Dermatology · Medicine
Porokeratosis
Also known as Porokeratosis · Porokeratosis of Mibelli (classic plaque type) · Disseminated superficial actinic porokeratosis (DSAP) · Linear porokeratosis · Porokeratosis palmaris et plantaris disseminata (PPPD) · Punctate porokeratosis · Cornoid lamella disorder
Porokeratosis is a clonal disorder of epidermal keratinisation, defined histologically by the PATHOGNOMONIC CORONOID LAMELLA — a vertical column of parakeratotic cells overlying a diminished granular layer with underlying dyskeratotic keratinocytes. Five classical variants are recognised: classic porokeratosis of Mibelli (large plaque, childhood onset), disseminated superficial actinic porokeratosis (DSAP, most common, sun-exposed adult skin, autosomal dominant with MVK/SLC17A9 mutations), linear porokeratosis along Blaschko's lines (highest malignant potential), porokeratosis palmaris et plantaris disseminata (palms/soles first), and punctate porokeratosis (1–2 mm keratotic spines on palms/soles). Clinical hallmark: annular plaque with a raised, thread-like, hyperkeratotic border (the clinical correlate of the cornoid lamella) and central atrophy. Malignant transformation to squamous cell carcinoma occurs in 5–15% of long-standing lesions, especially the linear and giant variants. Treatment is pathogenesis-directed: topical lovastatin plus cholesterol (JAMA Dermatology RCT 2023), topical 5-FU or imiquimod, cryotherapy, CO2 laser, photodynamic therapy; surgical excision for small or suspicious lesions.
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags
Overview & Definition
Porokeratosis is a heterogeneous group of clonal disorders of epidermal keratinisation that share a single unifying histological hallmark: the coronoid lamella. Clinically the disease presents as annular or serpiginous plaques with a raised, thread-like, hyperkeratotic peripheral ridge and central atrophy. The peripheral ridge you can feel with your fingertip is the clinical correlate of the histological cornoid lamella — appreciating that one-to-one correspondence is the single highest-yield clinical pearl the examiner can offer on this topic.[1][2]
The disorder was first described by Mibelli in 1893 under the name "porokeratosis" because he mistakenly attributed the lesions to a disorder of the eccrine sweat-pore ("poros"). The eponym stuck; the aetiology turned out to be entirely different. The condition is now understood to result from a clonal expansion of genetically abnormal keratinocytes that grow centrifugally and produce the characteristic ring of disordered cornification at the leading edge. Five classical clinical variants are recognised (see Classification), and several rarer patterns are described.[1]
Porokeratosis is more than a cosmetic problem because every long-standing lesion — and especially linear and giant porokeratosis — carries a measurable risk of malignant transformation to squamous cell carcinoma (SCC), with retrospective studies quoting a pooled transformation rate in the order of 5–15% depending on the variant and follow-up window.[10][11] Recognising the disease early, monitoring for change, and applying pathogenesis-directed therapy (topical lovastatin plus cholesterol, validated in a 2023 JAMA Dermatology randomised trial) are the three pillars of modern management.[3][4]
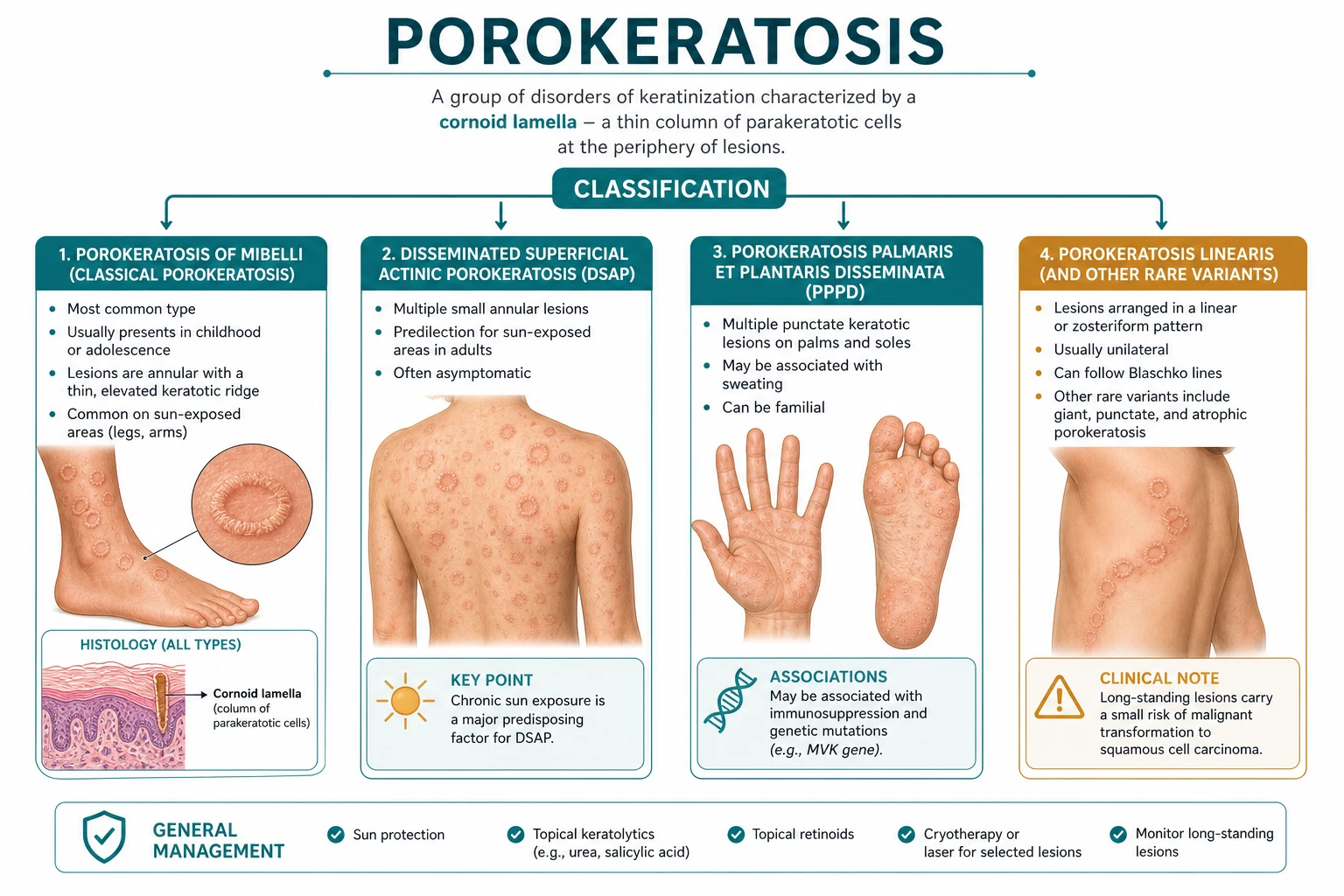
Classification
All five clinical variants share the same cornoid-lamella histology. The distinction is clinical (size, number, distribution, age of onset and inheritance pattern).[1][6]
- **Single or few large plaques** (1–20 cm), slowly expanding with prominent raised border.
- **Childhood onset**; M:F ≈ 3:1; can be sporadic or autosomal dominant.
- Limbs, trunk, face; lesions are often unilateral.
- Malignant transformation uncommon (<2%) but reported.
- **Most common variant**; multiple (often dozens–hundreds) small annular lesions 0.5–1 cm.
- **Sun-exposed skin** of adults (forearms, shins, V of neck, face); spares palms/soles.
- Worsens in summer; **autosomal dominant** with incomplete penetrance (MVK, SLC17A9, other loci).
- Female predominance after age 40; rare before puberty.
- Lesions follow **Blaschko's lines** in a linear or whorled unilateral distribution.
- Onset in childhood; reflects early somatic mosaicism in keratinocyte clones.
- **HIGHEST malignant transformation risk** (up to 7–11% in some series) → strict surveillance.
- Starts on **palms and soles** as small comedo-like plugs; spares flexures.
- Lesions **disseminate** centrifugally to trunk and limbs over years.
- Onset usually adolescence / early adulthood; rare, sporadic.
- 1–2 mm **discrete, keratotic spines or pits** on palms and soles (and sometimes digits).
- Often grouped; asymptomatic; can mimic punctate keratoderma or porokeratoma.
- May represent a limited form of PPPD confined to acral sites.
Rarer patterns worth recognising at the bedside
- Porokeratosis ptychotropica — well-demarcated erythematous-to-brown keratotic plaques of the gluteal/perianal flexural region; easily mistaken for inverse psoriasis, Bowen's disease, or chronic intertrigo. Strongly associated with malignant transformation. Dermoscopy shows peripheral keratotic ridge with central structureless area — confirmed on biopsy.[12]
- Genitogluteal porokeratosis — anatomically broader term covering ptychotropica, penile and scrotal variants. Often misdiagnosed for years as eczema or condyloma. Biopsy is mandatory in chronic intertriginous plaques.[5]
- Eruptive pruritic papular porokeratosis — acute, intensely itchy crops of small porokeratotic papules in adults; consider HIV, immunosuppression or paraneoplastic trigger.
- Giant porokeratosis — single plaque > 20 cm; very high SCC risk.[10]
- Follicular porokeratosis / porokeratoma — cornoid lamella localised to the follicular infundibulum; presents as a comedo-like or annular plaque with central keratotic plug.
- Vulval, penile and scrotal porokeratosis — under-recognised; biopsy any chronic penile plaque that does not respond to anti-fungal or anti-inflammatory therapy.
Epidemiology & Risk Factors
Porokeratosis is uncommon but not rare. DSAP is the most commonly diagnosed variant, accounting for the majority of adult cases; classic Mibelli and linear porokeratosis are encountered less often in a general dermatology clinic but are over-represented in tertiary paediatric and pigmented-skin cohorts respectively.[1][6]
Recognised precipitants
The single most reproducible environmental trigger is ultraviolet light, especially in DSAP — lesions emerge on sun-exposed skin, expand in summer and may improve with photoprotection. Other reproducible precipitants include:[6]
- Immunosuppression — solid-organ transplantation (renal, cardiac, hepatic), chronic lymphocytic leukaemia, HIV infection. The combination of long-term azathioprine, mycophenolate or ciclosporin and cumulative UV exposure produces a florid DSAP-like pattern that can be the first clinical clue to over-immunosuppression.
- Ionising radiation and electron-beam therapy — porokeratosis may arise within, or adjacent to, irradiated fields months to decades after treatment.
- Drug triggers — reported with antitumour antibiotics, thiazide diuretics (sun-sensitising), androgens, TRAIL-inducing agents; causality is not always proved.
- Long-term PUVA / narrowband UVB — paradoxically can both improve pre-existing DSAP and provoke new lesions.
- Chronic skin trauma — particularly relevant in linear porokeratosis (Koebner-like activation along Blaschko's lines).[3]
Heritability
Familial DSAP is autosomal dominant with incomplete penetrance. Two genes account for a substantial fraction of familial cases:[8]
- MVK (mevalonate kinase; chromosome 12q24) — also the gene mutated in mevalonate kinase deficiency and hyper-IgD syndrome, explaining why porokeratosis sits on the same biochemical pathway.[8][9][7]
- SLC17A9 (a vesicular nucleotide transporter; chromosome 6q24) — replicated in multiple Chinese and Japanese kindreds; encodes a transporter implicated in lysosomal ATP release and keratinocyte homeostasis.
Sporadic cases are mostly somatic mosaics, especially in the linear variant. Whole-exome sequencing has also implicated other pathway genes (PMVK, FDPS, GGPS1) but these are not part of routine diagnostic panels.[7]
When to suspect a paraneoplastic trigger
A sudden eruptive DSAP-like picture in an adult without UV or immunosuppression trigger is a recognised paraneoplastic dermatosis, most often linked to haematological malignancy, hepatocellular carcinoma and colorectal adenocarcinoma. In this setting the lesions may regress with treatment of the underlying malignancy. Onset in the context of cachexia, weight loss or B-symptoms should prompt screening.[2][6]
Pathophysiology
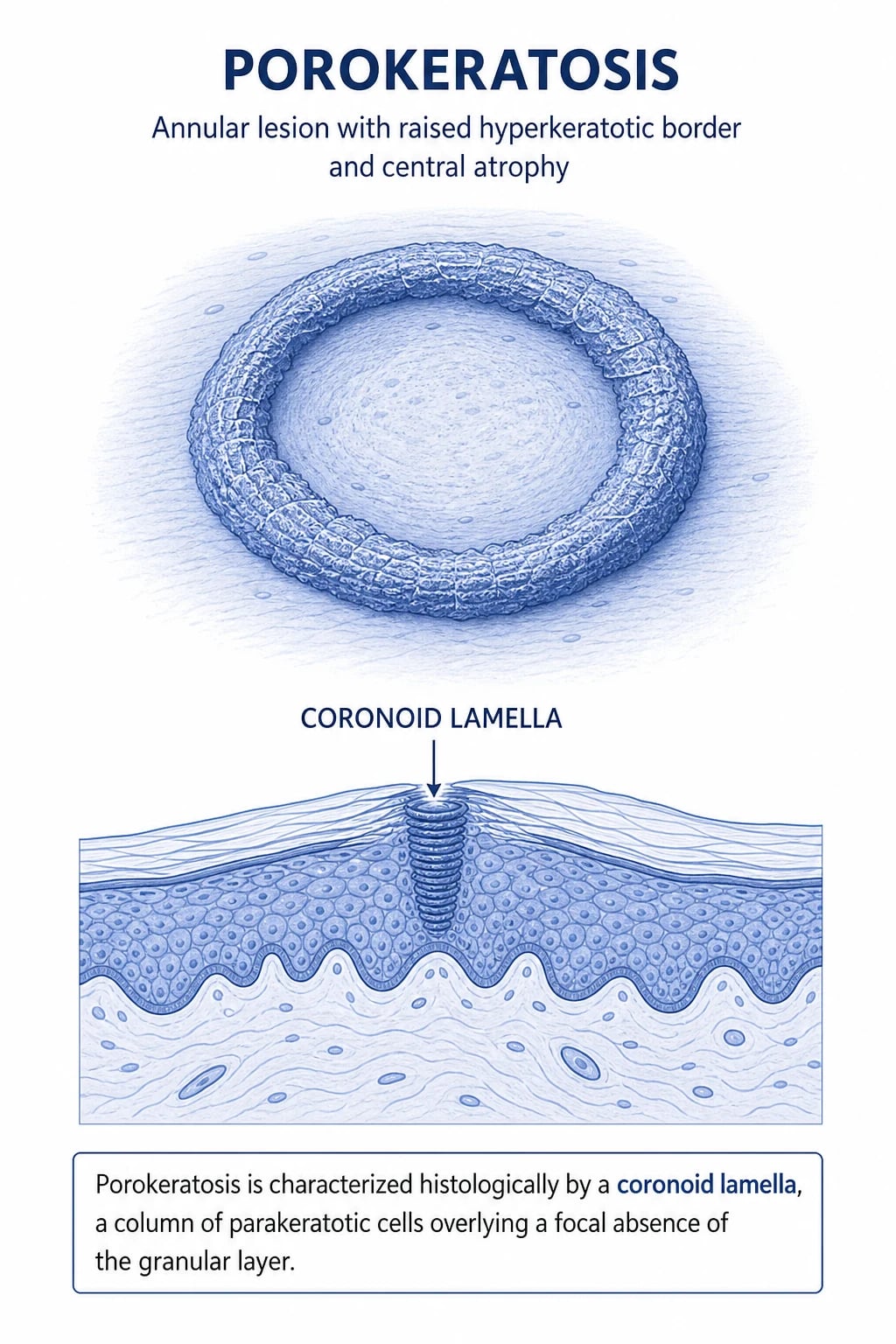
The cornoid lamella — what it is and what it isn't
The unifying histologic feature is the cornoid lamella (sometimes spelled coronoid in older literature). It is best described as a discrete vertical column of parakeratotic cells — nucleated, basophilic stratum corneum — that tilts obliquely away from the centre of the lesion and overlies a focal zone where the granular layer is absent or markedly thinned. Immediately deep to the absent granular layer sits a band of dyskeratotic, vacuolated keratinocytes with shrunken, brightly eosinophilic cytoplasm and pyknotic nuclei. The lamella is therefore a focal abnormality, not a diffuse one — the surrounding keratinocytes are often histologically normal.[1]
Why this matters clinically: the cornoid lamella is pathognomonic but is only found at the edge of the lesion. A punch biopsy taken from the centre will usually be reported as "non-specific" or "consistent with scar/atrophy" and miss the diagnosis. Always sample through the ridge of the lesion.[3]
The "poral" myth and the modern mechanistic model
Mibelli's original hypothesis (eccrine-duct hyperkeratosis) has been abandoned. Modern understanding, derived from loss-of-heterozygosity and X-chromosome inactivation studies on mosaic lesions, is that porokeratosis is a clonal proliferation of an abnormal keratinocyte stem cell that expands centrifugally at the expense of surrounding normal epidermis.[1][7]
Two molecular pathways are mechanistically important and they converge on a single therapeutic implication.[7]
- MVK encodes mevalonate kinase, a key enzyme of the mevalonate / cholesterol biosynthesis pathway.
- Loss-of-function mutations reduce downstream sterol/isoprenoid production, leading to abnormal keratinocyte differentiation and cornification.
- Pathway rescue = topical lovastatin (HMG-CoA reductase inhibitor) + cholesterol (pathogenesis-directed therapy).
- UV-induced p53 mutations in epidermal clones drive both the dyskeratotic phenotype and the long-term SCC risk.
- Immunosurveillance failure under iatrogenic immunosuppression permits clonal expansion — explains transplant DSAP.
- Reducing immunosuppression / strict photoprotection is a rational adjunct.
The MVK / mevalonate pathway in detail
The mevalonate pathway converts acetyl-CoA → HMG-CoA → mevalonate (the rate-limiting step catalysed by HMG-CoA reductase) → mevalonate-5-phosphate (catalysed by mevalonate kinase, MVK) → downstream isoprenoids (farnesyl-PP, geranylgeranyl-PP) and ultimately cholesterol. Statins inhibit HMG-CoA reductase upstream of MVK; in DSAP keratinocytes the problem is not statin-blockable upstream flow, but inefficient downstream conversion through a hypomorphic MVK enzyme. Adding lovastatin alone can paradoxically worsen the downstream deficit. Topical cholesterol bypasses the block, and the combination lovastatin + cholesterol is mechanistically rational.[7][3]
The biochemical link to inflammation is also instructive: loss of MVK function reduces prenylation of small GTPases (RhoA, Rac1), perturbs keratinocyte cytoskeleton and stress responses, and biases immunity toward IL-1β hyperactivation (as in mevalonate kinase deficiency) — providing a rationale for adjunctive anti-inflammatory therapy during flares.[7]
Malignant transformation pathway
Long-standing porokeratosis lesions accumulate UV-signature p53 mutations, oxidative DNA damage and loss of immunosurveillance. The clone that produced the cornoid lamella is the same clone that gives rise to SCC. Reported progression pathway:[10][11]
cornoid-lamella clone → dysplasia / actinic keratosis-like change → SCC in situ (Bowenoid) → invasive SCC.[6]
The risk is highest in the linear and giant variants (where the abnormal clone occupies a much larger body surface) and in organ-transplant recipients (where the clone survives unchecked).[7]
CORN
Clinical Presentation
The characteristic lesion
A well-circumscribed annular or serpiginous plaque with two cardinal features:[1]
- A raised, thread-like, hyperkeratotic peripheral ridge that can be appreciated by sliding a finger across the lesion's edge — this is the cornoid lamella.
- Central atrophy — slightly thinned, smooth, hypopigmented or pink, sometimes with a fine telangiectatic pattern in long-standing plaques.[1]
Additional features include:[1]
- Diameter ranging from millimetres (DSAP, punctate) to > 20 cm (porokeratosis of Mibelli, giant porokeratosis).
- Colour range: pink-red in active / inflamed lesions; brown-yellow at the ridge when hyperkeratotic; hypopigmented in long-standing atrophic centres.
- Slow centrifugal growth over months to years (1–2 mm/month is typical).
- Usually asymptomatic; mild pruritus in 10–30% and pain / tenderness only when fissured, eroded, eroded or malignant transformation occurs.
- Surface may be smooth or verrucous; excoriations indicate a symptomatic lesion.[10]
Variant-specific patterns
- **Distribution**: extensor forearms, shins, lateral upper arms, V of neck, face.
- **Number**: dozens to hundreds; **size**: 0.5–1 cm.
- **Time course**: summer worsening; lesions persist between seasons.
- **Symptoms**: usually asymptomatic; cosmetic concern is the commonest presentation.
- **Atypical**: pruritic, vesiculated, eroded or verrucous lesions have all been described.
- Streaks or whorls of typical annular porokeratotic lesions along **Blaschko's lines** — typically unilateral on a limb or hemitrun.
- May be present at birth or emerge in childhood; an adult-onset variant may follow Blaschko's lines within an area of prior inflammation.
- **Highest SCC risk — any new nodule, ulcer, or pain must be biopsied urgently.**
- **Gluteal / perianal flexural** plaques that are erythematous, keratotic and well demarcated; often undiagnosed for years.
- May extend onto natal cleft, inguinal folds and upper thighs.
- **Biopsy mandatory** — clinically mimics inverse psoriasis, Bowen's disease and chronic candidal intertrigo.
- **Punctate**: 1–2 mm keratotic spines or pits on palms/soles; can mimic pitted keratolysis, porokeratoma, or punctate keratoderma.
- **PPPD**: starts on palms/soles as small plugs and disseminates to trunk and limbs over years; rarely familial.
Atypical and easy-to-miss presentations
- Verrucous / hyperkeratotic DSAP — particularly in immunosuppressed transplant recipients; can resemble actinic keratoses or viral warts.
- Erosive or bullous DSAP — Nikolay sign can be positive, mimicking pemphigus or bullous impetigo.
- Inflammatory, pruritic, eruptive porokeratosis — consider drug trigger, HIV or paraneoplastic screen.
- Congenital linear porokeratosis — large body-surface involvement; SCC risk persists into adulthood.
- Porokeratoma — solitary, endophytic tumour-like lesion with cornoid lamella localised to the follicular ostium; clinicopathologically distinct from classic porokeratosis.[6]
Differential Diagnosis
The annular plaque with raised border invites a long list of diagnoses. The combination of palpable ridge, central atrophy, multiple lesions in typical distribution and cornoid lamella on biopsy is what secures the diagnosis.[10]
Three features that should drive the clinician straight to a biopsy:[8]
- Induration, ulceration, pain or rapid growth within a previously stable plaque — consider SCC until proved otherwise.[10][11]
- Unilateral, Blaschko-linear porokeratosis in a child — distinguish from linear lichen planus, linear psoriasis and inflammatory linear verrucous epidermal naevus (ILVEN).
- Persistent flexural (genitogluteal) plaque not responding to antifungals or topical steroids — biopsy to exclude porokeratosis ptychotropica or Bowen's disease.

Clinical & Bedside Assessment
Focused history (8 questions)
- Chronology — age of onset (Mibelli = childhood; DSAP = 30–50 y; PPPD = adolescence), evolution, seasonality.
- Distribution — sun-exposed only (DSAP); unilateral Blaschko-linear; palmar / plantar onset (PPPD).
- Symptoms — pruritus, pain, cosmetic concern, none.
- Sun exposure — occupation, outdoor hobbies, phototherapy, travel to tropical latitudes.
- Immunosuppression — organ transplant, HIV, haematological malignancy, biologics, azathioprine, ciclosporin, mycophenolate.
- Prior radiation or PUVA — fields and timing.
- Family history — first-degree relatives with similar plaques (suggests autosomal dominant DSAP).
- Systemic / B symptoms — weight loss, night sweats, change in bowel habit (paraneoplastic screen if positive).[11]
Full skin examination
A complete skin examination under good daylight (or a Wood's lamp for sub-clinical lesions in fair-skinned patients) is mandatory. Specifically examine:[1]
- Sun-exposed sites — face, neck, dorsal forearms, V of chest, anterior shins.
- Palms and soles — for punctate porokeratosis, PPPD, palmoplantar keratoderma mimics.
- Flexures — gluteal cleft, perianal area, groin, inframammary folds (porokeratosis ptychotropica, genitogluteal variants).
- Genitalia — penile, scrotal and vulval porokeratosis is under-recognised.
- Scalp and hair-bearing skin — follicular porokeratosis and porokeratoma.
- Mucous membranes — usually spared; if involved, broaden differential.[5]
Bedside sign worth knowing
The palpable ridge sign — sliding a gloved finger radially across the lesion will detect the hyperkeratotic ridge even when visual inspection is inconclusive. The ridge corresponds 1:1 to the cornoid lamella.[1]
Investigations
The single most useful diagnostic investigation is a skin biopsy through the ridge, examined with haematoxylin-eosin. No serum biomarkers exist; imaging is reserved for staging a confirmed malignant transformation or for paraneoplastic work-up.[1][6]
Histopathology — the five diagnostic features
- Vertical column of parakeratotic cells in the stratum corneum (the lamella itself).
- Absent or diminished granular layer directly under the column.
- Dyskeratotic, vacuolated keratinocytes in the spinous layer immediately beneath the column.
- Focal underlying dermal chronic inflammatory infiltrate of lymphocytes ± histiocytes (variable).
- Centrifugal cornoid-lamella formation when serial sections are examined — the lesion grows by laying down lamellae ahead of itself.[10]
Dermoscopy
Hand-held dermoscopy (polarised, 10×) is the single most useful bedside adjunct and in experienced hands correctly distinguishes porokeratosis from Bowen's disease, actinic keratosis and tinea:[13]
- Central area — homogeneous, structureless white-to-yellow or pink-red, often with fine linear vessels when atrophic.
- Peripheral ridge — a yellowish or whitish keratotic rim (the "ridge sign" of dermoscopy) studded with red, brown or black speckles representing the dilated capillaries and haemorrhage within the cornoid lamella and the underlying absent granular zone.[13]
- This pattern "peripheral keratotic ridge with central structureless area" is diagnostic of porokeratosis and can capture the disease at very early (< 5 mm) stages where the clinical ridge is not yet palpable.[12][13]
Reflectance confocal microscopy (RCM) and optical coherence tomography (OCT)
RCM and OCT are emerging non-invasive tools that visualise the cornoid lamella in vivo without biopsy. They are useful for monitoring and for ptychotropic / genital lesions where biopsy is technically awkward or repeated biopsies are undesirable.[12]
Paraneoplastic work-up
If a previously healthy adult presents with abrupt-onset eruptive porokeratosis, particularly the DSAP-like or eruptive-pruritic-papular patterns, screen for occult malignancy:[10]
- Bloods: full blood count, peripheral smear, ESR / CRP, liver function, LDH, calcium, faecal immunochemical test (FIT).
- Imaging: chest / abdominal CT; age- and sex-appropriate cancer screening (mammography, PSA, colonoscopy).
- HIV serology if not already performed.[12]
Malignancy staging when SCC is confirmed
If biopsy establishes invasive SCC within a long-standing porokeratosis lesion, perform lesion staging using AJCC 8th edition T staging for cutaneous SCC (high-risk features include size > 2 cm, depth > 6 mm, perineural invasion, immunosuppression, recurrent SCC), with imaging (USS of draining nodes ± MRI / CT) per AAD / BAD / NCCN guidance.[11]
Management — Resuscitation
Porokeratosis is almost never an emergency; the disease is chronic, indolent and most often asymptomatic. The "resuscitation" mindset applies in three scenarios:[2][10][11]
- Suspected malignant transformation — a previously stable porokeratosis plaque that becomes indurated, ulcerated, painful, haemorrhagic or rapidly growing requires urgent dermatology referral (2-week-wait / rapid-access pathway) for biopsy and SCC management. Photographic documentation (with a dated image) and an outline traced on tracing paper is an excellent triage tool in primary care.
- Extensive erosive / bullous porokeratosis — rarely reported as a painful, blistering flare in DSAP; admit for wound care, analgesia, fluid management and biopsies to exclude infection or pemphigus-like pathology.
- Paraneoplastic eruptive porokeratosis — the urgent work-up is the malignancy screen, not the skin disease.[10]
Outside these scenarios, all treatment is elective / definitive and delivered on an outpatient basis.[2]
Management — Definitive & Stepwise
Treatment is staged, with most clinicians following a conservative-to-aggressive ladder. Pathogenesis-directed therapy (lovastatin + cholesterol) sits at the centre of modern practice; procedural options are reserved for small numbers of lesions or refractory disease.[2][3][4]
Step 1 — Photoprotection and trigger avoidance
Step 2 — Topical pathogenesis-directed therapy
Step 3 — Topical keratolytics / antineoplastics
Step 4 — Procedural destructive therapy (single lesions)
Step 5 — Photodynamic therapy (PDT)
Step 6 — Systemic therapy (extensive / refractory disease)
Step 7 — Surgical / oncological management
Step 8 — Long-term surveillance and escalation
Topical 2% lovastatin + 2% cholesterol cream
Dose
Twice daily, 12–24 weeks
5-fluorouracil 5% cream
Dose
Apply twice daily × 4 weeks
Imiquimod 5% cream
Dose
Apply three nights/week × 6–12 weeks
Acitretin (oral retinoid)
Dose
0.5–1 mg/kg/day × 3–6 months
Cryotherapy (liquid nitrogen)
Dose
1–2 freeze-thaw cycles, 10–15 s per lesion
Ablative fractional CO2 laser
Dose
10–30 mJ per microbeam, single session
Photodynamic therapy (ALA or MAL-PDT)
Dose
3–4 h incubation, red light 37 J/cm², 1–2 sessions
Translating the ladder to real clinics
- Mild, recent-onset DSAP in a 45-year-old — photoprotection + topical lovastatin/cholesterol; review at 12 weeks.
- Extensive DSAP in a transplant recipient — discuss with transplant team about modest reduction in immunosuppression; add topical lovastatin/cholesterol + oral acitretin; consider field PDT.
- Punctate porokeratosis (asymptomatic) — observation, photoprotection; reserve destructive therapy for painful / functional lesions.
- Single small isolated plaque — liquid nitrogen cryotherapy, electrocautery or CO2 laser; review at 6 weeks for recurrence.
- Large linear porokeratosis or giant plaque — combine acitretin with topical lovastatin/cholesterol; mark baseline photographs; strict 3-monthly review for transformation.
- Paraneoplastic, eruptive DSAP in an adult — urgent malignancy screen; empiric photoprotection + topical 5-FU; treat the underlying cancer.[3]
Specific Subtypes & Scenarios
Porokeratosis of Mibelli
Few large plaques arising in childhood on a limb or trunk; M:F 3:1; up to half are familial (autosomal dominant). Slow centrifugal growth, prominent ridge, deep central atrophy. Management is procedural for small lesions (cryotherapy, excision or CO2 laser); extensive disease may warrant oral acitretin. Malignant transformation rate is low but reported, particularly in long-standing giant Mibelli plaques.[1]
Disseminated superficial actinic porokeratosis (DSAP)
The commonest variant. Multiple small (5–10 mm) annular papules and plaques on sun-exposed skin of adults, particularly women aged 30–50; spares palms/soles. Sporadic or familial; MVK and SLC17A9 mutations account for a substantial fraction of familial cases. UV is the dominant environmental trigger — photoprotection is therapeutic. Treatment is the topical / systemic ladder above; the lovastatin + cholesterol regimen should be the cornerstone because of the mechanistic rationale.[6][4][7]
Linear porokeratosis
Linear or whorled distribution along Blaschko's lines — the most useful way to remember this variant's distribution. Reflects somatic mosaicism in an early keratinocyte precursor cell. Onset is usually in childhood; the disease may extend across a limb or hemitrun, raising cosmetic and functional concerns. Carries the highest SCC risk of all variants (7–11% in some series); lifelong surveillance is mandatory. Treatment combines pathogenesis-directed topical therapy with periodic procedural ablation of thickened plaques; prophylactic excision of stable plaques in adults can be justified in selected cases.[11]
Porokeratosis palmaris et plantaris disseminata (PPPD)
Palms and soles first; lesions later spread to trunk and proximal extremities. Often resistant to topical therapy because of the thick stratum corneum at acral sites; physical modalities (PDT, CO2 laser) and oral retinoids are preferred. Familial forms exist but the disorder is mostly sporadic.[3]
Punctate porokeratosis
1–2 mm keratotic papules or pits on palms and soles; may be limited to digits. Can be a forme fruste of PPPD. Generally asymptomatic; treat only if symptomatic (pain on walking, gripping). Topical keratolytics (urea 20–40%, salicylic acid 10–20%) and careful curettage of larger spines work well.[4]
Porokeratosis ptychotropica
Gluteal/perianal flexural plaques, easily dismissed as chronic intertrigo, inverse psoriasis or candidiasis. Biopsy mandatory in any chronic flexural plaque unresponsive to antifungals. Malignant transformation risk is real; successful treatments reported with topical lovastatin/cholesterol, CO2 laser and PDT.[12]
Transplant-associated and HIV-associated porokeratosis
A DSAP-like eruption is a recognised cutaneous marker of iatrogenic immunosuppression after solid-organ transplantation and in HIV infection. The lesions may be the first clinical clue to over-immunosuppression; reduction in immunosuppression in consultation with transplant physicians can ameliorate the disease. Topical therapy is first-line; systemic retinoids reserved for extensive disease.[3]
Paraneoplastic porokeratosis
Abrupt-onset eruptive DSAP-like picture in an adult without UV or immunosuppression trigger; screen for haematological, hepatic and gastrointestinal malignancy as above. Treating the underlying tumour often induces lesional regression.[11]
Complications & Pitfalls
Disease-related
- Squamous cell carcinoma — the most important complication. Annual transformation rate is small overall but climbs in linear/giant/transplant-related variants; biopy any change.[10][11]
- Cosmetic disfigurement — particularly when extensive DSAP affects the face and V of neck or when linear porokeratosis covers a large surface in childhood.
- Functional impairment — PPPD and punctate porokeratosis on weight-bearing soles can interfere with gait and footwear.
- Pruritus and excoriation — particularly in eruptive pruritic papular porokeratosis; superinfection is uncommon but reported.
- Psychosocial morbidity — visible DSAP and flexural porokeratosis are sources of embarrassment, social withdrawal and reduced quality of life; address explicitly.
Treatment-related
- Topical 5-FU and imiquimod — local inflammation, crusting and post-inflammatory hyperpigmentation. The "treatment-injury" reaction is expected, but excessive reaction warrants a treatment pause and review of diagnosis.
- Cryotherapy — blistering, hypopigmentation and atrophic scarring; counsel explicitly.
- CO2 laser — post-inflammatory hyper- or hypopigmentation, particularly in skin of colour; combine with strict sun avoidance and consider topical antioxidants to mitigate.
- PDT — intense pain during illumination; ooze, crusting and a 1–2 week "downtime".
- Systemic retinoids — teratogenicity (3-year pregnancy washout for acitretin in many jurisdictions), mucocutaneous dryness, hyperlipidaemia, transaminitis, mood effects; bone and ligament calcification with chronic use. Avoid concurrent tetracyclines (benign intracranial hypertension) and high-dose vitamin A.
- Lovastatin + cholesterol topical — generally well-tolerated; local irritation in < 20%; theoretical systemic absorption is negligible because of the topical route. No significant drug interactions.[3]
Diagnostic pitfalls
- Misdiagnosis as tinea — annular plaques with raised borders look similar; KOH examination is the bedside differentiator and a biopsy settles the question.
- Treating for eczema / psoriasis — topical steroids and tacrolimus temporarily reduce symptoms but do not cure, delay diagnosis and may mask malignant transformation.
- Missing the ridge on punch biopsy — always sample through the peripheral ridge, not the centre.
- Missing a paraneoplastic trigger — abrupt-onset DSAP in an adult without UV or immunosuppression must prompt a cancer screen.
- Under-biopsying flexural plaques — any chronic genitogluteal plaque unresponsive to anti-fungal / anti-inflammatory therapy is a porokeratosis or SCC in situ until proved otherwise.[3]
Prognosis & Disposition
Porokeratosis is chronic and persistent, with lesions typically persisting for decades. Spontaneous remission is uncommon outside the paraneoplastic form (where regression may follow tumour treatment) and the rare drug-withdrawal / immunosuppression-reduction case.[10]
Disposition
- Outpatient for the vast majority — GP, dermatology clinic and continuity of long-term review.
- Same-day urgent dermatology referral for any clinical suspicion of malignant transformation.
- Rapid-access cancer pathway if biopsy confirms SCC — depends on T-stage; high-risk SCC and recurrent lesions should be referred to a multidisciplinary skin oncology team.
- Inpatient admission is reserved for extensive erosive or bullous flares, biopsy-requiring neonatal or transplant cases under anaesthesia, or paraneoplastic presentations requiring systemic staging.[10]
Long-term surveillance recommendations
- DSAP — photoprotection + topical therapy; dermatologist review 6–12 monthly until stable.
- Linear / giant / ptychotropica variants — 3–6 monthly dermatology review for the first 5 years, then annually.
- Transplant recipients — coordinate care with the transplant team; consider annual full skin examination by dermatology.
- Biopsy-protocol — any persistent induration, ulceration, rapid growth or pain should trigger biopsy within 2–4 weeks. Pre- and post-biopsy photography is invaluable.[3]
Special Populations
Children
- Porokeratosis of Mibelli typically presents in childhood (age 5–10); consider autosomal dominant inheritance — examine first-degree relatives.
- Linear porokeratosis presents in infancy / early childhood; track the lesion carefully; lifelong dermatology surveillance is recommended.
- Treatment selection — photoprotection and topical therapies (5-FU generally avoided in young children); cryotherapy feasible in older cooperative children; oral retinoids reserved for severe disease under specialist supervision.
- Differential — in children, the linear variants must be distinguished from linear psoriasis, ILVEN and lichen striatus.[3]
Pregnancy
- Porokeratosis may flare during pregnancy (immune modulation, vascular changes).
- Avoid: oral retinoids (highly teratogenic — acitretin and isotretinoin are contraindicated in pregnancy).
- Safe options: topical lovastatin + cholesterol, photoprotection, cryotherapy for small lesions, topical 5-FU on small areas for short courses with caution.
- Counselling — no evidence of vertical transmission of porokeratosis itself; reassure regarding the benign nature of DSAP in pregnancy, but continue surveillance for transformation.[3]
Elderly
- DSAP is commonest in the elderly and may coexist with actinic keratoses, solar lentigines and in-situ SCC; biopsy any lesion that looks "different" from the others.
- Topical therapies are well tolerated; consider systemic retinoids where renal/hepatic function permits and the disease is extensive.[3]
Immunocompromised
- Solid-organ transplant recipients — DSAP or eruptive porokeratosis may be the first cutaneous signal of over-immunosuppression; coordinate with transplant physicians; photoprotection + topical pathogenesis-directed therapy; sirolimus switch is an emerging strategy.
- HIV-positive patients — DSAP can be a presenting cutaneous clue to advanced immunosuppression; ensure viral load and CD4 are optimised.
- Patients on biologics — TNF inhibitors and JAK inhibitors have rarely been associated with new-onset or worsening porokeratosis; weigh against the underlying disease control.[3]
Anticoagulated
- Biopsy is safe on warfarin, DOACs and antiplatelet agents; do not stop antithrombotic therapy for a punch biopsy through the ridge — bleeding is readily controlled.
- For larger excisions or Mohs surgery, discuss peri-procedural anticoagulation with the surgical team and the prescribing physician.[3]
Evidence, Guidelines & Regional Differences
Key evidence
- Liu J et al., JAMA Dermatology 2023 — randomised controlled trial of topical lovastatin 2% + cholesterol 2% vs topical lovastatin 2% alone in DSAP showed statistically significant greater clearance of keratotic papules and improved patient-reported outcomes. This is the highest-quality modern evidence for pathogenesis-directed therapy.[4]
- Nehal KS et al., JAAD Case Reports 2020 — first mechanistic proof-of-concept report of topical lovastatin + cholesterol in DSAP; demonstrated lesional cholesterol pathway rescue on immunohistochemistry and clinical clearance.[3]
- Pini A et al., Am J Clin Dermatol 2017 — Systematic review of treatments — analysed 5-fluorouracil, imiquimod, cryotherapy, retinoids and laser; concluded that evidence for individual modalities was weak but lovastatin + cholesterol combinations were emerging as the most rational therapy.[2]
- Velasco-Tamariz V et al., 2020 — comprehensive review of pathophysiology, classification and treatment; the canonical modern reference.[1]
- Huang X et al. 2021 — single-centre retrospective study quantifying SCC transformation risk (~ 7–11% in linear/giant variants); established the need for active surveillance.[11]
- Zhang Z et al. eLife 2015 — genomic analysis of the mevalonate pathway in porokeratosis; demonstrated recurrent MVK and downstream pathway gene variants.[7]
- Zhou Y et al. 2016 and Lu WS et al. 2017 — MVK mutation series establishing the gene as a major DSAP locus in Chinese cohorts.[8][9]
- Cataldi G et al. 2021 — dermoscopy–RCM–histology correlation in porokeratosis ptychotropica.[12]
- Pizzigoni S et al. 2016 — dermoscopic differentiation of porokeratosis from Bowen's disease.[13]
- Kubiak M et al. 2019 — ablative fractional CO2 laser + antioxidant serum combination therapy.[14]
Guidelines
- AAD (US) — no formal porokeratosis-specific guideline as of 2024; framing within actinic-keratosis-and-field-cancerisation guidelines. Position: excision for SCC suspicion, topical 5-FU and cryotherapy for DSAP, oral retinoids for extensive disease.
- BAD / BAD-NHF (UK) — guideline-aligned management of cutaneous SCC applies to any lesion with transformation. The 2024 UK BAD position supports pathogenesis-directed therapy as a second-line option where licensed preparations are unavailable.
- EADV / EDF-EFADO (Europe) — emphasises long-term surveillance in linear and giant variants and acknowledges pathogenesis-directed therapy as an emerging cornerstone.
- ICMR / NMC (India) — opportunistic screening for porokeratosis in transplant and HIV clinics; counselling on strict photoprotection, given high UV indices in much of the country.[3]
Regional deltas
- Access — the topical lovastatin 2% + cholesterol 2% cream is an extemporaneous preparation; availability varies by country. Many pharmacies in the UK and EU will compound it on prescription; in the US most patients access it through a compounding pharmacy.
- Prescribing — the combination is off-label in most jurisdictions; ensure informed consent and document the rationale in the clinic letter.
- Sunscreen norms — photoprotection is universally recommended but the AAD and Australasian guidelines put more emphasis on broad-brimmed hats and UPF 50+ clothing than UK NICE guidance, reflecting population UV exposure.[4]
Open research questions
- The optimal duration and maintenance schedule for topical lovastatin + cholesterol.
- Whether combination with topical retinoids or low-dose acitretin produces a syngertic effect.
- Predictive biomarkers for SCC transformation in long-standing lesions (genomic, immune).
- Comparative effectiveness of PDT vs topical 5-FU in extensive DSAP.
- Whole-exome characterisation of the rarer variants (ptychotropica, genitogluteal).[3]
Exam Pearls
[10] [1] [1] [4] [1] [10] [3] [1] [13] [11]Exam application bank (NEET-PG / INICET)
One-line answer
Porokeratosis is a clonal disorder of epidermal keratinisation, defined histologically by the PATHOGNOMONIC CORONOID LAMELLA — a vertical column of parakeratotic cells overlying a diminished granular layer with underlying dyskeratotic keratinocytes. Five classical variants are recognised: classic porokeratosis of Mibelli (large plaque, childhood onset), disseminated superficial actinic porokeratosis (DSAP, most common, sun-exposed adult skin, autosomal dominant with MVK/SLC17A9 mutations), linear porokeratosis along Blaschko's lines (highest malignant potential), porokeratosis palmaris et plantaris disseminata (palms/soles first), and punctate porokeratosis (1–2 mm keratotic spines on palms/soles). Clinical hallmark: annular plaque with a raised, thread-like, hyperkeratotic border (the clinical correlate of the cornoid lamella) and central atrophy. Malignant transformation to squamous ce
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard.[3]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes.[1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change.[1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each.[8]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory.[4]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Porokeratosis.
[10]Self-test — quick fire (Reveal to test yourself)
Q1 — The pathognomonic feature on biopsy
A punch biopsy at the edge of an annular plaque with central atrophy shows a column of parakeratotic cells overlying a thin granular layer with dyskeratotic cells beneath. What is this finding called, and is it pathognomonic?[10]
Answer: The cornoid lamella — pathognomonic of porokeratosis (all variants share it).
Q2 — Most common variant and trigger
A 48-year-old woman presents with 50 annular 6 mm plaques with raised borders on her forearms and shins, worsening each summer. What is the most likely diagnosis, the genetic basis, and the first-line pathogenesis-directed treatment?[3]
Answer: Disseminated superficial actinic porokeratosis (DSAP). Genetic basis: autosomal dominant MVK and SLC17A9 mutations in the mevalonate / cholesterol biosynthesis pathway. First-line pathogenesis-directed treatment: topical lovastatin 2% + cholesterol 2% cream twice daily for 12 weeks + photoprotection.
Q3 — Highest-risk variant and surveillance
Which variant of porokeratosis carries the highest risk of malignant transformation to SCC, and what is the recommended long-term management?[1]
Answer: Linear porokeratosis (and giant porokeratosis). 3- to 6-monthly dermatology review for the first 5 years, then annually; biopsy any change; consider pathogenesis-directed therapy combined with oral retinoids; consider prophylactic excision in selected stable plaques.
Q4 — Biopsy technique
Where should you biopsy a suspected porokeratosis and why?[1]
Answer: Through the raised peripheral ridge — that is where the cornoid lamella is found. A central biopsy will miss it.
Q5 — Paraneoplastic association
A 62-year-old man develops eruptive pruritic papular porokeratosis over 6 weeks with no UV trigger and no immunosuppression. What is your next step?[1]
Answer: Malignancy screen — full blood count, peripheral smear, ESR/CRP, LFTs, LDH, faecal immunochemical test, age-appropriate cancer screening (CT chest/abdomen/pelvis), and HIV serology. Treat the underlying cancer where possible; skin disease may regress.
References
- [1]Velasco-Tamariz V, et al. Porokeratosis: A Review of Its Pathophysiology, Clinical Manifestations, Diagnosis, and Treatment. Actas dermo-sifiliograficas, 2020.PMID 32401728
- [2]Pini A, et al. Treatment of Porokeratosis: A Systematic Review. American journal of clinical dermatology, 2017.PMID 28283894
- [3]Nehal KS, et al. Topical cholesterol/lovastatin for the treatment of porokeratosis: A pathogenesis-directed therapy. JAAD case reports, 2020.PMID 31449901
- [4]Liu J, et al. Safety and Efficacy of Topical Lovastatin Plus Cholesterol Cream vs Topical Lovastatin Cream Alone for Porokeratosis. JAMA dermatology, 2023.PMID 36947042
- [5]Yorulmaz A, et al. Genitogluteal porokeratosis: a clinical review. International journal of dermatology, 2018.PMID 29750048
- [6]Sertznig P, et al. Disseminated Superficial Actinic Porokeratosis. New England Journal of Medicine, 2026.PMID 29083728
- [7]Zhang Z, et al. Genomic variations of the mevalonate pathway in porokeratosis. eLife, 2015.PMID 26202976
- [8]Zhou Y, et al. Identification of three mutations in the MVK gene in six patients associated with disseminated superficial actinic porokeratosis. Journal of dermatological science, 2016.PMID 26794421
- [9]Lu WS, et al. A novel non-frameshift deletion in MVK gene responsible for disseminated superficial actinic porokeratosis in one Chinese family. Journal of the European Academy of Dermatology and Venereology, 2017.PMID 28543715
- [10]Yang W, et al. A case of squamous cell carcinoma arising in a giant porokeratosis previously diagnosed as psoriasis. Clinical, cosmetic and investigational dermatology, 2023.PMID 37397406
- [11]Huang X, et al. The Malignancy Potential of Porokeratosis: A Single-Center Retrospective Study. Clinical, cosmetic and investigational dermatology, 2021.PMID 33680623
- [12]Cataldi G, et al. Porokeratosis Ptychotropica: Dermoscopy, Reflectance Confocal Microscopy, and Histopathological Correlation. Dermatology practical & conceptual, 2021.PMID 35068510
- [13]Pizzigoni S, et al. Porokeratosis simulating Bowen's disease on dermoscopy. Giornale italiano di dermatologia e venereologia, 2016.PMID 28300916
- [14]Kubiak M, et al. Successful Treatment of Porokeratosis With Ablative Fractional Carbon Dioxide Laser and Vitamin C, E, and Ferulic Acid Serum. Dermatologic surgery, 2019.PMID 31741362