Dermatology · Medicine
Sarcoidosis
Also known as Sarcoidosis · Cutaneous sarcoidosis · Besnier-Boeck-Schaumann disease
Sarcoidosis is a multisystem granulomatous disease of unknown cause characterised by non-caseating ('naked') granulomas in multiple organs — lungs (90%), lymph nodes, skin (25-30%), eyes. Cutaneous lesions are divided into specific (granulomatous on biopsy: papules, plaques, lupus pernio, scar sarcoidosis) and non-specific (reactive: erythema nodosum). Lupus pernio (chronic violaceous indurated plaques on nose/cheeks) is the most disfiguring cutaneous form and signals chronic pulmonary + upper respiratory tract disease. Löfgren syndrome (erythema nodosum + bilateral hilar lymphadenopathy + ankle arthritis) is an acute, self-limiting presentation with good prognosis. Diagnosis requires the triad of compatible clinical + radiological + histological findings, excluding TB and fungal infection. Management: topical/intralesional corticosteroids and hydroxychloroquine for cutaneous disease; oral corticosteroids for organ-threatening systemic disease; methotrexate and TNF inhibitors (infliximab, adalimumab) for refractory cases.
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags

Definition and Epidemiology
Sarcoidosis is a multisystem granulomatous disease of unknown cause characterised by the formation of non-caseating ('naked') granulomas in multiple organs. The lung and lymph nodes are most commonly affected (~90%), but the skin, eyes, heart, nervous system, liver, spleen, bones, and kidneys may all be involved.[1][6]
- Incidence: varies geographically; higher in Black/African-American, Scandinavian, and Irish populations.[6]
- Age/sex: bimodal (25-35 and 45-65); slight female predominance.
- Cutaneous involvement: 25-30% of patients; may be the presenting feature and the most accessible site for biopsy.[3][4]
Pathophysiology
Unknown antigen (possibly infectious — mycobacteria, Propionibacterium acnes, environmental, occupational — silica, beryllium) triggers an exaggerated Th1-cell-mediated immune response in genetically susceptible individuals (HLA-DRB1*03, HLA-B8, BTNL2 associations). Activated CD4+ T-cells and macrophages accumulate and organise into non-caseating granulomas. Activated macrophages produce 1,25-dihydroxyvitamin D (causing hypercalcaemia) and angiotensin-converting enzyme (ACE) (elevated in serum; used as a non-specific activity marker).[1]
[1]Quick numbers for the examiner
Cutaneous Manifestations
Cutaneous lesions are divided into specific (granulomatous on biopsy, confirming systemic sarcoidosis) and non-specific (reactive; erythema nodosum is the prototype).[3][4]
[1]GRANULOMA - sarcoidosis features
Epithelioid cells, multinucleated giant cells, sparse lymphocytic cuff; no necrosis
1-alpha hydroxylase in macrophages converts 25-OH to 1,25-(OH)2 vit D; nephrolithiasis
PPD negative (anergy); helps differentiate from TB; BCG contraindicated
Sterile granulomas; rule out TB, fungal, sarcoid-like reactions (drugs, malignancy)
Anterior uveitis most common; emergency to prevent vision loss
BHL on CXR Scadding stage 1; symmetric; egg-shell calcification
Limited cutaneous SSc overlap; smooth muscle atrophy
Cardiac sarcoid: heart block, arrhythmia, SCD; ECG + cardiac MRI
Serum ACE 60% sensitive, 70% specific; tracks disease activity
Specific Subtypes & High-Yield Scenarios [1]
- Lupus pernio: violaceous indurated plaques on the nose, cheeks, ears, lips, fingers; strong predictor of chronic systemic sarcoidosis with upper airway and bone involvement; most treatment-resistant cutaneous form.
- Scar/tattoo sarcoidosis: granulomatous infiltration of old scars, tattoos, venepuncture sites; valuable diagnostic clue; Do NOT tattoo a sarcoidosis patient (it will reactivate).
- Darier-Roussy subcutaneous sarcoidosis: painless firm subcutaneous nodules on the trunk or extremities; deep granulomas in the subcutis; can mimic panniculitis or lymphoma.
- Mucosal sarcoidosis: oral, nasal, conjunctival; oral involvement in approximately 5-10% of patients; can affect salivary glands (xerostomia).
- Nail sarcoidosis: rare; nail dystrophy, clubbing, onycholysis, subungual hyperkeratosis; usually indicates chronic disease.
- Erythema nodosum as part of Lofgren syndrome: acute presentation; excellent prognosis; bilateral hilar lymphadenopathy + ankle arthritis + EN + fever; 90% resolve within 2 years.
- Heerfordt syndrome (uveoparotid fever): anterior uveitis + parotid gland enlargement + CN VII (facial nerve) palsy + fever; rare but characteristic.
- Mimickers: cutaneous TB, granuloma faciale, granulomatous rosacea, sarcoidal granuloma annulare, drug-induced granulomas (interferon), berylliosis, foreign body granuloma.
- Paraneoplastic sarcoidosis: case reports of sarcoid-like granulomas triggered by malignancy (haematological > solid); investigate if atypical course.
- Blau syndrome / Early-onset sarcoidosis: autosomal dominant NOD2 mutations; triad of granulomatous arthritis, uveitis, skin rash in children; can mimic JIA. [1]
Complications & Pitfalls
- Cardiac sarcoidosis is the most-feared complication (sudden cardiac death from ventricular arrhythmia or complete heart block). ECG and cardiac MRI at diagnosis. Holter monitoring; consider ICD if indicated.
- Neurosarcoidosis can present with isolated CN VII palsy (often bilateral — "facial diplegia"); always exclude Lyme disease, GBS, sarcoidosis.
- Hypercalcaemia (10-20%): from 1,25-dihydroxyvitamin D; nephrocalcinosis, nephrolithiasis, renal failure; avoid vitamin D and sun exposure.
- Uveitis (10-30%): anterior > posterior; can lead to synechiae, glaucoma, cataract, vision loss; urgent ophthalmology.
- Pulmonary fibrosis (Scadding stage 4): restrictive pattern; right heart failure; lung transplant may be needed.
- Pitfall: misdiagnosis as TB leading to unnecessary anti-tuberculous therapy; always send AFB and TB culture from any granulomatous biopsy.
- Drug-induced sarcoidosis: interferon, immune checkpoint inhibitors, TNF inhibitors (paradoxical), allopurinol; review medications. [1]
Special Populations
- Children: rare; usually presents with skin, joint, eye involvement; Blau syndrome with NOD2 mutations; lung involvement less common.
- Pregnancy: may improve during pregnancy (Th2 shift); flare postpartum; consider drug safety (methotrexate contraindicated; azathioprine relatively safe; biologics data limited).
- Ethnicity: African-American patients have higher incidence, more severe disease, more extrapulmonary involvement (eye, skin); Scandinavian and Irish populations also high.
- Elderly: late-onset sarcoidosis (>50 yr) tends to be more chronic; higher risk of cardiac involvement.
- Immunocompromised: can develop sarcoidosis-like granulomas as immune reconstitution (immune reconstitution inflammatory syndrome IRIS) in HIV patients starting ART; in transplant recipients on immunosuppression withdrawal. [1]
Evidence, Guidelines & Regional Differences (Extended)
- ATS/ERS/WASOG 1999 statement on sarcoidosis: foundational classification criteria and assessment.
- British Thoracic Society (BTS) 2008: interstitial lung disease guideline including sarcoidosis.
- WASOG (World Association of Sarcoidosis and Other Granulomatous disorders): organ assessment instrument; international registry.
- First-line therapy: systemic corticosteroids (prednisone 0.5-1 mg/kg/day, taper over 6-12 months).
- Steroid-sparing agents: methotrexate (most common second-line), azathioprine, mycophenolate mofetil, hydroxychloroquine (especially for cutaneous and hypercalcaemia), minocycline, doxycycline.
- Biologics: infliximab and adalimumab for refractory disease; small RCTs show benefit for cutaneous and pulmonary disease; biosimilars available.
- Repository corticotropin (ACTH) gel: FDA-approved for sarcoidosis (rare use).
- JAK inhibitors and anti-IL-12/23 (ustekinumab): under investigation.
- Regional differences: US uses more methotrexate; Europe more azathioprine; India more hydroxychloroquine for cutaneous disease. [1]
Exam Pearls (Extended)
[1]Specific cutaneous lesions
- Papules and plaques: red-brown to violaceous; commonly on face, neck, shoulders, extensor limbs; may show apple-jelly colour on diascopy (similar to cutaneous TB). Often annular or serpiginous.
- Lupus pernio: the most characteristic and disfiguring cutaneous manifestation — chronic, violaceous, indurated plaques on the nose, cheeks, ears, lips, fingers. Strongly associated with chronic pulmonary sarcoidosis, upper respiratory tract involvement, and bone cysts. Usually resistant to treatment.[3]
- Scar sarcoidosis: granulomatous infiltration of old surgical/trauma scars, tattoos, or venepuncture sites — a valuable diagnostic clue (sarcoidosis 'seeks out' scars).
- Subcutaneous nodules (Darier-Roussy sarcoidosis): painless firm subcutaneous nodules on trunk/extremities; granulomas in subcutis.
- Ulcerative, verrucous, hypopigmented, ichthyosiform, and alopecic variants are rarer.
Non-specific cutaneous lesions
- Erythema nodosum (EN): tender red nodules on shins; the commonest non-specific reactive lesion. Part of Löfgren syndrome (see below). Histology shows septal panniculitis (NOT granulomatous), so EN does not confirm sarcoidosis on biopsy.[5]
Systemic Features and Named Syndromes

Pulmonary (90%)
- Bilateral hilar lymphadenopathy (BHL) — the radiological hallmark.
- Interstitial lung disease — reticulonodular infiltrates, fibrosis (upper-lobe predominant).
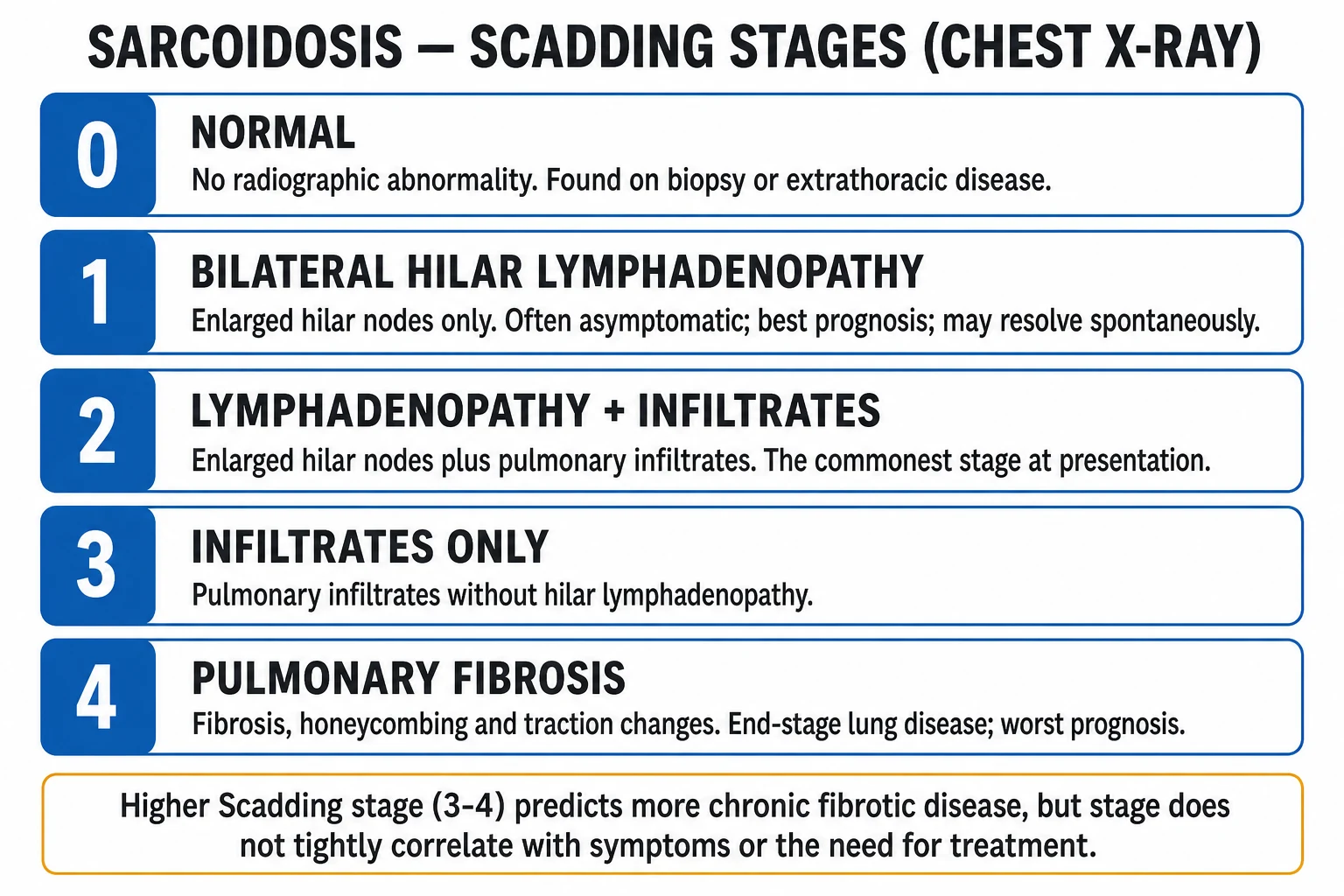
- Scadding chest X-ray stages (0-4): Stage 0 = normal; Stage 1 = BHL only; Stage 2 = BHL + infiltrates; Stage 3 = infiltrates only (no BHL); Stage 4 = pulmonary fibrosis.[1]
- Symptoms: dry cough, dyspnoea.
Named syndromes
- Löfgren syndrome (acute sarcoidosis): the triad of erythema nodosum + bilateral hilar lymphadenopathy + ankle arthritis/peri-arthritis ± fever. Self-limiting; usually resolves within 2 years; excellent prognosis; often needs no treatment (NSAIDs ± colchicine). HLA-DRB1*03 association.[3][5]
- Heerfordt syndrome (uveoparotid fever): uveitis + parotid gland enlargement + facial nerve (CN VII) palsy + fever. May be a presenting feature.[1]
Other organ involvement
- Ocular: anterior uveitis (common), conjunctival nodules, dry eye (keratoconjunctivitis sicca); uveitis may cause visual loss.
- Cardiac: arrhythmias, conduction defects (heart block), cardiomyopathy; sudden cardiac death risk — ECG and cardiac MRI warranted in all newly diagnosed patients.[2]
- Neurological (neurosarcoidosis): cranial nerve palsies (especially VII), aseptic meningitis, hypothalamic/pituitary involvement (diabetes insipidus), seizures.
- Renal/endocrine: hypercalcaemia and hypercalciuria (from 1,25-dihydroxyvitamin D production by activated macrophages); nephrocalcinosis, nephrolithiasis, renal impairment.
- Hepatic/splenic: granulomatous hepatitis, hepatosplenomegaly; abnormal LFTs.
- Bone: cystic radiolucent lesions (especially phalanges of hands); often coexists with lupus pernio.
- Salivary/lacrimal glands: bilateral enlargement (may mimic Sjögren).
Histopathology

The histological hallmark is the non-caseating ('naked') granuloma:[3][4]
- Compact collection of epithelioid macrophages and multinucleated giant cells (Langhans or foreign-body type).
- Sparse surrounding lymphocytic infiltrate — the "naked" appearance (contrasts with the dense lymphocytic rim of tuberculosis granulomas).
- No central necrosis/caseation — the key distinction from tuberculosis and fungal infections.
- Asteroid bodies (star-shaped eosinophilic inclusions) and Schaumann bodies (concentric laminated calcifications) may be present within giant cells but are non-specific (also seen in berylliosis, TB, foreign-body reactions).
- Special stains (Ziehl-Neelsen for AFB, PAS/GMS for fungi) are negative — essential to exclude infectious mimics. [1]
Investigations and Diagnosis
Diagnosis requires the triad: (1) compatible clinical/radiological picture; (2) histological evidence of non-caseating granulomas in ≥1 tissue; (3) exclusion of other granulomatous diseases (TB, fungal infection, foreign-body reaction, Crohn's).[1][3]
- Skin biopsy of a specific lesion (papule/plaque/scar) — the most accessible tissue; high yield.
- Chest X-ray / HRCT chest — BHL, interstitial infiltrates, fibrosis.
- Pulmonary function tests — restrictive pattern + reduced DLCO.
- Serum ACE — often elevated but non-specific (also elevated in TB, lymphoma, thyroid disease); useful for monitoring disease activity, not diagnosis.
- Serum calcium + 24-hour urinary calcium — detect hypercalcaemia/hypercalciuria.
- FBC — lymphopenia common; LFTs, renal function, electrolytes.
- ECG ± cardiac MRI — screen for cardiac sarcoidosis.
- Ophthalmology assessment — slit-lamp for uveitis.
- Bronchoalveolar lavage — CD4:CD8 ratio >3.5 supports diagnosis.
- Gallium-67 scan — historic; "panda" sign (bilateral parotid + lacrimal uptake) and "lambda" sign (bilateral hilar + right paratracheal uptake); rarely used now. [1]
Differential Diagnosis
- Cutaneous papules/plaques: tuberculosis (lupus vulgaris — caseating granulomas; positive AFB), fungal infections (chromoblastomycosis, sporotrichosis), granuloma annulare (mucin, palisading granulomas), cutaneous Crohn's, foreign-body granuloma, lymphoma cutis, rosacea.
- Erythema nodosum (septal panniculitis): streptococcal infection, drugs (OCP, sulphonamides), inflammatory bowel disease, Behçet, pregnancy, malignancy.[5]
- Bilateral hilar lymphadenopathy: lymphoma, TB, metastatic malignancy, pneumoconiosis.
Management
Cutaneous sarcoidosis
- Localised disease: topical corticosteroids (potent) or intralesional triamcinolone injections.[3]
- Disfiguring/extensive disease: hydroxychloroquine (antimalarial; first-line systemic for cutaneous disease); oral corticosteroids (short course) for severe or rapid control.[3][7]
- Steroid-sparing/refractory: methotrexate (most commonly used steroid-sparing agent), azathioprine, mycophenolate mofetil, leflunomide.[7]
- Refractory lupus pernio: TNF-α inhibitors (infliximab, adalimumab) — particularly effective for lupus pernio and refractory cutaneous disease.[3][7]
Systemic sarcoidosis

- Löfgren syndrome: self-limiting; NSAIDs ± colchicine; corticosteroids rarely needed.[3]
- Symptomatic or progressive pulmonary disease: oral corticosteroids (prednisolone 20-40 mg/day, taper over 6-24 months) — first-line.[2]
- Cardiac, neuro, ocular (sight-threatening), or hypercalcaemia with renal involvement: corticosteroids mandatory.[2]
- Steroid-sparing: methotrexate, azathioprine, mycophenolate mofetil.[2][7]
- Refractory: TNF-α inhibitors (infliximab, adalimumab); rituximab.[7]
Organ-Specific Management: Pulmonary, Cardiac & Neurosarcoidosis (Extended)
When sarcoidosis threatens a vital organ, treatment is mandatory, sustained, and escalates along a defined steroid-to-steroid-sparing-to-biologic pathway. The ERS 2021 clinical practice guidelines stratify therapy by organ involvement and disease chronicity; topical and cutaneous strategies are insufficient when the lung parenchyma, conduction system, or central nervous system is involved.[2]
Pulmonary sarcoidosis is the most common organ-threatening manifestation (90%) and the principal driver of morbidity. Asymptomatic stage I disease (BHL alone) and self-limiting Löfgren syndrome generally do not require corticosteroids — observation alone is appropriate, with serial spirometry, 6-minute walk test, and chest imaging every 3-6 months.[2] Indications for treatment include: (1) symptomatic parenchymal disease with cough/dyspnoea, (2) declining FVC or FEV1 (typically > 10% relative fall over 6-12 months), (3) progressive radiological change from stage I/II to stage III/IV, (4) significant gas transfer (DLCO) impairment, or (5) pulmonary hypertension. First-line treatment is oral prednisolone 20-40 mg/day for 2-4 weeks, then a slow taper to 5-10 mg/day over 6-12 months. Relapse on taper is common (up to 50%) and signals the need for a steroid-sparing agent. Second-line steroid-sparing agents include methotrexate 10-25 mg weekly (most evidence), azathioprine 100-200 mg/day, mycophenolate mofetil 1-2 g/day, and leflunomide; hydroxychloroquine is particularly useful when there is concomitant hypercalcaemia or cutaneous disease.[2][7] Third-line biologics for refractory or fibrotic pulmonary sarcoidosis include infliximab 5 mg/kg at weeks 0, 2, 6, then every 4-8 weeks, and adalimumab 40 mg subcutaneously weekly or biweekly; both have RCT evidence in pulmonary and cutaneous disease.[7] Antifibrotic therapy (nintedanib, pirfenidone) is under investigation for stage IV fibrosis; lung transplantation remains the definitive option for end-stage disease. Pulmonary rehabilitation, supplemental oxygen, smoking cessation, vaccination (influenza, pneumococcal, COVID-19), and screening for pulmonary hypertension by echocardiography are all part of comprehensive care.
Cardiac sarcoidosis is the leading cause of sarcoidosis-related mortality (up to 25% of sarcoid deaths in some series) due to sudden cardiac death from ventricular tachycardia or complete heart block.[2] All newly diagnosed patients warrant a 12-lead ECG, Holter monitoring for 24-48 hours, and a transthoracic echocardiogram; cardiac MRI with late gadolinium enhancement (LGE) is the gold-standard non-invasive test, with sensitivity ~95% for active myocardial inflammation when combined with T2-weighted oedema imaging. FDG-PET (with dietary preparation to suppress physiological myocardial glucose uptake) is used to assess active inflammation, monitor treatment response, and guide biopsy site if endomyocardial sampling is required. Diagnostic criteria follow the Japanese Ministry of Health (JMH) and Heart Rhythm Society (HRS) 2014 consensus, requiring histological confirmation from cardiac tissue OR a combination of clinical, imaging, and ECG criteria. Treatment principles: oral corticosteroids are mandatory — typically prednisolone 30-60 mg/day for 2-4 weeks, slow taper to 5-10 mg/day over 6-12 months. Early steroid therapy improves outcomes; delays more than 6 months from symptom onset reduce ejection fraction recovery. Steroid-sparing agents (methotrexate, azathioprine, mycophenolate) are added when relapses occur during taper; infliximab is particularly effective for active myocardial sarcoidosis (CHASM-CS trial evidence). Device therapy: permanent pacemaker (PPM) for high-grade AV block; implantable cardioverter-defibrillator (ICD) for secondary prevention after VT/VF arrest, or primary prevention if LVEF remains ≤35% despite optimal medical therapy, or in patients with extensive LGE on cardiac MRI. Anti-arrhythmic therapy (amiodarone, sotalol) may be required for recurrent VT; catheter ablation is adjunctive for VT storm. Heart transplantation is an option for refractory end-stage cardiac sarcoidosis — outcomes comparable to other aetiologies.
Neurosarcoidosis affects approximately 5% of patients and is potentially devastating because granulomas infiltrate cranial nerves, meninges, hypothalamus, pituitary, spinal cord, and peripheral nerves.[2] The cranial nerves most commonly involved are CN VII (facial palsy — most frequent, often bilateral, "facial diplegia", mimicking Lyme disease, GBS, and Bell's palsy), CN II (optic neuropathy with visual loss), CN VIII (sensorineural hearing loss/vertigo), and CN V (facial numbness). Clinical patterns: (1) cranial neuropathy (50% — most common); (2) meningeal disease (aseptic meningitis, chronic meningitis); (3) parenchymal brain or spinal cord lesions (mass-like or diffuse); (4) hypothalamic-pituitary involvement (diabetes insipidus, panhypopituitarism, hyperprolactinaemia); (5) peripheral neuropathy (small-fibre neuropathy with burning pain and autonomic dysfunction). Diagnostic workup includes contrast-enhanced MRI brain (and spine if symptoms suggest), CSF analysis (lymphocytic pleocytosis, elevated protein, low glucose in ~25%; CSF ACE insensitive), and tissue confirmation where feasible (meningeal or brain biopsy, sometimes Kveim-style blind biopsy). Serum ACE and chest CT help but are non-specific. Treatment of neurosarcoidosis mandates higher corticosteroid doses than pulmonary disease — typically IV methylprednisolone 500-1000 mg/day for 3-5 days followed by oral prednisolone 1 mg/kg/day (max 60-80 mg), tapered slowly over 12-24 months given the high relapse rate (up to 50%). Steroid-sparing agents should be introduced early — methotrexate, azathioprine, or mycophenolate; infliximab 5 mg/kg is highly effective for refractory neurosarcoidosis and is increasingly used as early steroid-sparing second-line. Adjunctive therapy: antiepileptics for seizures, desmopressin for diabetes insipidus, hormone replacement for pituitary failure. Prognosis is variable: cranial neuropathy alone often responds well; parenchymal brain/spinal disease has higher relapse and morbidity.
HANDLE - organ-threatening sarcoidosis first-line therapy
Prednisolone 1 mg/kg/day for neuro; 30-60 mg/day cardiac; 20-40 mg/day pulmonary
Methotrexate 1st-line; azathioprine, mycophenolate alternatives
Slow taper over 6-24 months; relapse risk 30-50% on rapid taper
ICD for cardiac (LVEF ≤35% or VT/VF); PPM for complete heart block
ECG, Holter, echo, spirometry, MRI every 6-12 months; relapse can occur years later
Infliximab 5 mg/kg or adalimumab 40 mg weekly for refractory organ disease
Sarcoidosis organ-specific management: escalation timeline
Clinical pearls — organ-threatening sarcoidosis
- Cardiac sarcoidosis is the #1 cause of sarcoidosis-related death; never dismiss palpitations or syncope in a known sarcoidosis patient.
- The chest X-ray can be normal in cardiac sarcoidosis (only 5% have BHL at cardiac presentation); cardiac MRI with LGE is the imaging gold standard.
- Neurosarcoidosis presenting as bilateral CN VII palsy is a classic fellowship exam vignette — exclude Lyme, GBS, sarcoid, lymphoma.
- Infliximab is the biologic of choice for refractory cardiac, neuro, and lupus pernio disease; adalimumab is an alternative if infliximab fails.
- Infliximab is contraindicated in active TB — always screen with QuantiFERON/IGRA before starting anti-TNF therapy.
- Steroid-induced side effects (osteoporosis, diabetes, hypertension, cataracts) are substantial — add bone protection (calcium, vitamin D, bisphosphonate) and monitor glucose/BP from day one.
- Methotrexate is teratogenic — both men and women need contraception; switch to azathioprine before conception.
- Cardiac sarcoidosis can present years after initial diagnosis — maintain a low threshold for ECG and cardiac MRI in any sarcoidosis patient with new cardiac symptoms.
- Pulmonary hypertension in sarcoidosis (group 5) requires right heart catheter confirmation and specific therapy (endothelin antagonists, PDE5 inhibitors).
- Watch for paradoxical sarcoidosis when using anti-TNF for other indications (rheumatoid arthritis, IBD) — new granulomatous disease may emerge.
Specific Drug Doses (Extended)
Sarcoidosis — specific drug doses for cutaneous and systemic disease
| Drug | Indication | Starting dose | Maintenance / taper notes | Monitoring |
|---|---|---|---|---|
| Prednisone (oral) | First-line for organ-threatening disease (cardiac, neuro, sight-threatening uveitis, hypercalcaemia); severe cutaneous | 0.5–1 mg/kg/day (typical max 40–60 mg/day); IV methylprednisolone 500–1000 mg/day × 3–5 days for acute cardiac/neuro presentation | Taper by 5–10 mg every 2–4 weeks to 10–15 mg/day by month 3, then by 1–2.5 mg/month; total duration 6–24 months; never stop abruptly | BP, glucose, weight, electrolytes; bone protection (calcium, vitamin D, bisphosphonate if >3 months); baseline DEXA; ocular pressure |
| Hydroxychloroquine | First-line systemic for chronic cutaneous sarcoidosis and hypercalcaemia; steroid-sparing for skin | 200–400 mg/day (≤ 6.5 mg/kg ideal body weight/day to minimise retinopathy) | Onset 3–6 months; combine with TCS or systemic agent for rapid control; ophthalmology review after 5 years (or sooner if risk factors) | Baseline + annual ophthalmology (spectral-domain OCT, visual fields); G6PD; LFTs; QTc if combined QT-prolonging drugs |
| Methotrexate | Most commonly used steroid-sparing agent (cutaneous and pulmonary); weekly dosing | 15 mg once weekly (oral or subcutaneous); folic acid 5 mg the day after (or 1 mg daily except MTX day) | Onset 6–12 weeks; max 25 mg/week; renal/hepatic dose adjust; teratogenic — both partners need contraception | Baseline + monthly FBC, LFT, creatinine; hepatitis B/C, HIV serology; CXR if pulmonary symptoms; consider pneumocystis prophylaxis if on high dose |
| Azathioprine | Steroid-sparing alternative to MTX (CNS, pulmonary, cutaneous); preferred in pregnancy | 1–3 mg/kg/day (typically 100–200 mg/day); start low if TPMT low/intermediate | Onset 3–6 months; TPMT genotype/activity mandatory before starting to avoid life-threatening myelosuppression | FBC + LFT weekly × 4 weeks, then fortnightly × 2 months, then monthly; TPMT activity; consider NUDT15 in East Asian patients |
| Mycophenolate mofetil (MMF) | Steroid-sparing for refractory cutaneous and neurosarcoidosis | 1–1.5 g twice daily (2–3 g/day total); start 500 mg BD and up-titrate over 2–4 weeks | Often combined with low-dose steroid or calcineurin inhibitor; teratogenic — contraception | FBC + LFT monthly × 6 months then 3-monthly; pregnancy test; GI side-effects common |
| Infliximab (anti-TNF-α chimeric mAb, IV) | Refractory lupus pernio, cardiac sarcoidosis, neurosarcoidosis, chronic cutaneous disease | 5 mg/kg IV at weeks 0, 2, 6 (induction), then every 4–8 weeks | Premedicate with paracetamol + antihistamine; check QuantiFERON/IGRA before starting (TB reactivation risk); weigh biosimilar use; concomitant MTX/azathioprine reduces anti-drug antibodies | Infusion reactions (acute and delayed); screen for hepatitis B; monitor LFTs; assess for paradoxical sarcoidosis |
| Adalimumab (anti-TNF-α fully human mAb, SC) | Alternative to infliximab for refractory cutaneous and systemic disease; preferred where SC administration is convenient | 40 mg SC every 2 weeks (some protocols use 40 mg weekly for refractory disease); 80 mg loading dose may be used | Pre-screen for TB, hepatitis B; rotate injection sites; check anti-drug antibodies if loss of response | Injection-site reactions; infection surveillance; LFTs; paradoxical sarcoidosis |
| Repository corticotropin (ACTH) gel | Refractory sarcoidosis (FDA-approved; rarely used outside US) | 40–80 U SC every 1–2 weeks | Acts via melanocortin and steroid-independent immunomodulation; expensive | Hyperglycaemia, hypertension, fluid retention; monitor as for chronic steroids |
| Minocycline / doxycycline | Mild cutaneous sarcoidosis (small-series evidence); anti-inflammatory rather than antimicrobial effect | Minocycline 100 mg BD or doxycycline 100 mg BD | Often combined with hydroxychloroquine; avoid in pregnancy and in children younger than 12 years | LFTs; drug rash, eosinophilia, systemic symptoms (DRESS); photosensitivity |
| Colchicine | Adjunct for erythema nodosum and acute inflammatory sarcoid arthralgia | 0.6 mg BD–TDS | Avoid in renal impairment; adjust for drug interactions (CYP3A4 inhibitors) | FBC, creatinine; counsel on diarrhoea, neuromyopathy with prolonged use |
- Prednisone 0.5–1 mg/kg/day is the cornerstone of organ-threatening sarcoidosis; for cardiac and neurosarcoidosis the upper end is preferred (often preceded by IV methylprednisolone 500–1000 mg/day × 3–5 days), and the taper extends over 6–24 months to limit relapse (relapse rate 30–50% on rapid withdrawal).
- Methotrexate 15 mg weekly (with folic acid) is the most widely used steroid-sparing agent across dermatology, rheumatology, and pulmonology sarcoid clinics; full onset takes 6–12 weeks and it is teratogenic, so both partners need contraception and a washout of ≥ 3 months before conception.
- Hydroxychloroquine 200–400 mg/day (weight-dosed ≤ 6.5 mg/kg ideal body weight) is first-line systemic therapy for chronic cutaneous sarcoidosis — particularly lupus pernio — and for sarcoid-related hypercalcaemia; retinal screening is required after 5 years of use (sooner with renal disease or tamoxifen co-administration).
- Azathioprine 1–3 mg/kg/day is a key alternative steroid-sparing agent and is the preferred immunosuppressant in pregnancy; TPMT activity must be checked before initiation because homozygous TPMT deficiency causes life-threatening pancytopenia.
- Mycophenolate 1–1.5 g twice daily (2–3 g/day total) is increasingly used for refractory cutaneous and neurosarcoidosis; it is contraindicated in pregnancy (teratogenic) and commonly causes GI side-effects.
- Infliximab 5 mg/kg IV at weeks 0, 2, 6 then every 4–8 weeks is the biologic of choice for refractory lupus pernio, cardiac sarcoidosis, and neurosarcoidosis; QuantiFERON/IGRA screening for latent TB is mandatory before the first infusion.
- Adalimumab 40 mg SC every 2 weeks (40 mg weekly for refractory disease) is the principal subcutaneous alternative to infliximab; it carries the same TB-reactivation and paradoxical-sarcoidosis risks and is preferred where home administration matters. [1]
Prognosis
- Löfgren syndrome: excellent; resolves within 2 years in most; HLA-DRB1*03 predicts good outcome.[3]
- Lupus pernio / chronic pulmonary fibrosis: chronic, progressive; poorer prognosis.[3]
- Cardiac sarcoidosis: leading cause of sarcoidosis-related death (sudden cardiac death); requires screening and treatment.[2]
- Mortality 1-5%; main causes: respiratory failure, cardiac involvement, neurosarcoidosis.[6]
Evidence and Guidelines
- ERS 2021 clinical practice guidelines: corticosteroids first-line for organ-threatening sarcoidosis; steroid-sparing agents for chronic disease; TNF inhibitors for refractory.[2]
- Treatment review (2020): multidisciplinary approach; tailoring to organ involvement.[7]
- Cutaneous sarcoidosis reviews (2023, 2024): morphological classification; hydroxychloroquine and MTX as main systemic agents; TNF inhibitors for lupus pernio.[3][4]
Exam Pearls
[1]Red Flags
Exam application bank (NEET-PG / INICET)
One-line answer
Sarcoidosis is a multisystem granulomatous disease of unknown cause characterised by non-caseating ('naked') granulomas in multiple organs — lungs (90%), lymph nodes, skin (25-30%), eyes. Cutaneous lesions are divided into specific (granulomatous on biopsy: papules, plaques, lupus pernio, scar sarcoidosis) and non-specific (reactive: erythema nodosum). Lupus pernio (chronic violaceous indurated plaques on nose/cheeks) is the most disfiguring cutaneous form and signals chronic pulmonary + upper respiratory tract disease. Löfgren syndrome (erythema nodosum + bilateral hilar lymphadenopathy + ankle arthritis) is an acute, self-limiting presentation with good prognosis. Diagnosis requires the triad of compatible clinical + radiological + histological findings, excluding TB and fungal infection. Management: topical/intralesional corticosteroids and hydroxychloroquine for cutaneous disease; or
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard. [1]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes. [1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change. [1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each. [1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory. [1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Sarcoidosis.
[1]References
- [1]Sève P, Pacheco Y, Durupt F, et al. Sarcoidosis: A Clinical Overview from Symptoms to Diagnosis Cells, 2021.PMID 33807303
- [2]Baughman RP, Valeyre D, Korsten P, et al. ERS clinical practice guidelines on treatment of sarcoidosis Eur Respir J, 2021.PMID 34140301
- [3]Ezeh N, Caplan A, Rosenbach M, et al. Cutaneous Sarcoidosis Dermatol Clin, 2023.PMID 37236714
- [4]Abdelghaffar M, Hwang E, Damsky W. Cutaneous Sarcoidosis Clin Chest Med, 2024.PMID 38245372
- [5]Pérez-Garza DM, Chavez-Alvarez S, Ocampo-Candiani J, et al. Erythema Nodosum: A Practical Approach and Diagnostic Algorithm Am J Clin Dermatol, 2021.PMID 33683567
- [6]Rossides M, Darlington P, Kullberg S, et al. Sarcoidosis: Epidemiology and clinical insights J Intern Med, 2023.PMID 36872840
- [7]Gerke AK. Treatment of Sarcoidosis: A Multidisciplinary Approach Front Immunol, 2020.PMID 33329511