter changes, vascular malformations or infarcts.
- Electroencephalography (EEG) if seizures are suspected.
- Developmental assessment by a paediatrician or neurodevelopmental team. [1]
Management — Acute Priorities
Most IP patients do not require acute resuscitation, but two situations demand urgent action. The first, retinal disease, is the most common and preventable cause of serious morbidity. The second, neurological crisis, is less common but must be recognised promptly. [1]
Urgent ophthalmology referral
Any newborn with suspected IP should have dilated indirect fundoscopy by an ophthalmologist as soon as possible after birth. Retinal vascular abnormalities can progress rapidly to retinal detachment and blindness, but this complication is preventable with early laser photocoagulation or cryotherapy of the avascular peripheral retina. The rationale for laser treatment is the same as in retinopathy of prematurity: ablation of the avascular peripheral retina removes the source of angiogenic drive and reduces the risk of tractional detachment. Ongoing surveillance is required until retinal vascularisation is complete, usually until 3 to 5 years of age.[3]
Parents should be warned that early laser treatment may be required on the first or second ophthalmology visit. If treatment is delayed, vitreoretinal surgery may become necessary, but visual outcomes are poor once tractional detachment has developed. The examination may ask why ophthalmology is the most urgent referral: the answer is that retinal detachment is silent in the neonatal period and preventable only if treated before it occurs. [1]
Seizures or altered consciousness
If a patient with IP presents with seizures, manage with standard paediatric or adult emergency protocols: airway protection, oxygen, glucose correction, and anticonvulsant therapy (for example, benzodiazepines for acute control, then levetiracetam or sodium valproate depending on age and seizure type). Arrange urgent brain MRI and neurological referral. [1]
Management — Definitive and Stepwise
There is no curative treatment for IP. Management is multidisciplinary, supportive and surveillance-based, coordinated by dermatology or paediatrics. The goal is to prevent preventable complications (especially blindness), manage active symptoms, and support development. Each specialty has a defined role. [1]
Dermatological management
- Stage 1 vesicular lesions — gentle wound care, emollients, and topical corticosteroids for inflammation. Mid-potency topical steroids such as mometasone furoate 0.1% cream or hydrocortisone 1% may be used for short courses on limited areas. Secondary bacterial infection should be treated with appropriate antibiotics; topical mupirocin 2% or oral flucloxacillin are common choices depending on severity. Avoid aggressive debridement. Systemic corticosteroids are rarely needed and should be reserved for severe, widespread inflammatory flares.
- Stage 2 verrucous lesions — emollients, topical keratolytics such as salicylic acid 3-6% or urea 10%, and sometimes gentle curettage if lesions are troublesome. Most resolve spontaneously. Oral retinoids are not standard and should be avoided in infants.
- Stage 3 hyperpigmentation — usually fades spontaneously; sunscreen and cosmetic camouflage if needed. Bleaching agents such as hydroquinone are generally not recommended in children and are unnecessary because the pigment resolves naturally.
- Stage 4 atrophic streaks — emollients, sun protection, and cosmetic camouflage. No treatment reverses the atrophy. Laser or surgical treatments are not standard and may worsen scarring.
- Nail and hair changes — supportive care; cosmetic measures for alopecia or nail dystrophy. Nail surgery is rarely indicated and may be complicated by poor healing. [1]
Ophthalmological management
- Dilated indirect fundoscopy at diagnosis, then repeated at intervals dictated by findings (often every 1-3 months initially, then every 6-12 months until vascular maturity). If the retina is fully vascularised and stable, follow-up can be spaced out; if avascular retina is present, surveillance continues until the peripheral retina is vascularised.
- Laser photocoagulation or cryotherapy for avascular peripheral retina to prevent retinal detachment. Laser is usually performed under general anaesthesia in neonates and infants. Cryotherapy is an alternative in some centres.
- Surgical repair if retinal detachment occurs, though visual prognosis is guarded once detachment has developed. Vitrectomy and scleral buckling may be required.
- Correction of strabismus, cataracts and other treatable ocular findings. Strabismus surgery should be planned after retinal disease is controlled. [1]
Dental management
- Paediatric dental referral when the first teeth erupt. Early assessment allows planning of space maintenance, orthodontics and restorative care.
- Restorative work for peg-shaped teeth, space maintainers or orthodontics for hypodontia, and implants or bridges in adulthood. The goal is to preserve function, speech and aesthetics.
- Feeding and speech support if dental anomalies are severe. [1]
Neurological management
- Anticonvulsants for seizures, chosen by seizure type and age. Infantile spasms may respond to vigabatrin or adrenocorticotropic hormone (ACTH); focal seizures may be treated with levetiracetam, sodium valproate or carbamazepine. The choice in infants must balance efficacy against toxicity and licensing.
- Developmental therapy (physiotherapy, occupational therapy, speech therapy) for motor or cognitive delay. Early intervention improves functional outcomes even when structural brain injury is fixed.
- Neurosurgical or stroke-unit input for cerebral infarction or vascular malformations. [1]
Genetic counselling
- Explain X-linked dominant inheritance with male lethality. Use a clear family tree if possible.
- An affected mother has a 50% risk of transmitting the mutation to each daughter (who will be affected) and each son (who will usually die in utero). Because male conceptuses are often lost, the family may show excess female offspring and a history of male miscarriages.
- Offer prenatal diagnosis if the familial mutation is known. Chorionic villus sampling at 11-14 weeks or amniocentesis at 15-18 weeks can identify the mutation. Sexing of the fetus is usually offered first, because male fetuses carrying the mutation are almost always affected lethally and may be managed differently.
- Screen at-risk female relatives for subtle skin, dental or ocular findings. Adult female relatives may not know they have the condition until examined carefully. [1]
IP management priorities
Specific Subtypes and Scenarios
Classic IP
Most patients have classic IP with variable expressivity of the four skin stages and extracutaneous features. The severity of skin disease does not always predict the severity of ocular or neurological disease, so every patient needs complete screening regardless of how mild the rash appears. A mother with minimal skin findings can still have a daughter with severe retinal disease, because the randomness of X-inactivation means that the phenotype cannot be predicted from the family history alone. [1]
IP-immunodeficiency overlap (NEMO-related)
Patients with hypomorphic NEMO mutations may present with anhidrotic ectodermal dysplasia with immunodeficiency (EDA-ID) rather than classic IP. Features include conical teeth, sparse hair, anhidrosis or hypohidrosis, and recurrent bacterial infections (especially Staphylococcus aureus, Streptococcus pneumoniae, Haemophilus influenzae and mycobacteria). Some patients have a combined phenotype with both IP skin disease and immunodeficiency, sometimes called IP-immunodeficiency overlap. Management requires immunology input, immunoglobulin replacement, prophylactic antibiotics and vaccination planning. Live vaccines are generally contraindicated until immune status is clarified. [1]
Affected males
Surviving males are rare. Possible explanations include: [1]
- Klinefelter syndrome (47,XXY) — the second X chromosome allows random X-inactivation and mosaicism similar to females. Such patients may show the full IP phenotype and need endocrine follow-up for hypogonadism, infertility, and the other long-term health implications of Klinefelter syndrome.
- Postzygotic somatic mosaicism — the mutation occurs after fertilisation, so only some tissues carry the mutation; the phenotype can be mild or patchy. If the germline is affected, the male can transmit the mutation to daughters. [1]
Familial recurrence risk
A mother with IP should be offered prenatal diagnosis because: [1]
- Each daughter has a 50% chance of being affected. The phenotype in the daughter will be independent of the mother's severity because of random X-inactivation.
- Each son has a 50% chance of inheriting the mutation and usually dying in utero. A male fetus that survives may have Klinefelter syndrome or mosaicism and should be evaluated carefully.
- A history of recurrent male miscarriages in the maternal lineage is a classic clue.
- Paternal gonadal mosaicism can transmit an apparently de novo mutation to multiple offspring from an unaffected father. [1]
Complications and Pitfalls
Retinal detachment and blindness
This is the most serious and most preventable complication. Failure to perform early dilated fundoscopy is the major pitfall. Once tractional retinal detachment occurs, visual recovery is poor. Laser photocoagulation of the avascular peripheral retina is the standard preventive treatment, analogous to treatment for retinopathy of prematurity. Parents should be educated about the importance of attending all ophthalmology appointments, even when the infant appears well.[3]
Neurological disability
Seizures, intellectual disability, motor delay and stroke-like cerebral infarcts can cause lifelong disability. Neurological injury is usually established early, so early identification and rehabilitation are important. [1]
Dental complications
Severe hypodontia and peg-shaped teeth can cause malocclusion, speech difficulties, feeding problems and psychosocial distress. Early dental referral and orthodontic planning improve outcomes. [1]
Diagnostic pitfalls
- Treating neonatal IP as herpes simplex — vesicular IP can be misdiagnosed and treated with acyclovir, delaying the correct diagnosis. Always test for HSV if the clinical picture is uncertain, but recognise that Blaschkoid distribution + eosinophilia strongly favours IP.
- Missing adult IP — stage 4 streaks alone may be dismissed as hypomelanosis of Ito or post-inflammatory change. A history of neonatal blistering, dental anomalies, ocular findings or family history should raise suspicion.
- Assuming mild skin disease means mild systemic disease — severe retinal or neurological disease can occur in patients with minimal skin findings. [1]
Prognosis and Disposition
The prognosis of IP depends mainly on the presence and severity of ocular and neurological involvement. With modern multidisciplinary care, most patients have a normal lifespan. Quality of life is influenced by cosmetic appearance, visual outcome, neurological disability, and the burden of long-term follow-up. [1]
- Cutaneous disease — all four stages usually resolve; stage 4 atrophic streaks and vertex alopecia are the lifelong residual signs. The skin disease does not predispose to skin cancer.
- Ocular disease — early detection and laser treatment can preserve vision; retinal detachment carries a poor visual prognosis. Lifelong ophthalmology follow-up is often recommended, although the frequency decreases once the retina is fully vascularised and stable.
- Neurological disease — established deficits are usually permanent, but early developmental intervention improves functional outcomes. Seizures can usually be controlled with anticonvulsants.
- Dental disease — manageable with modern restorative and orthodontic care. Hypodontia may require implants or bridges in adulthood.
- Reproductive counselling — allows informed family planning and prenatal diagnosis. Many affected women have healthy children with appropriate genetic counselling. [1]
Most patients can be managed as outpatients with coordinated multispecialty follow-up. Newborns require urgent ophthalmology assessment and often a paediatric neurology baseline review. Disposition planning should include a clear follow-up schedule, genetic counselling referral, and written instructions for parents about red flags (new seizures, visual changes, infection of skin lesions). [1]
Special Populations
Neonates
Neonatal IP presents with vesicles and bullae. The priorities are: [1]
- Exclude serious infection (herpes simplex, bacterial sepsis) when clinically indicated. This is important because neonatal HSV is an emergency, but it should not delay recognition of IP if the clinical picture is classic.
- Perform urgent ophthalmology referral within days of birth.
- Obtain dermatology review and skin biopsy.
- Arrange genetic testing. [1]
Parents need clear education about the four stages, the variable expressivity, and the importance of screening. They should be given a written plan for follow-up appointments. [1]
Pregnancy and family planning
An affected mother has a 50% risk for each pregnancy. Prenatal diagnosis by chorionic villus sampling at 11-14 weeks or amniocentesis at 15-18 weeks is available when the familial mutation is known. Non-invasive prenatal testing for fetal sex and, in some jurisdictions, for the specific mutation may be offered. Fathers with gonadal mosaicism can transmit the disease even if clinically unaffected, so the family history should be interpreted carefully. Recurrent affected offspring from an unaffected father strongly suggest paternal mosaicism. [1]
Rare affected males
Males with Klinefelter syndrome or mosaicism should be managed with the same multidisciplinary approach, with added endocrine and fertility counselling if Klinefelter is present. Immunodeficiency should be excluded if infections are recurrent. [1]
Immunocompromised or immunodeficiency overlap
Patients with NEMO-related immunodeficiency require immunology follow-up, infection surveillance, prophylactic antibiotics and consideration of immunoglobulin replacement. Live vaccines may be contraindicated depending on immune status. [1]
Evidence, Guidelines and Regional Differences
Management of IP is based on expert consensus and observational cohorts because the condition is too rare for large randomised trials. The following principles are widely accepted and are reflected in major international resources: [1]
- Early ophthalmology screening is the highest-yield intervention. All newborns with suspected IP should have a dilated fundoscopic examination as soon as possible after birth. This recommendation is consistent across GeneReviews, NORD, the British Association of Dermatologists and the American Academy of Dermatology.[3]
- Genetic testing with IKBKG/NEMO analysis is recommended to confirm the diagnosis and enable family counselling. The common exon 4-10 deletion should be tested by deletion/duplication analysis, not just sequencing.[5]
- Multidisciplinary care involving dermatology, ophthalmology, paediatric dentistry, neurology and genetics is standard of care.
- Regional differences relate mainly to the timing of the first ophthalmology examination and the threshold for laser treatment. Some centres perform fluorescein angiography in neonates and treat even small avascular zones aggressively, while others rely on serial indirect ophthalmoscopy until peripheral retinal vascularisation is documented. All agree that early detection is essential. In resource-limited settings, the first examination may be delayed; telemedicine and retinal imaging may help bridge gaps, but direct examination by an experienced ophthalmologist remains the gold standard.
- No disease-modifying therapy exists. Anti-inflammatory or anti-apoptotic agents targeting the NF-kappaB pathway remain experimental and are not standard of care.
Exam Pearls
[1]Exam application bank (NEET-PG / INICET)
One-line answer
Incontinentia pigmenti (IP; Bloch-Sulzberger syndrome) is an X-linked dominant genodermatosis caused by mutation in the IKBKG/NEMO gene (Xq28). It is lethal in most hemizygous males and almost exclusively affects females. The hallmark is a 4-stage Blaschkoid cutaneous eruption (vesicular, verrucous, hyperpigmented, atrophic/hypopigmented) with extracutaneous involvement of teeth, eyes, CNS, hair and nails.
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard. [1]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes. [1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change. [1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each. [1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory. [1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Incontinentia Pigmenti.
[1]References
- [1]Rosser T. Incontinentia pigmenti Semin Pediatr Neurol, 2024.PMID 39389657
- [2]Cammarata-Scalisi F, Fusco F, Ursini MV. Incontinentia Pigmenti Actas Dermosifiliogr (Engl Ed), 2019.PMID 30660327
- [3]Islam YFK, Khurshid SG. Incontinentia pigmenti and the eye Curr Opin Ophthalmol, 2022.PMID 35819905
- [4]Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti An Bras Dermatol, 2014.PMID 24626645
- [5]Scheuerle AE, Ursini MV. Incontinentia Pigmenti 1993.PMID 20301645