Dermatology · General Medicine
Pemphigus Vulgaris & Bullous Pemphigoid
Also known as Pemphigus vulgaris · Bullous pemphigoid · Pemphigus · Pemphigoid · Autoimmune blistering disease · Mucous membrane pemphigoid · Pemphigoid gestationis
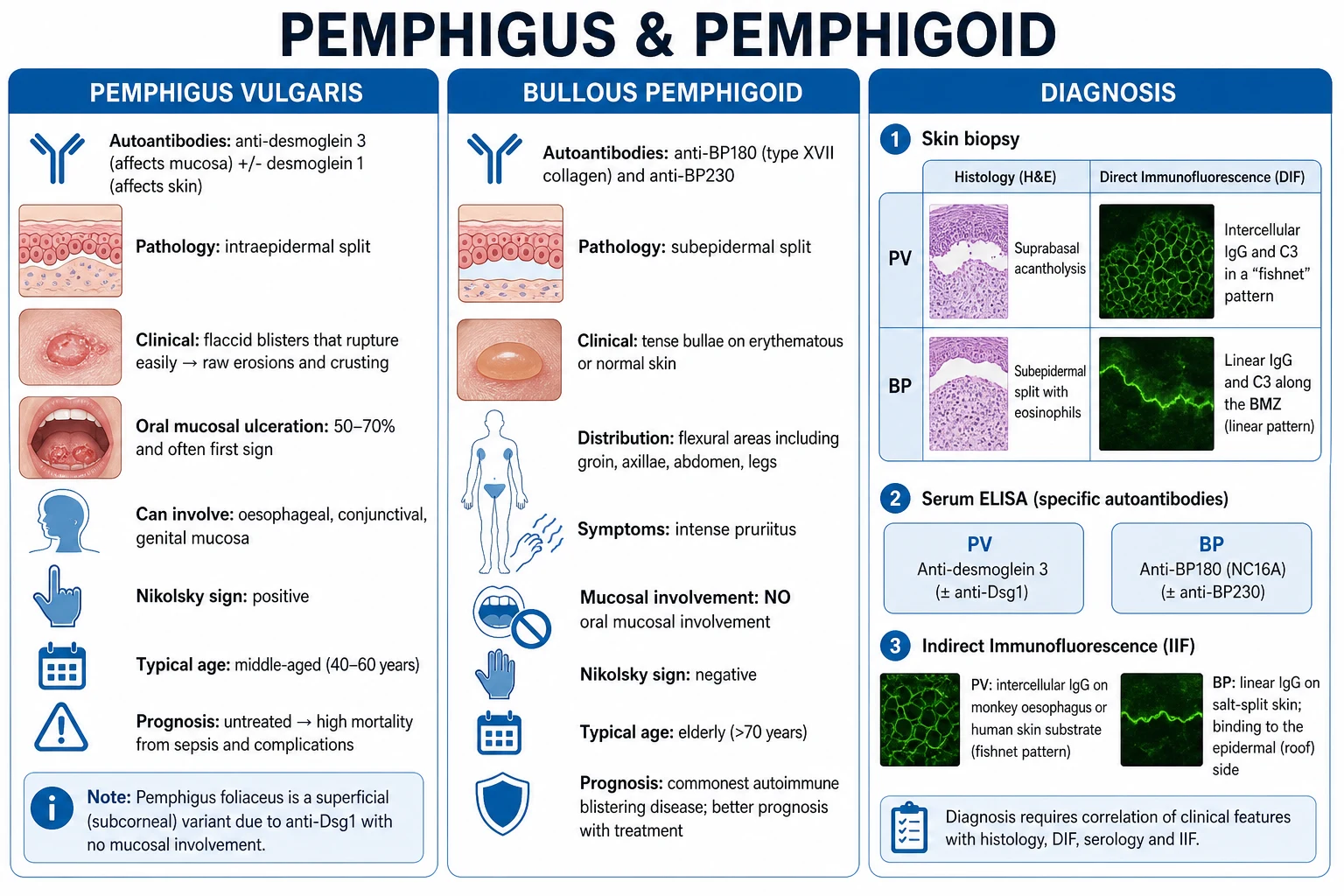
Pemphigus vulgaris (PV) and bullous pemphigoid (BP) are the two archetype autoimmune blistering diseases of the skin and mucosa, distinguished by the level of the split and the target antigen. PV: pathogenic IgG against desmoglein 3 (Dsg3, mucosal) ± desmoglein 1 (Dsg1, cutaneous) → suprabasal intraepidermal acantholysis → flaccid blisters, raw erosions, oral ulceration, positive Nikolsky sign; affects middle-aged adults (40 to 60), Mediterranean/Jewish/Indian predilection; potentially fatal without immunosuppression. BP (commonest autoimmune blistering disease overall, and especially in the elderly): IgG against BP180 (BPAG2) and BP230 (BPAG1) of the hemidesmosome → subepidermal split → tense bullae on flexural skin, pruritic, oral mucosa spared, Nikolsky negative. Diagnosis rests on a triad: skin biopsy H&E + peri-lesional direct immunofluorescence + serum ELISA. DIF: PV = intercellular fishnet IgG/C3; BP = linear BMZ IgG/C3. Salt-split skin: BP binds roof, EBA binds floor. Treatment revolutionised by rituximab — now first-line for moderate-severe PV (PEMPHIX) and refractory BP — and by doxycycline + potent topical steroid as the safer first-line BP strategy (BLISTER).
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags
Overview & Definition
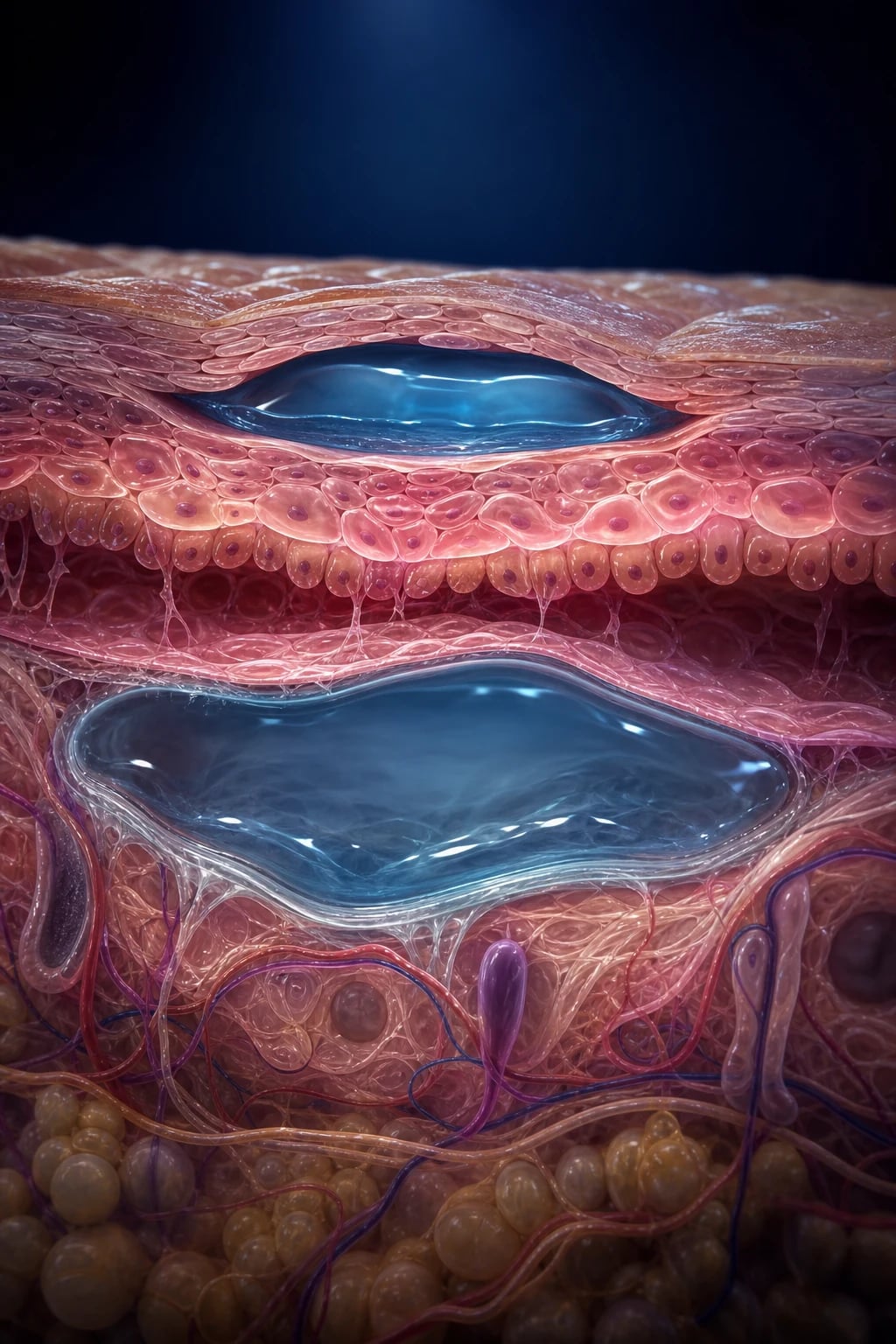
The pemphigus and pemphigoid group together constitute the archetype autoimmune blistering diseases of the skin and mucous membranes. They are unified by a single pathogenic mechanism — autoantibodies directed against structural proteins of the skin, producing a clinically visible blister — and they are distinguished from one another by two axes that the candidate must master: the anatomical level of the split (intraepidermal vs subepidermal) and the target antigen (desmogleins in pemphigus; hemidesmosomal antigens BP180 and BP230 in pemphigoid). These two axes in turn dictate the clinical picture, the immunofluorescence pattern, the natural history, and the treatment ladder.[7]
Pemphigus (from the Greek pemphix, "blister") is defined by intraepidermal split caused by IgG autoantibodies against the cadherin-family desmosomal proteins desmoglein 1 and desmoglein 3, producing acantholysis (loss of keratinocyte-to-keratinocyte adhesion). The blisters are flaccid, lie high in the epidermis (so the roof is thin and ruptures easily), and the resulting raw erosions are extensive. Mucosal involvement is prominent, because mucosal epithelium expresses desmoglein 3 but not desmoglein 1 (see Pathophysiology). Untreated pemphigus is uniformly fatal within years — historically a death sentence, now treatable but still carrying a 5 to 10 percent mortality from complications of the disease and its treatment.[1][7]
Pemphigoid ("resembling pemphigus") is defined by a subepidermal split — the blister floor is the intact basal lamina and the roof is the full-thickness epidermis, producing a thick-walled, tense, long-lasting bulla. The autoantibodies target the hemidesmosomal proteins BP180 (BPAG2, a transmembrane collagen) and BP230 (BPAG1, a cytoplasmic plakin) of the dermoepidermal junction. Because the split is beneath the entire epidermis, mucosa is typically spared and the clinical course is more indolent. Bullous pemphigoid is the single commonest autoimmune blistering disease overall, and is the dominant blistering disorder of the elderly.[2][5]
The clinical skill, the viva answer, and the MCQ discriminator all reduce to a single pivot — recognising the blister type and confirming it with biopsy and immunofluorescence, then treating promptly with an immunosuppressive ladder in which rituximab has moved from salvage to first-line for moderate-severe pemphigus vulgaris (PEMPHIX, 2017) and is now widely used in refractory pemphigoid. The single highest-yield laboratory pearl of the entire chapter: on salt-split skin, BP antibodies bind the epidermal roof while epidermolysis bullosa acquisita antibodies bind the dermal floor.[3][4]
Classification
The two broad families subdivide into clinically important variants. Both axes must be remembered because they shift the differential, the immunofluorescence pattern, and the treatment ladder. [1]
The pemphigus group
- Pemphigus vulgaris (PV) — the commonest and most severe form; IgG against Dsg3 (mucosal-dominant type) or Dsg3 + Dsg1 (mucocutaneous type). Accounts for approximately 70 percent of all pemphigus. Includes the variant pemphigus vegetans, in which vegetating, fungoid masses form in intertriginous areas (Hallopeau and Neumann subtypes).
- Pemphigus foliaceus (PF) — superficial, IgG against Dsg1 only; subcorneal split; spares mucosa entirely (Dsg3 is intact and compensates in mucosa); presents with scaly, crusted erosions on seborrhoeic areas (face, scalp, upper trunk). Includes the endemic fogo selvagem (Brazilian PF, linked to black fly exposure and a putative environmental antigen).
- Pemphigus erythematosus (Senear-Usher) — localised PF with lupus-like features; anti-Dsg1 plus ANA; centromedial facial seborrhoeic rash.
- Paraneoplastic pemphigus (PNP) — IgG against desmoplakin, envoplakin, periplakin, BP230, and Dsg3; non-healing painful mucositis, polymorphous cutaneous lesions, and underlying non-Hodgkin lymphoma, chronic lymphocytic leukaemia, Castleman disease, or other neoplasms. Stormy course, high mortality.
- Drug-induced pemphigus — thiol drugs (captopril, penicillamine) are the classic cause (direct biochemical disruption of desmogleins); non-thiol drugs (rifampicin, NSAIDs, cephalosporins, carbamazepine, penicillins) cause immune-mediated pemphigus, usually foliaceus-like.
- IgA pemphigus — IgA against desmocollin 1/2; subcorneal pustular or intraepidermal neutrophilic pattern; responds dramatically to dapsone. [1]
The pemphigoid group
- Bullous pemphigoid (BP) — the prototype; elderly; tense pruritic flexural bullae; anti-BP180/BP230; subepidermal with eosinophils.
- Mucous membrane pemphigoid (MMP, formerly cicatricial pemphigoid) — predominantly mucosal (oral, ocular, laryngeal, genital); scarring is the hallmark; conjunctival scarring causes symblepharon, synechiae, and blindness; anti-BP180 (especially the C-terminal domain), laminin-332, or β4 integrin; urgent ophthalmology referral is non-negotiable.
- Pemphigoid gestationis (PG, herpes gestationis) — pregnancy or immediate postpartum; intensely pruritic urticarial plaques starting periumbilically, progressing to vesicles and bullae; anti-BP180 NC16A (same target as BP, but the IgG is complement-fixing and the inflammatory response is amplified in pregnancy).
- Linear IgA bullous dermatosis (LABD) — linear IgA along the BMZ; string-of-pearls or cluster-of-jewels annular pattern; both childhood (chronic bullous disease of childhood) and adult forms; vancomycin is the classic drug trigger; responds to dapsone.
- Epidermolysis bullosa acquisita (EBA) — IgG against type VII collagen (anchoring fibrils); trauma-prone sites (hands, feet, elbows, knees), milia and scarring are hallmarks; salt-split binding to the dermal floor is the discriminator from BP.
- Anti-p200 pemphigoid — IgG against laminin γ1; clinical overlap with BP and EBA. [1]
Epidemiology & Risk Factors
Pemphigus vulgaris is rare (annual incidence 0.1 to 0.5 per 100,000 in most populations, up to 3 per 100,000 in endemic regions) but has striking ethnic and geographic predilection: it is markedly more common in Mediterranean, Ashkenazi Jewish, and Indian/South Asian populations, and in endemic Brazil (fogo selvagem). The peak age is 40 to 60 years, with women marginally over-represented. HLA-DRB1*0402 and HLA-DQB1*0503 are strongly associated, supporting a T-cell-driven, MHC-II-restricted autoimmune mechanism.[7]
Bullous pemphigoid is the commonest autoimmune blistering disease in the developed world (annual incidence 20 to 40 per 100,000 in those older than 80 years) and its incidence is rising as the population ages. It affects the elderly (median age 75 to 80), with a slight male predominance. A striking and exam-relevant association is with neurological disease: patients with dementia, Parkinson disease, multiple sclerosis, stroke, and epilepsy have a significantly elevated risk of BP — thought to reflect cross-reactivity between BP180 isoforms expressed in brain and skin. Other associations include psoriasis, chronic lymphocytic leukaemia, UV radiation, radiotherapy, and certain drugs (loop diuretics, PD-1/PD-L1 checkpoint inhibitors, spironolactone).[5]
Mucous membrane pemphigoid is rare (1 to 2 per million per year), affects postmenopausal women more than men, and is dominated by its ocular complications. Pemphigoid gestationis complicates approximately 1 in 10,000 to 1 in 50,000 pregnancies (in the second or third trimester or immediately postpartum), recurs in subsequent pregnancies, and can occur with molar pregnancy or choriocarcinoma.[9]
Drug-induced pemphigus risk: captopril, penicillamine (thiol drugs — direct biochemical effect, may remit on withdrawal), rifampicin, NSAIDs, cephalosporins, penicillins, carbamazepine (non-thiol, immune-mediated). Drug-induced BP has been linked to loop diuretics, aldosterone antagonists, DPP-4 inhibitors, and PD-1/PD-L1 checkpoint inhibitors. [1]
Pathophysiology
Pemphigus vulgaris — the desmoglein compensation hypothesis
The molecular mechanism of PV is the textbook exemplar of how an autoantibody causes disease by direct loss of cell adhesion. Pathogenic IgG (predominantly IgG4 subclass, with IgG1 contributing) binds the extracellular domain of desmoglein 3 and/or desmoglein 1 — transmembrane cadherins that mediate adhesion at desmosomes. Binding has at least three downstream consequences: (1) steric hindrance — the antibody physically blocks desmoglein trans-interaction; (2) intracellular signalling — p38MAPK, Rho-GTPase, c-Myc, and EGFR cascades are activated, causing basal cell shrinkage and reorganisation; (3) complement activation and plasminogen-plasmin cascade amplifies the split. The result is suprabasal acantholysis: the keratinocytes of the lower spinous layer detach from one another but remain tethered to the basement membrane, producing the pathognomonic "row of tombstones" appearance with a single layer of basal cells lining the blister floor.[7][8]
The desmoglein compensation hypothesis explains why Dsg3 antibodies alone cause mucosal disease but Dsg3 + Dsg1 antibodies cause mucocutaneous disease. Dsg3 is expressed in the basal and immediate suprabasal layers of both skin and mucosa; Dsg1 is expressed throughout the upper epidermis in skin but only superficially in mucosa. A patient with anti-Dsg3 alone has lost adhesion in the lower layer of mucosa (Dsg1 cannot compensate in mucosa because it is not expressed there in sufficient quantity), but in skin the upper Dsg1 holds the epidermis together — hence mucosal-dominant PV. Adding anti-Dsg1 strips compensation and produces mucocutaneous PV. Pemphigus foliaceus (anti-Dsg1 only) spares mucosa because Dsg3 compensates there, and produces only superficial (subcorneal) skin lesions because the upper epidermis depends on Dsg1 alone.[8]
Bullous pemphigoid — the subepidermal split
BP autoantibodies target two hemidesmosomal proteins: BP180 (BPAG2, type XVII collagen) — a transmembrane collagen whose non-collagenous NC16A ectodomain is the dominant antibody target — and BP230 (BPAG1) — a cytoplasmic plakin that anchors keratin intermediate filaments to the hemidesmosome. Binding triggers complement activation (C5a → eosinophil and neutrophil chemotaxis), mast-cell degranulation, and release of matrix metalloproteinases (MMP-9, MMP-2) and elastase that cleave BP180, severing the hemidesmosome and producing a subepidermal blister filled with eosinophils. The split lies beneath the entire epidermis, so the roof is thick (the full viable epidermis), producing a tense, durable bulla that lasts for days — and the oral mucosa is spared because the split is beneath, not within, the mucosa.[5]
The tense bulla of BP and the flaccid blister of PV are the direct mechanical consequence of the split level: an intraepidermal split has a thin, friable roof that ruptures at minor trauma (PV), whereas a subepidermal split has a thick, robust roof (BP). The Nikolsky sign follows the same logic — only when the split is high in the epidermis does lateral pressure on apparently normal skin shear off the superficial layer. [1]
Pemphigoid gestationis
In PG the antibody targets the same NC16A domain of BP180 as in BP, but is typically an IgG1 subclass that fixes complement robustly, and the hormonal-immune milieu of pregnancy amplifies inflammation. Placental expression of MHC-II molecules (shared with the BP180 ectodomain) is thought to be the trigger; the disease frequently flares at delivery and recurs, often earlier and more severely, in subsequent pregnancies.[9]
Clinical Presentation
Pemphigus vulgaris
The classical PV patient is a middle-aged Mediterranean/Jewish/Indian individual presenting with oral erosions that precede cutaneous blisters by weeks to months. Oral involvement occurs in 50 to 70 percent at onset and ultimately in 90 percent, making isolated oral disease a common first presentation — often misattributed to aphthous ulcers, oral candidiasis, or poorly fitting dentures.[1][2]
- Mucosal lesions: painful, shallow, irregular erosions of the buccal mucosa, palate, gingiva (desquamative gingivitis), lips, tongue, oropharynx, larynx (hoarseness), conjunctiva, oesophagus (dysphagia), and genital mucosa. They rarely form intact blisters because the thin intraepidermal roof is destroyed by minor friction. Desquamative gingivitis — diffuse erythema and sloughing of the attached gingiva — is a common and easily missed pattern.
- Cutaneous lesions: flaccid, flabby blisters arising on normal or erythematous skin, rupturing within hours to leave large, raw, weeping erosions that crust and extend. The blisters spread laterally with pressure (Asboe-Hansen sign). Any site may be involved but the scalp, face, chest, axillae, and groins predominate. Nikolsky sign positive.
- Atypical presentations: isolated oral disease; vulvar or penile erosions mistaken for sexually transmitted infection; nail involvement (onychodystrophy, paronychia); ocular PV mimicking conjunctivitis. [1]
Bullous pemphigoid
The BP patient is elderly (typically over 70), often with comorbid neurological disease, presenting with intense pruritus weeks to months before blisters appear. A prodromal phase of eczematous, urticarial, or excoriated plaques is common and frequently misdiagnosed as eczema or scabies. [1]
- Cutaneous lesions: tense bullae (1 to 3 cm) on normal or erythematous skin, symmetrically distributed on flexural surfaces — inner thighs, groins, axillae, lower abdomen, flexor forearms, and lower legs. The blisters do not rupture easily and may contain clear, haemorrhagic, or purulent fluid. Pruritus is intense. Crusts and erosions follow rupture, but milia are uncommon (unlike EBA). Nikolsky sign negative.
- Mucosal involvement is uncommon (10 to 30 percent, mild, transient, non-scarring) — distinguishing BP from MMP. The oral mucosa is typically spared — a high-yield discriminator.
- Atypical variants: localised BP (lower legs in women, pretibial), vesicular BP (mimicking DH), nodular/prurigo-like BP, paediatric BP, mucosal-dominant BP (overlap with MMP). [1]
Mucous membrane pemphigoid
MMP presents predominantly with mucosal lesions, with scarring as the defining feature. Oral involvement (90 percent) is most common — desquamative gingivitis is the single most frequent sign, with blistering and erosions of the palate and buccal mucosa. Ocular involvement (50 to 70 percent) is the feared complication: chronic conjunctivitis, subepithelial fibrosis, shortening of the fornices, symblepharon (adhesion of lid to globe), synechiae, trichiasis, corneal abrasion, and progressive blindness — often bilateral and irreversible. Laryngeal and oesophageal involvement can cause stridor, stenosis, and dysphagia. Cutaneous lesions (25 percent) are scattered tense bullae that heal with scarring and milia. Skin Nikolsky is negative. [1]
Pemphigoid gestationis
PG classically begins in the second or third trimester (less often immediately postpartum) with intense pruritus and urticarial plaques classically beginning in the periumbilical area, often with periumbilical targetoid lesions. The eruption spreads to the trunk, buttocks, and proximal extremities, evolving into tense vesicles and bullae. Mucosa is spared. It typically remits spontaneously weeks to months postpartum, but flares at delivery are common and it recurs earlier and more severely in subsequent pregnancies. The neonate may develop transient blistering from transplacental antibody.[9]
Pemphigus foliaceus
PF produces superficial, scaly, crusted erosions on seborrhoeic areas (scalp, face, upper chest, back) with "cornflake" scale and no mucosal involvement. Patients rarely demonstrate intact blisters because the split is subcorneal and the roof is the thin stratum corneum. [1]
Exam application bank (NEET-PG / INICET)
One-line answer
Pemphigus vulgaris (PV) and bullous pemphigoid (BP) are the two archetype autoimmune blistering diseases of the skin and mucosa, distinguished by the level of the split and the target antigen. PV: pathogenic IgG against desmoglein 3 (Dsg3, mucosal) ± desmoglein 1 (Dsg1, cutaneous) → suprabasal intraepidermal acantholysis → flaccid blisters, raw erosions, oral ulceration, positive Nikolsky sign; affects middle-aged adults (40 to 60), Mediterranean/Jewish/Indian predilection; potentially fatal without immunosuppression. BP (commonest autoimmune blistering disease overall, and especially in the elderly): IgG against BP180 (BPAG2) and BP230 (BPAG1) of the hemidesmosome → subepidermal split → tense bullae on flexural skin, pruritic, oral mucosa spared, Nikolsky negative. Diagnosis rests on a triad: skin biopsy H&E + peri-lesional direct immunofluorescence + serum ELISA. DIF: PV = intercellula
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard. [1]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes. [1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change. [1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each. [1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory. [1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Pemphigus Vulgaris & Bullous Pemphigoid.
Differential Diagnosis
The differential of an autoimmune blistering disease is wide, because blistering is a final common pathway for many insults. The high-yield strategy is to triage by blister type (flaccid vs tense), by mucosal involvement, and by Nikolsky sign, then to confirm with DIF and salt-split IIF. [1]
PV or BP — the four-question discriminator
BLISTER
The complete differential also includes aphthous ulcers, erythema multiforme, oral lichen planus, oral candidiasis, herpes simplex, acute contact dermatitis, porphyria cutanea tarda, bullous diabeticorum, bullous tinea, and insect bites. In any blistering disorder, the single diagnostic step is skin biopsy for H&E plus peri-lesional biopsy for DIF; serology (ELISA, IIF) confirms. [1]
Clinical & Bedside Assessment
A structured bedside examination of a patient with suspected autoimmune blistering disease serves two purposes — to recognise the blister type and its distribution, and to triage severity (extensive skin loss, mucosal involvement, airway compromise). [1]
Step 1 — recognise the blister. Document the morphology (flaccid vs tense; intact vs ruptured; vesicle, bulla, pustule, erosion, crust, scale), distribution (flexural, seborrhoeic, acral, mucosal, photo-distributed), and the number and size of lesions. Examine the scalp, postauricular skin, axillae, groins, and the entire oral mucosa (buccal, palate, gingiva, tongue, oropharynx). Evert the eyelids to inspect the conjunctiva (MMP). Photograph with a ruler in frame. [1]
Step 2 — elicit the Nikolsky sign. Apply firm lateral (sliding) pressure with a thumb or cotton-tipped applicator to apparently normal skin 1 to 2 cm from an active lesion. Positive Nikolsky = the superficial epidermis shears off, leaving a moist, glistening erosion. A positive Nikolsky indicates an intraepidermal split and is the single most useful bedside discriminator for pemphigus vulgaris (also positive in TEN, staphylococcal scalded skin syndrome, and severe contact dermatitis — never diagnostic alone). [1]
Step 3 — elicit the Asboe-Hansen sign (Nikolsky's indirect sign). Apply vertical pressure to the intact roof of an existing blister. Positive Asboe-Hansen = the blister extends laterally onto adjacent, previously unblistered skin. Specific for pemphigus; negative in BP. [1]
Step 4 — Tzanck smear. Gently scrape the floor of a fresh blister with a scalpel blade, smear on a glass slide, fix, and stain with Giemsa, Wright, or H&E. Acantholytic Tzanck cells — large, rounded keratinocytes with enlarged hyperchromatic nuclei and a perinuclear halo — are characteristic of pemphigus, herpesvirus infection, and Darier disease. The Tzanck smear is rapid and cheap but does NOT distinguish pemphigus from HSV (hence the dictum "Tzanck is a cytology, not a diagnosis"). It has been largely supplanted by biopsy + DIF but remains a viva favourite. [1]
Step 5 — screen for systemic associations and paraneoplastic disease. Examine lymph nodes, liver, spleen (for paraneoplastic pemphigus with lymphoma/CLL/Castleman); examine for neurological disease in suspected BP (dementia, Parkinson, stroke); assess nutritional status (extensive oral erosions cause malnutrition); check temperature, pulse, BP, and urine output for systemic deterioration. [1]
Step 6 — grade severity. The Pemphigus Disease Area Index (PDAI) grades activity in skin, scalp, mucosa, and damage; the Autoimmune Bullous Skin Disorder Intensity Score (ABSIS) incorporates body surface area and oral involvement. These scores guide treatment intensity and are research endpoints in the pemphigus trials.[3]
Investigations
The diagnosis rests on a triad of tests performed together — never on a single test alone: [1]
- Skin biopsy for H&E — from the edge of a fresh blister (including intact blister and adjacent perilesional skin). 4 mm punch, in formalin.
- Peri-lesional biopsy for direct immunofluorescence (DIF) — 4 mm punch from normal-appearing skin immediately adjacent to an active lesion (the perilesional skin, not the blister itself, which is often false-negative due to degradation of immunoreactants), in Michel's medium or Zeus transport (NOT formalin).
- Serum ELISA and/or indirect immunofluorescence (IIF) — to detect circulating antibody and quantify titre. [1]
Histology (H&E)
- PV: suprabasal intraepidermal split with a single row of basal keratinocytes adherent to the basement membrane — the "row of tombstones" — and free-floating rounded acantholytic cells (Tzanck cells) within the blister cavity. The split is high in the epidermis.
- BP: subepidermal blister with no acantholysis, typically filled with eosinophils (and neutrophils). The intact epidermis forms the roof.
- MMP: subepidermal split with mixed inflammatory infiltrate; subepithelial fibrosis in chronic lesions.
- PF: subcorneal split (very superficial) with neutrophilic or eosinophilic infiltrate.
- DH: neutrophilic microabscesses at the tips of dermal papillae. [1]
Direct immunofluorescence (DIF) — the diagnostic centrepiece
DIF on perilesional skin is the single highest-yield test and the most commonly examined. [1]
DIF patterns — the 'fishnet vs line' discriminator
PATTERNS
- PV: intercellular fishnet (honeycomb) IgG ± C3 throughout the epidermis and mucosal epithelium.
- BP: continuous linear IgG and/or C3 along the basement membrane zone (BMZ).
- MMP: identical to BP — linear IgG/C3 along the BMZ (BP180 is the dominant target; laminin-332 and β4 integrin in some subtypes).
- PG: identical to BP — linear C3 (and often IgG) along the BMZ.
- LABD: linear IgA along the BMZ.
- DH: granular IgA at the tips of dermal papillae.
- EBA: linear IgG along the BMZ — only distinguished from BP by salt-split binding pattern. [1]
Indirect immunofluorescence (IIF) and salt-split skin
IIF detects circulating antibody in the patient's serum applied to a substrate. PV is best detected on monkey oesophagus or guinea pig oesophagus (intercellular staining); BP is best detected on human skin split through the lamina lucida (salt-split skin), where IgG binds the epidermal roof. EBA antibody binds the dermal floor — the highest-yield laboratory pearl in the chapter. MMP may show binding to both roof and floor or to the floor alone (laminin-332 MMP).[7]
ELISA — quantification and monitoring
- PV: anti-Dsg3 and anti-Dsg1 ELISA — titres correlate with disease activity and predict relapse. A rising anti-Dsg3 titre heralds a flare, prompting escalation before clinical deterioration.
- BP: anti-BP180 NC16A ELISA — titres correlate with disease activity and survival; anti-BP230 ELISA is supportive.
- PG: anti-BP180 NC16A, identical antigen to BP.
- MMP: anti-BP180 (often C-terminal), anti-laminin-332, anti-α6β4 integrin. [1]
Adjunct investigations
- FBC, ESR, CRP, U&E, LFTs, albumin — baseline before immunosuppression; track eosinophilia (BP).
- TPMT activity before azathioprine (low activity → myelosuppression).
- G6PD level before dapsone (risk of haemolysis).
- Hepatitis B, hepatitis C, HIV, QuantiFERON-TB before rituximab (reactivation risk).
- Serum IgG and immunoglobulins before and during rituximab (hypogammaglobulinaemia risk).
- DEXA scan, HbA1c, lipid profile, BP, weight — steroid safety baseline.
- Swab of any secondarily infected erosion for bacterial culture. [1]
Management — Resuscitation
Autoimmune blistering disease becomes a medical emergency when skin loss is extensive — extensive PV with body surface area involvement greater than 30 percent behaves like a partial-thickness burn, with the same risks of fluid and electrolyte loss, hypothermia, hypoproteinaemia, secondary infection, and sepsis.[1]
Resuscitation bundle for severe/widespread pemphigus: [1]
- Admit to a burn unit or ICU if BSA involved is over 30 percent, electrolytes deranged, sepsis suspected, or airway/laryngeal involvement present.
- Intravenous access and fluid resuscitation using a burn-style formula (e.g., 2 to 4 mL/kg/%BSA in the first 24 h, balanced crystalloid), titrated to urine output 0.5 to 1.0 mL/kg/h.
- Electrolyte surveillance — sodium, potassium, calcium, magnesium, phosphate every 6 to 12 h; correct hypoalbuminaemia; watch for hyponatraemia (insensible loss, SIADH from pain).
- Wound care — non-adherent dressings (paraffin gauze, silicone), warm ambient temperature (30 to 32 °C to reduce insensible loss), aseptic technique and reverse isolation, daily inspection for secondary infection.
- Pain control — paracetamol, judicious opioids (avoid NSAIDs if possible); topical anaesthetics (lidocaine viscous, benzydamine mouthwash) for oral erosions.
- Nutrition — early enteral feeding; supplement calories, protein, vitamins; percutaneous endoscopic gastrostomy if oral disease prevents intake.
- Infection surveillance — daily temperature, inflammatory markers; swab of suspicious lesions; treat Staphylococcus aureus and Gram-negative sepsis empirically with anti-staphylococcal agents.
- DVT prophylaxis — LMWH (immobility + inflammation = high risk).
- Oral care — soft diet, antiseptic mouthwashes (chlorhexidine, benzydamine), nystatin or clotrimazole for oral candidiasis prophylaxis, topical anaesthetics before meals.
- Ophthalmology review if any conjunctival involvement — to prevent symblepharon and corneal ulceration. [1]
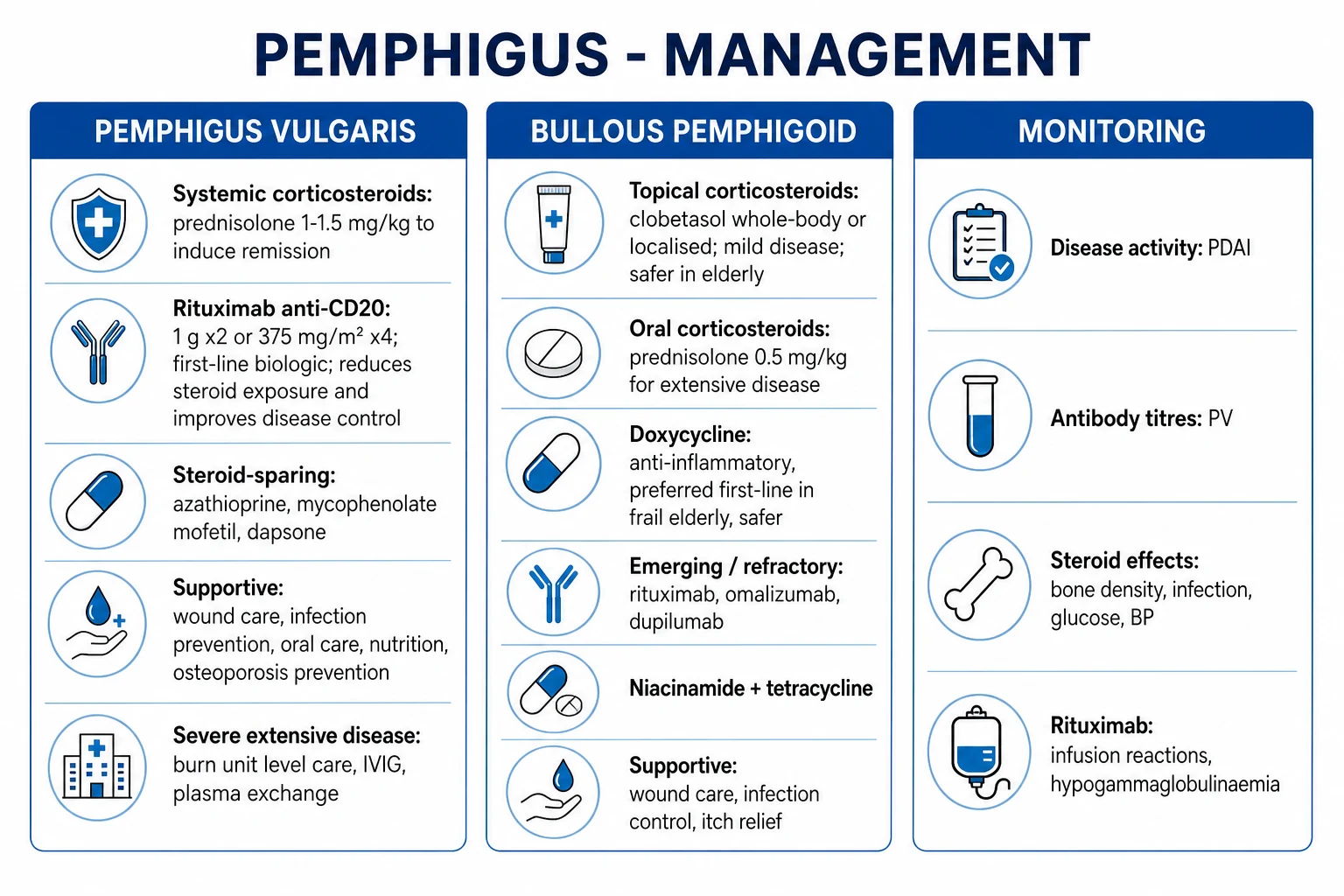
Management — Definitive & Stepwise
Definitive management differs sharply between PV and BP because of differences in natural history, age, and severity. The pivotal recent evidence — PEMPHIX (2017) for PV and BLISTER (2017) for BP — has shifted first-line therapy. [1]
Pemphigus vulgaris — first-line is now rituximab + short-course prednisone (PEMPHIX)
The PEMPHIX trial (Joly et al, Lancet 2017) randomised patients with newly diagnosed moderate-to-severe PV to rituximab 1000 mg IV at days 1 and 15, plus a short prednisone taper (0.5–1.0 mg/kg/day tapering over 3–6 months) versus prednisone 1.0–1.5 mg/kg/day alone. The rituximab arm was superior: 89.5 percent of rituximab patients versus 53.3 percent of prednisone-only patients achieved sustained remission off-therapy at 24 months; rituximab halved the cumulative steroid dose and reduced serious adverse events.[3]
Stepwise ladder for PV: [1]
[1]Adjunctive measures: [1]
- Topical therapy — high-potency topical corticosteroids (clobetasol) to localised erosions; bland emollients.
- Oral care — topical steroids (betamethasone, clobetasol rinses), topical tacrolimus, anaesthetic mouthwashes.
- Steroid-safety bundle — bone protection (calcium + vitamin D, bisphosphonate if DEXA-confirmed osteoporosis or long-term steroids); PJP prophylaxis (co-trimoxazole 480 mg/day if on long-term ≥20 mg prednisolone + a second immunosuppressant); gastric protection (PPI if symptomatic or high risk); glycaemic and BP surveillance; ocular monitoring (cataract, glaucoma).
- Rituximab pre-infusion checks — HBV/HCV/HIV screen (reactivation), QuantiFERON-TB, baseline immunoglobulins; pre-medicate with paracetamol, antihistamine, hydrocortisone 100 mg IV; monitor for infusion reactions. [1]
Escalation triggers in PV: failure to achieve disease control (no new lesions) within 4 to 6 weeks of starting steroids; rising anti-Dsg3 titre on serial ELISA; extensive mucosal or cutaneous disease at presentation; relapse on tapering. [1]
Bullous pemphigoid — first-line is doxycycline + topical clobetasol (BLISTER)
The BLISTER trial (Williams et al, Lancet 2017) randomised elderly patients with new BP to doxycycline 200 mg/day versus prednisolone 0.5 mg/kg/day as initial therapy. Doxycycline was non-inferior for blister control at 6 weeks (74 percent of doxycycline patients versus 91 percent of prednisolone patients had ≤3 blisters, within the predefined 37 percent non-inferiority margin) and was markedly safer — fewer serious, life-threatening, and fatal adverse events at 12 months. The European S2K guideline (Borradori 2022) recommends topical clobetasol 30 g/day whole-body application (where feasible) or oral doxycycline 200 mg/day as first-line, reserving oral prednisolone 0.5 mg/kg/day (NOT the 1 mg/kg used in PV) for refractory or severe disease.[4][5]
Stepwise ladder for BP: [1]
[1]Why doxycycline? It is anti-inflammatory (inhibits matrix metalloproteinases, including those that cleave BP180), has a favourable safety profile in the elderly (compared with high-dose steroids), is cheap and oral, and provides anti-staphylococcal cover for secondarily infected lesions. Photosensitivity and oesophageal irritation are practical concerns — advise sun protection and taking with water while upright. [1]
Adjunctive measures in BP: antihistamines for pruritus (sedating at night — chlorphenamine, hydroxyzine; non-sedating by day — cetirizine, loratadine); bland emollients; compression bandaging for associated venous disease; frailty assessment, fall prevention, osteoporosis prophylaxis, glycaemic and BP monitoring for the elderly. [1]
Mucous membrane pemphigoid — early aggressive therapy to prevent scarring
MMP demands early, aggressive immunosuppression because the scarring is irreversible. The European S3 guideline (Schmidt 2021) stratifies therapy by severity.[6]
- Mild disease (oral only, no ocular/laryngeal): topical clobetasol gel/rinses, topical tacrolimus, doxycycline 200 mg/day, dapsone (after G6PD testing).
- Moderate-severe (ocular, laryngeal, oesophageal, genital): systemic prednisolone 0.5–1.0 mg/kg/day + cyclophosphamide 1–2 mg/kg/day OR rituximab 1000 mg × 2 (now preferred in ocular disease); IVIG for refractory.
- Severe/rapidly progressive ocular disease: dapsone + rituximab, urgent ophthalmology, subconjunctival triamcinolone in selected cases; mitomycin C and amniotic membrane transplantation for established symblepharon.
- Laryngeal involvement: ENT, tracheostomy if airway threatened, intralesional steroid. [1]
Specific Subtypes & Scenarios
Pemphigus foliaceus is generally milder than PV; first-line is potent topical steroids (clobetasol) for limited disease, antimalarials (hydroxychloroquine 200–400 mg/day) for sun-exposed disease, dapsone for the IgA/neutrophilic variants, low-dose prednisolone (0.5 mg/kg/day) for widespread disease, and rituximab for refractory PF.[7]
Paraneoplastic pemphigus (PNP) demands treatment of the underlying neoplasm (lymphoma, CLL, Castleman) plus systemic steroids and rituximab or cyclophosphamide; the mucositis is characteristically refractory, the lung involvement (bronchiolitis obliterans) is often fatal, and overall mortality is high. [1]
Drug-induced pemphigus — withdraw the offending drug (captopril, penicillamine, rifampicin, NSAIDs); thiol-drug cases may resolve without immunosuppression, while non-thiol-drug cases behave like idiopathic PV and require standard therapy. [1]
IgA pemphigus responds dramatically to dapsone (50–150 mg/day, after G6PD testing), reflecting its neutrophilic mechanism; alternatively colchicine, acitretin, or phototherapy. [1]
Linear IgA bullous dermatosis — dapsone is first-line; withdraw the offending drug (vancomycin); colchicine, sulfasalazine, IVIG for refractory. [1]
Pemphigoid gestationis (PG) — manage jointly with obstetrics. Topical clobetasol for mild localised disease; oral prednisolone 0.5–1.0 mg/kg/day for moderate-severe. Avoid rituximab, mycophenolate, methotrexate, cyclophosphamide (teratogenic); avoid anti-Ro serology confusion with neonatal lupus. Monitor for foetal growth restriction, prematurity, and transient neonatal blistering (from transplacental antibody). The disease typically remits weeks to months postpartum but recurs, often earlier and more severely, in subsequent pregnancies.[9][10]
Complications & Pitfalls
Disease-related complications of PV include secondary bacterial infection (Staphylococcus aureus, Streptococcus, Gram-negative), sepsis, fluid and electrolyte loss, hypoalbuminaemia, malnutrition (from oral disease and dysphagia), laryngeal and oesophageal strictures, ocular scarring (in ocular PV), anaemia of chronic disease, and venous thromboembolism. Untreated PV is uniformly fatal within years. [1]
Treatment-related complications dominate modern mortality. Corticosteroids: infection (including reactivation of TB, HBV, herpes), hyperglycaemia and new-onset diabetes, hypertension, osteoporosis and vertebral fracture, avascular necrosis of the femoral head, peptic ulcer disease, cataract and glaucoma, psychiatric disturbance (steroid psychosis, mood elevation), adrenal suppression, skin atrophy and striae, growth retardation in children. Azathioprine: hepatotoxicity, myelosuppression (especially if TPMT-deficient), pancreatitis. Mycophenolate: myelosuppression, GI upset, teratogenicity. Cyclophosphamide: haemorrhagic cystitis (mesna), bladder cancer, myelosuppression. Rituximab: infusion reactions, late-onset neutropenia, hypogammaglobulinaemia (cumulative infection risk), HBV reactivation, PML (rare), vaccine impairment (live vaccines contraindicated; non-live vaccines should ideally be given before infusion or 4 weeks before next cycle). Dapsone: haemolysis (especially G6PD-deficient), methaemoglobinaemia, agranulocytosis, dapsone hypersensitivity syndrome (DRESS — at 4–8 weeks). [1]
BP-specific complications in the elderly: delirium from systemic steroids, falls and fragility fractures, opportunistic pneumonia, opportunist infections, cardiovascular decompensation — all driving the 20 to 40 percent one-year mortality of BP that is largely iatrogenic.[5]
Pitfalls: [1]
- Nikolsky sign is not pathognomonic — positive in PV, TEN, SSSS, and severe contact dermatitis; never diagnostic alone.
- Tzanck smear is not a diagnosis — acantholytic cells also occur in HSV, zoster, and Darier disease.
- Missing MMP — desquamative gingivitis labelled as "oral lichen planus" for months; by the time symblepharon develops, sight is permanently threatened.
- Treating PV like a burn with surgical debridement — the skin is fragile and debridement extends the wound; pathergy is not a feature of PV, but secondary infection risk is high.
- Stopping rituximab abruptly — relapse is common; titrate to B-cell count and anti-Dsg3 titre.
- Inadequate steroid-safety bundle — every patient on long-term steroids needs bone, gastric, glycaemic, and infection prophylaxis.
- Inadequate pre-rituximab screening — HBV, HCV, HIV, TB, immunoglobulins.
- Misattributing PG to polymorphic eruption of pregnancy (PEP) — PEP spares the periumbilicus (starts in striae), PG starts periumbilically; PEP is more common and milder; PG is intensely pruritic and bullous.[9]
Prognosis & Disposition
PV: untreated, near-uniformly fatal within 5 years from sepsis or electrolyte complications; treated, 5 to 10 percent mortality at 5 years, predominantly from steroid complications and infection. The PEMPHIX trial reported 89.5 percent sustained remission off-therapy at 24 months with first-line rituximab. Long-term control is now achievable for most patients.[3]
BP: typically self-limiting over 3 to 6 years (treatment can be tapered and stopped), but 1-year mortality is 20 to 40 percent in the elderly, driven by steroid complications, comorbidity, and neurological disease. The BLISTER trial demonstrated that doxycycline-first strategies halve serious adverse events without compromising blister control.[4][5]
MMP: chronic, relapsing; prognosis dominated by ocular scarring (irreversible) — early aggressive therapy is essential. [1]
PG: typically resolves weeks to months postpartum; recurs, often earlier and more severely, in subsequent pregnancies; neonatal risk of transient blistering.[9]
Dispositions: severe/widespread PV — burn unit or ICU; ocular involvement — urgent ophthalmology; laryngeal involvement — ENT; suspected PNP — haematology-oncology; rituximab therapy — day-case infusion unit; long-term management — specialist dermatology with shared-care GP. [1]
Long-term monitoring: serial anti-Dsg3/Dsg1 ELISA in PV (rising titre heralds relapse, prompting escalation before clinical deterioration); PDAI/ABSIS scoring; B-cell counts and immunoglobulins on rituximab; DEXA, HbA1c, lipids, BP on steroids; FBC, LFTs on azathioprine/MMF. [1]
Special Populations
The frail elderly (BP): prefer topical clobetasol (where feasible to apply) and doxycycline over oral prednisolone; if steroids are essential, use the lowest effective dose (0.5 mg/kg or less), with frailty assessment, fall prevention, bone protection, and delirium surveillance. Polypharmacy review is essential — review diuretics, anticholinergics, anticoagulants. [1]
Pregnancy and breastfeeding:
- Pemphigoid gestationis: topical clobetasol for mild disease; oral prednisolone 0.5–1.0 mg/kg/day (category A in pregnancy, safe) for moderate-severe; avoid rituximab (B-cell depletion in neonate), mycophenolate, methotrexate, cyclophosphamide (all teratogenic); monitor foetus for growth restriction and prematurity.[9][10]
- PV in pregnancy (rare): systemic steroids first-line; azathioprine relatively safe (category B); rituximab avoided if possible but used for severe refractory disease; neonatal pemphigus from transplacental anti-Dsg3 is transient, resolving as maternal antibody clears (weeks to months).[10]
- Breastfeeding: prednisolone, azathioprine, and rituximab are generally compatible; mycophenolate and cyclophosphamide are not.
Children: paediatric PV is rare but follows the same immunopathology; weight-based steroid dosing (1–2 mg/kg/day) with attention to growth suppression and bone health (DEXA, calcium, vitamin D); paediatric BP and chronic bullous disease of childhood (linear IgA disease, dapsone-responsive) are distinct entities. [1]
HIV and immunocompromised: PV may be more severe and refractory; rituximab is effective but requires careful screening (HBV reactivation risk); watch for opportunistic infection. [1]
Transplant patients and concurrent immunosuppression: check drug interactions — azathioprine and calcineurin inhibitors share CYP/hypoxanthine pathways; TPMT testing is mandatory before azathioprine; monitor for cumulative immunosuppression (infection, lymphoma). [1]
Anticoagulated patients: extensive skin loss and immobility raise VTE risk — LMWH prophylaxis; warfarin/DOACs continue, but beware drug interactions with azathioprine (steroid-sparing agents rarely interact) and antibiotics used for secondary infection. [1]
Evidence, Guidelines & Regional Differences
[1]Landmark trials: [1]
- PEMPHIX (Joly 2017, Lancet) — first-line rituximab + short-term prednisone vs prednisone alone for moderate-severe PV: rituximab superior (89.5% vs 53.3% sustained remission off-therapy at 24 months; fewer serious adverse events).[3]
- BLISTER (Williams 2017, Lancet) — doxycycline 200 mg vs prednisolone 0.5 mg/kg initial therapy for BP: doxycycline non-inferior for blister control at 6 weeks with markedly fewer serious, life-threatening, and fatal adverse events.[4]
- European S2K (Borradori 2022) and S3 MMP (Schmidt 2021) — current European guidelines standardising BP and MMP management.[5][6]
- FDA approval of rituximab for PV (2018) — first disease-specific biologic approval for pemphigus vulgaris.
Controversies and emerging therapies: [1]
- Maintenance rituximab dosing — 500 mg at months 6 and 12 versus treat-to-flare; emerging evidence favours proactive maintenance to extend remission.
- Combination therapy — rituximab + IVIG versus rituximab alone for refractory disease.
- Janus kinase (JAK) inhibitors (rituximab-resistant PV) — emerging case series.
- Complement inhibitors (eculizumab, avacopan) — niche for complement-driven PV.
- Omalizumab and dupilumab — emerging for refractory BP (IgE and type 2 inflammation roles).
- Novel anti-CD19 chimeric antigen receptor T cells (CAR-T) — early-phase for severe refractory autoimmune blistering disease.[7]
Exam Pearls
[1]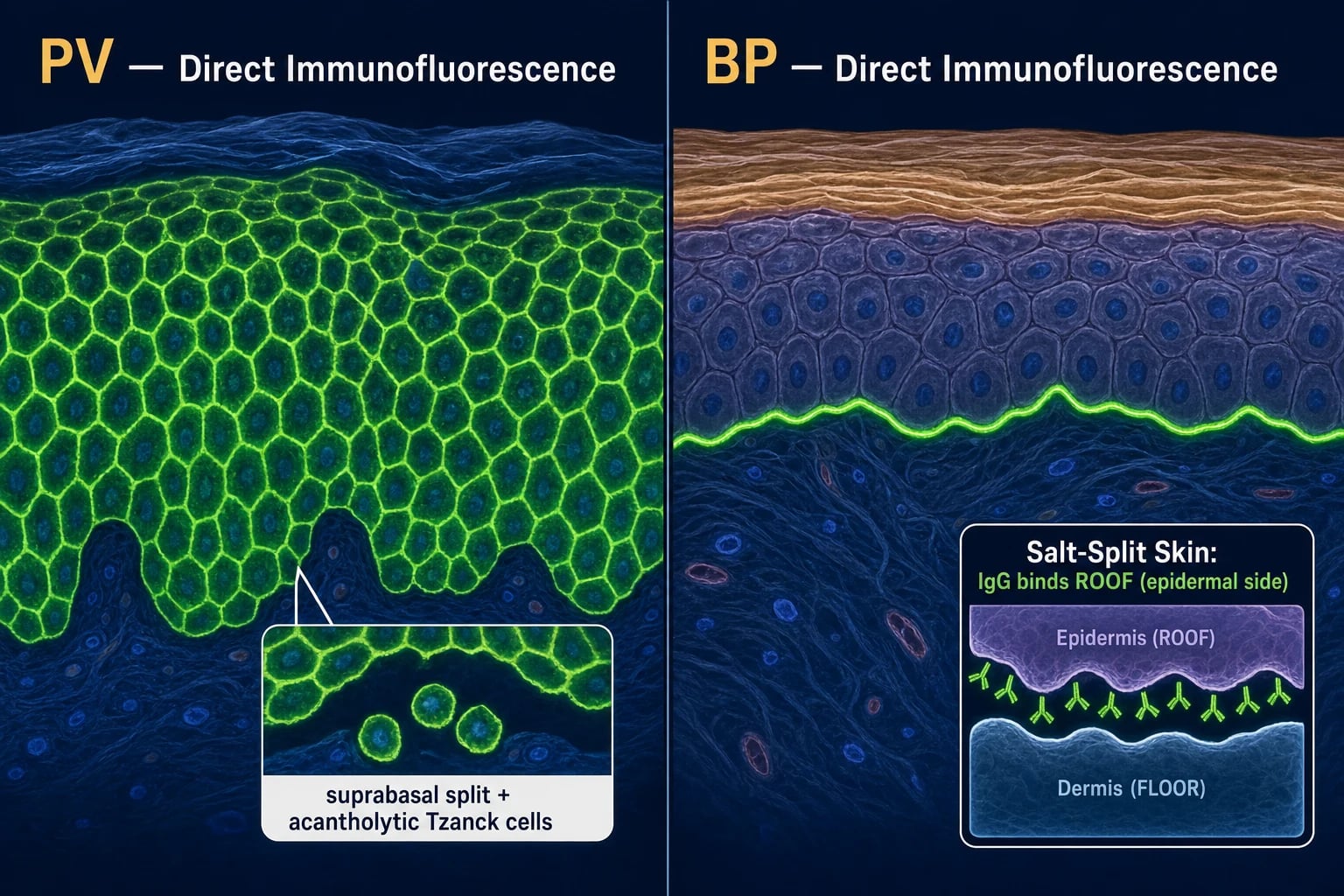
References
- [1]Alramadhan SA, Islam MN. Vesiculobullous Lesions of the Oral Cavity Oral Maxillofac Surg Clin North Am, 2023.PMID 37019505
- [2]Hussain MH, Tanweer F, Sakagiannis G. Pemphigus Vulgaris and Bullous Pemphigoid of the Upper Aerodigestive Tract: A Review Article and Novel Approaches to Management ORL J Otorhinolaryngol Relat Spec, 2021.PMID 33902048
- [3]Joly P, Maho-Vaillant M, Prost-Squarcioni C, et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial Lancet, 2017.PMID 28342637
- [4]Williams HC, Wojnarowska F, Kirtschig G, et al. Doxycycline versus prednisolone as an initial treatment strategy for bullous pemphigoid: a pragmatic, non-inferiority, randomised controlled trial Lancet, 2017.PMID 28279484
- [5]Borradori L, Amagai M, Schmidt E, et al. Updated S2 K guidelines for the management of bullous pemphigoid initiated by the European Academy of Dermatology and Venereology (EADV) J Eur Acad Dermatol Venereol, 2022.PMID 35766904
- [6]Schmidt E, Tasanen K, Maciej P, et al. European Guidelines (S3) on diagnosis and management of mucous membrane pemphigoid, initiated by the European Academy of Dermatology and Venereology - Part II J Eur Acad Dermatol Venereol, 2021.PMID 34309078
- [7]Zhou MH, Feldman RJ, Huang A. Acantholytic disorders: Update on pathophysiology, diagnosis, and management J Am Acad Dermatol, 2026.PMID 41730429
- [8]Sielski L, Sinha AA. Desmoglein compensation hypothesis fidelity assessment in Pemphigus Front Immunol, 2022.PMID 36211362
- [9]Wilson BH, Pomeranz MK, Castelo-Soccio L. Dermatologic Conditions of Pregnancy Prim Care, 2025.PMID 40835281
- [10]Shah T, Doot RK, Messenger AG, et al. Critical analysis of pemphigus vulgaris in pregnancy Front Immunol, 2026.PMID 41983144