Dermatology · Medicine
Tuberous sclerosis complex
Also known as Tuberous sclerosis complex (TSC) · Bourneville disease · Epiloia · Tuberous sclerosis
Tuberous sclerosis complex (TSC) is an autosomal dominant, multi-organ, hamartomatous neurocutaneous disorder caused by loss-of-function mutations in TSC1 (hamartin) or TSC2 (tuberin) leading to constitutive mTORC1 activation. Fellowship-level assessment requires the 2012/2021 International TSC Diagnostic Criteria reproduced verbatim, the full cutaneous tetrad (ash-leaf macule, adenoma sebaceum, shagreen patch, Koenen tumour), multi-organ surveillance (SEGA, cardiac rhabdomyoma, renal angiomyolipoma, pulmonary LAM, retinal hamartomas), infantile-spasm management with vigabatrin, mTOR-inhibitor pharmacology (everolimus systemic, sirolimus/rapamycin topical for facial angiofibromas and oral for LAM), TAND, and the TSC2-PKD1 contiguous gene syndrome.
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags
Overview and Definition
Tuberous sclerosis complex (TSC) is one of the canonical neurocutaneous syndromes examined across dermatology, paediatrics, neurology, nephrology, respiratory medicine and clinical genetics. It is an autosomal dominant, multi-organ, hamartomatous disorder caused by loss-of-function mutations in one of two tumour-suppressor genes — TSC1 (encoding hamartin, chromosome 9q34) or TSC2 (encoding tuberin, chromosome 16p13.3) — resulting in constitutive activation of the mTORC1 signalling pathway.[1][18]
The clinical hallmark is the formation of benign hamartomas — disorganised but mature overgrowths composed of cell types native to the tissue — in virtually every organ system: brain (cortical tubers, subependymal nodules, subependymal giant cell astrocytoma [SEGA]), heart (cardiac rhabdomyoma), kidneys (renal angiomyolipoma, cysts), lungs (lymphangioleiomyomatosis [LAM]), skin (adenoma sebaceum, ash-leaf macules, shagreen patch, Koenen tumours) and eye (retinal hamartomas).[1][2]
Three historical synonyms remain in the literature and are still tested in viva:[1]
- Bourneville disease — named after Désiré-Magloire Bourneville, the French neurologist who described the cortical tubers in 1880.
- Epiloia — an older term from the 1920s compressing "epilepsy", "low intelligence" and "adenoma sebaceum" into a single word; it appears in many older textbooks and Indian examination answer keys.
- Tuberous sclerosis — the literal description of the potato-like hardening of cerebral gyri (cortical tubers with gliosis) that Bourneville identified at post-mortem.[13]
Critical distinction from neurofibromatosis. TSC is not a variant of neurofibromatosis. Both are autosomal dominant neurocutaneous disorders with multi-organ involvement, but they are caused by mutations in different genes (NF1 on 17q11.2 encoding neurofibromin; TSC1/TSC2 on 9q34/16p13 encoding hamartin/tuberin), the molecular pathways differ (TSC = mTOR; NF1 = Ras/MAPK), the cutaneous features differ (café-au-lait and neurofibromas vs ash-leaf and angiofibromas), and the major organ complications differ (NF1 = optic glioma, MPNST, pheochromocytoma; TSC = SEGA, AML, LAM).[1]
Classification
The most clinically useful classifications divide TSC by aetiology, genetic status, and organ-specific phenotype.[1][2]
The 2012 International TSC Diagnostic Criteria (updated 2021) divide findings into major and minor criteria and are reproduced verbatim in the Investigations section of this topic.[2][3]
Epidemiology and Risk Factors
The birth incidence of TSC is approximately 1 in 6,000 to 1 in 10,000 live births, and the population prevalence is approximately 1 in 20,000, reflecting the lower end of the incidence range once premature mortality is taken into account.[1][2] Two-thirds of cases are caused by de novo mutations with unaffected parents, and approximately one-third are inherited as autosomal dominant; the lower-than-expected family clustering reflects the fact that mildly affected parents (a single angiofibroma or an isolated seizure in adulthood) are often unrecognised.[1]
Penetrance is near-complete by age 20: by then almost every individual with a pathogenic TSC1 or TSC2 variant has at least one diagnostic feature, but expressivity is strikingly variable, even within the same family, owing to differences in somatic second-hit events (see Pathophysiology).[1][16]
Sex-specific patterns matter for two organs:[1]
- LAM is almost exclusively a disease of women of reproductive age, with a clinical prevalence of 30–40% in adult women with TSC; LAM in men is rare but reported.[1][8]
- Autism spectrum disorder is more common in boys than in girls (around 50% of affected boys vs 20–30% of affected girls), paralleling the male predominance of idiopathic autism.[1]
The principal causes of premature death in TSC are sudden unexpected death in epilepsy (SUDEP), renal failure and Wunderlich haemorrhage, complications of LAM (pneumothorax, respiratory failure), and obstructive hydrocephalus from SEGA in untreated patients.[1][2] Modern mTOR-inhibitor therapy is changing this trajectory, but organised surveillance and prompt intervention remain essential.[2]
Pathophysiology
The mTOR pathway — named for the mammalian target of rapamycin — is the central node that the hamartin-tuberin complex normally restrains.[1][18]
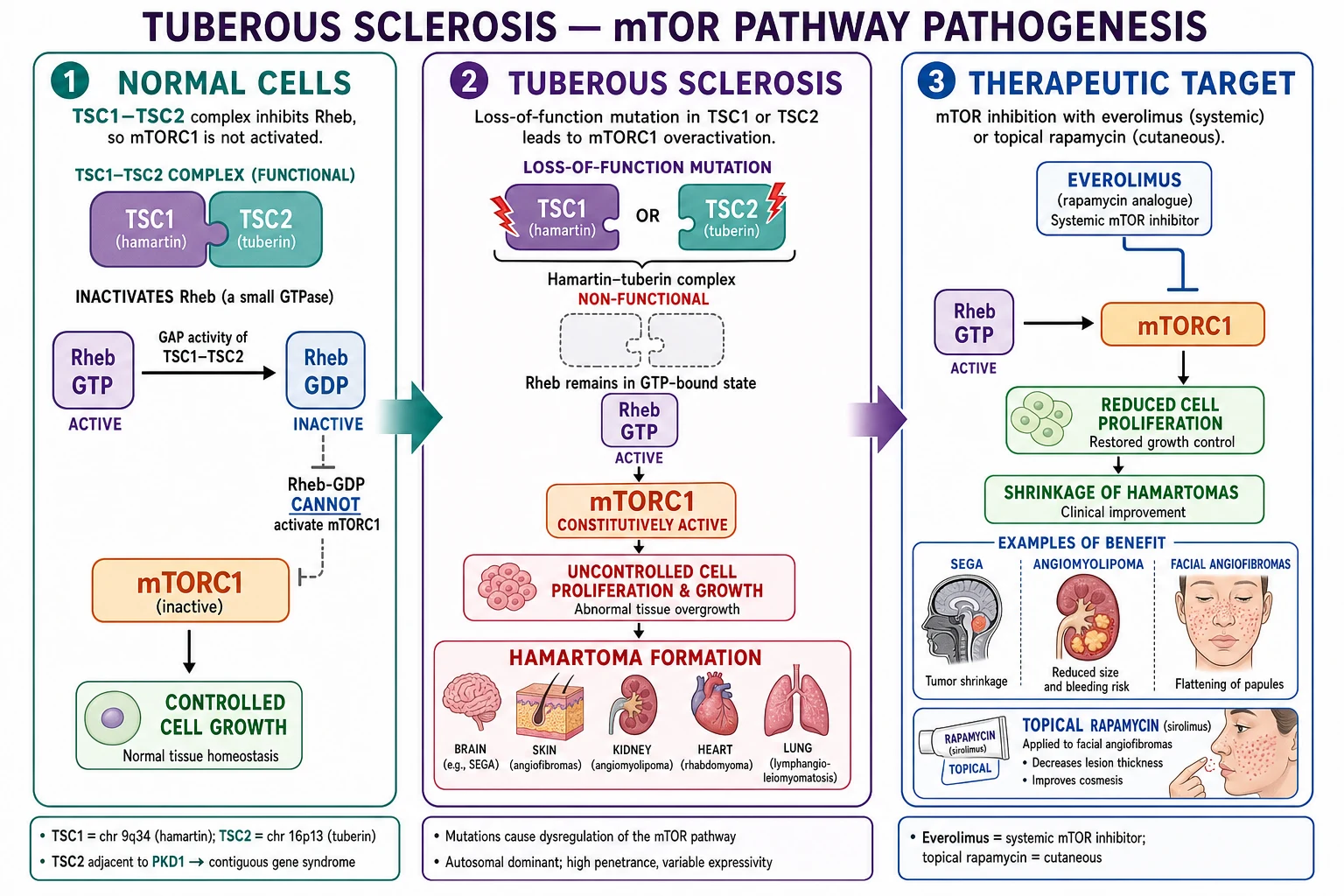
The hamartin-tuberin (TSC1-TSC2) complex
In a normally signalling cell, the proteins hamartin (encoded by TSC1) and tuberin (encoded by TSC2) form a heterodimeric complex that acts as a GTPase-activating protein (GAP) for the small GTPase Rheb (Ras homolog enriched in brain). In its GTP-bound state Rheb activates mTORC1; the GAP function of the hamartin-tuberin complex accelerates the hydrolysis of Rheb-GTP to Rheb-GDP, switching Rheb off and keeping mTORC1 quiescent in the absence of growth-factor signalling.[1][18]
When growth factors or nutrients arrive, AKT phosphorylates tuberin and releases the GAP complex from Rheb, allowing Rheb-GTP to accumulate and mTORC1 to switch on, driving translation (through S6K1 and 4EBP1), autophagy suppression, and cell growth.[1]
Loss-of-function in TSC
In TSC, a germline loss-of-function mutation inactivates one allele of either TSC1 or TSC2. Because the gene behaves as a tumour suppressor (Knudson two-hit model), a somatic second hit (loss of the remaining wild-type allele) in a susceptible cell removes the last functional GAP activity in that lineage. mTORC1 is then constitutively active, S6K1 and 4EBP1 drive unchecked translation, and the affected cell clones expand into a hamartoma — a benign overgrowth composed of cells native to the tissue (cortical neurons, renal tubular epithelium, vascular smooth muscle, adipocytes, fibroblasts).[1][11][20]
The mTOR cascade in TSC
HamTUB-RhebG
TSC1 gene product (chromosome 9q34)
Forms heterodimer with tuberin
GAP activity switches Rheb off
TSC2 gene product (chromosome 16p13)
When mutated → S6K1 / 4EBP1 drive translation
Multi-organ — brain, heart, kidney, lung, skin, eye
Why hamartomas and not carcinomas
The lesions in TSC are hamartomas — disorganised but mature collections of native-cell types — and are biologically benign, with rare documented malignant transformation (chiefly renal cell carcinoma arising in long-standing AML, and rare SEGA-type glial tumours). The rarity of malignant conversion reflects the fact that mTORC1 activation is a proliferative and survival signal, not a full oncogenic driver; a second "second hit" beyond TSC1/TSC2 loss is required to convert a hamartoma to a carcinoma, and such additional hits are uncommon.[1][11]
Therapeutic corollary: mTOR inhibitors
The molecular logic explains why everolimus (oral 40-O-(2-hydroxyethyl) rapamycin) and sirolimus (rapamycin itself) are effective in TSC. Both are rapalogs that bind FKBP12, and the rapamycin-FKBP12 complex allosterically inhibits mTORC1, recreating — pharmacologically — the brake that the hamartin-tuberin complex exerts biologically.[5][7][8][9][10]
Clinical Presentation — Cutaneous, Neurological and Systemic
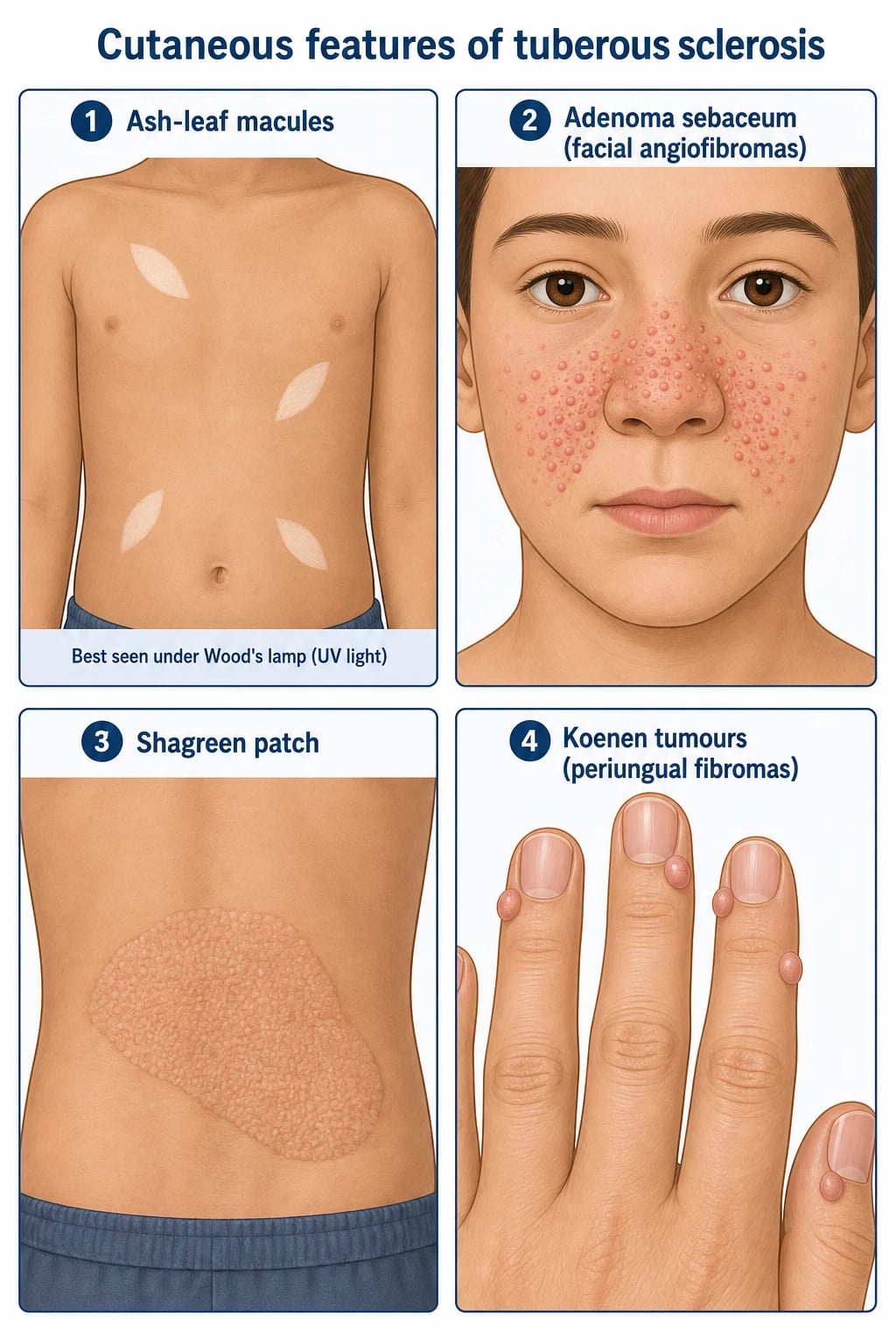
TSC touches virtually every organ system and the diagnosis can be made from the skin examination alone in a substantial minority of patients. The eight cutaneous features, with age-related onset, are listed in the Compare box below.[1][2][12]
Cutaneous tetrad
Typical age-by-age cutaneous evolution
The single most useful bedside test that confirms an ash-leaf macule is Wood's lamp examination at 365 nm. The macule appears as a sharply demarcated area of reduced fluorescence, with surrounding normal skin giving a bluish background.[1][12] Three or more hypomelanotic macules is a major criterion; two or fewer is non-diagnostic and requires other features or genetic testing.[2][3]
Neurological presentation
Neurological involvement is the principal source of morbidity and drives most TSC mortality.[1][13]
- Epilepsy occurs in 80–90% of patients; onset is typically in infancy or early childhood, with infantile spasms (West syndrome) as the first seizure type in roughly 30–50% of cases.[1][10] Focal seizures follow, often drug-refractory.
- Autism spectrum disorder affects up to 50% of children with TSC; intellectual disability affects approximately half; behavioural and psychiatric features ("TSC-Associated Neuropsychiatric Disorders", TAND) are near-universal and require structured screening.[1]
- Cortical tubers are hamartomatous malformations on MRI and are the pathological substrate for seizures; subependymal nodules line the lateral ventricles; subependymal giant cell astrocytoma (SEGA) arises near the foramen of Monro in 5–15% of patients and can obstruct CSF flow.[1][2]
- Subependymal nodules may show serial growth, and a nodule that enlarges >1 cm, enhances, or sits adjacent to the foramen of Monro should be re-imaged and followed as a SEGA.[2][5]
Cardiac, renal, pulmonary, ocular and dental features
- Cardiac rhabdomyoma is detected in roughly 50% of infants and is often the presenting sign in utero or in the neonatal period; the lesions are usually multiple, intramural, and regress spontaneously through childhood, requiring no intervention in most cases unless they cause arrhythmia or outflow obstruction.[1]
- Renal angiomyolipoma (AML) occurs in 70–80% of adults with TSC; they are mesenchymal tumours composed of blood vessels, smooth muscle and adipose tissue and carry a risk of spontaneous (Wunderlich) haemorrhage once they exceed 3–4 cm or develop aneurysms ≥ 5 mm.[1][11][7]
- Lymphangioleiomyomatosis (LAM) is the pulmonary manifestation, almost exclusively in adult women, characterised by diffuse, thin-walled cystic lung destruction, recurrent pneumothorax (in roughly 50% of affected women), chylous pleural effusions, and progressive airflow obstruction.[1][8][15]
- Retinal hamartomas are present in 30–50% of patients and are usually asymptomatic; rare complications include vitreous haemorrhage and visual loss.[1]
- Dental enamel pitting is a minor diagnostic criterion and is found on focused dental examination in most adults.[2][3]
Differential Diagnosis
The differential diagnosis of TSC is anchored on the index lesion — ash-leaf macule, facial angiofibroma, shagreen patch, Koenen tumour, or multi-system hamartomas — and on excluding the principal mimics.[1][12]
Clinical and Bedside Assessment
The structured assessment of a patient with suspected TSC has three sequential objectives: (1) confirm the diagnosis, (2) stage the disease across organ systems, (3) initiate therapy and surveillance.[1][2]
General inspection. Examine the entire skin surface in a well-lit room, including scalp, hair-bearing scalp (look for forehead plaques under the hairline), face, trunk and limbs, palms and soles, and nails. Inspect the oral cavity for enamel pits and gingival fibromas.[1]
Wood's lamp examination. With the room darkened and the lamp held 10–15 cm from the skin, sweep across the trunk and limbs to identify hypomelanotic macules and confetti lesions. Document the number, size and distribution of macules.[2][12]
Multidisciplinary baseline staging. Every newly diagnosed patient — child or adult — should have a structured baseline evaluation, ideally in a TSC clinic co-ordinating dermatology, paediatric or adult neurology, nephrology, pulmonology (for adult women), cardiology, ophthalmology, clinical genetics, and neuropsychology.[2][4] The minimum staging panel is:
- Brain MRI with gadolinium (cortical tubers, subependymal nodules, SEGA).[2][4]
- EEG in infants and any patient with suspected seizures.[13]
- Echocardiogram in infants and any patient with signs or symptoms of cardiac involvement; many rhabdomyomas regress spontaneously and only need to be followed if symptomatic.[2]
- Renal MRI or contrast-enhanced CT in adults (more sensitive than ultrasound for small fat-poor AML); ultrasound is acceptable in children.[2][11]
- Chest CT (high-resolution) in adult women at diagnosis and every 5–10 years for LAM surveillance; reserve for men unless symptomatic.[8][15]
- Ophthalmology review for retinal hamartomas.[2]
- Neuropsychology assessment for TAND — autism, intellectual disability, ADHD, anxiety, mood, behavioural difficulties.[1][2]
Investigations — The 2012/2021 International TSC Diagnostic Criteria
The 2012 International TSC Diagnostic Criteria — updated in 2021 and reproduced in summary below — are the global diagnostic reference.[2][3] A definite diagnosis is established by either a genetic criterion OR a clinical criterion (two major features, or one major + two or more minor features). A possible diagnosis is established by either one major feature or two or more minor features.[2][3]
A. Genetic criterion
Identification of a pathogenic mutation in TSC1 or TSC2 by validated molecular genetic testing is sufficient to make a definite diagnosis — independent of clinical features.[2][3] Note: a pathogenic mutation is one that is clearly protein-truncating (nonsense, frameshift, canonical splice-site, large deletion) or missense with demonstrated loss of function; variants of uncertain significance (VUS) do not satisfy this criterion alone.[2]
B. Clinical criteria — MAJOR features
- Hypomelanotic macules (≥3), at least 5 mm in diameter.[2][3]
- Facial angiofibromas (≥3) or forehead fibrous plaque.[2][3]
- Fibrous cephalic plaque. (Updated 2021 wording; previously a sub-bullet of angiofibromas.)[2]
- Ungual fibromas (≥2) — Koenen tumours.[2][3]
- Shagreen patch (connective-tissue naevus).[2][3]
- Multiple retinal hamartomas.[2][3]
- Cortical dysplasias (cortical tubers and cerebral white-matter radial migration lines on MRI).[2][3]
- Subependymal nodules (≥2).[2][3]
- Subependymal giant cell astrocytoma (SEGA).[2][3]
- Cardiac rhabdomyoma.[2][3]
- Lymphangioleiomyomatosis (LAM).[2][3]
- Renal angiomyolipoma (≥2).[2][3]
C. Clinical criteria — MINOR features
- Confetti skin lesions.
- Dental enamel pits (>3).
- Intraoral fibromas (≥2).
- Retinal achromic patch.
- Multiple renal cysts.
- Non-renal hamartomas.
- Sclerotic bone lesions.[2]
(Source: Northrup H et al. Pediatric Neurology 2021, PMID 34399110.[2])

Imaging findings by organ
- Brain MRI: cortical tubers (T2/FLAIR hyperintense gyral cores with subcortical white-matter signal change), subependymal nodules (small T2 hyperintense nodules along the lateral ventricles), SEGA (an enhancing mass >1 cm at or near the foramen of Monro, often with cystic components), and radial migration lines (curvilinear T2/FLAIR hyperintensities from the ventricle to the cortex).[1][2]
- Renal imaging: ultrasound is the first-line screen; MRI is preferred in adults because it can distinguish AML (with fat content) from renal cell carcinoma (without fat) and identify aneurysms ≥5 mm.[11][19]
- Cardiac echocardiogram: intramural, often multiple, echogenic masses in the ventricular wall or septum; usually maximal at birth and regressing through childhood.[1]
- Chest CT (high-resolution): thin-walled, diffusely distributed cysts 2–5 mm in diameter with normal intervening lung in LAM; in advanced disease, cysts coalesce and the lungs enlarge.[1][8]
Genetic testing — when, how, and yield
Genetic testing is indicated in any patient with suspected TSC who wishes confirmation, who is considering prenatal diagnosis in a future pregnancy, or who has an atypical phenotype; it is NOT required when the clinical criteria are unequivocally met, although most patients and families now request it for prognosis and cascade screening.[2][3]
- Sequencing of TSC1 and TSC2 (Sanger or NGS panel, often through a virtual neurocutaneous panel) detects point mutations and small indels in roughly 75–85% of patients.
- Deletion/duplication analysis (MLPA, array CGH, or NGS-based CNV calling) is required to detect the TSC2-PKD1 contiguous deletion and other large rearrangements, which together account for a small but important fraction of cases.[2][18]
Biochemical biomarker for LAM
Serum VEGF-D is elevated in TSC-LAM and sporadic LAM and is now incorporated into the diagnostic algorithm: a level ≥ 800 pg/mL in a woman with compatible CT findings confirms LAM and avoids lung biopsy.[15]
Management — Emergencies
Four emergencies dominate the acute management of TSC and should be in every on-call induction.[2][11][8]
Management — Definitive and Stepwise
Management of TSC is multidisciplinary, organ-by-organ, lifelong, and uses an escalating ladder from observation and topical therapy to systemic mTOR inhibition.[2][4][5][7][9][10]
mTOR inhibitors — the cornerstone
Everolimus is the systemic mTOR inhibitor of choice for SEGA, renal AML ≥3 cm, and TSC-associated refractory focal-onset seizures. It is given orally, titrated to a trough level of 5–15 ng/mL, and is approved by the FDA (2010, 2012, 2017) and EMA for these three indications.[5][7][10]
- EXIST-1 (Franz 2013, Lancet) — everolimus reduced SEGA volume by ≥50% in 35% of patients vs 0% on placebo; the open-label extension showed sustained response at 5 years.[5][6]
- EXIST-2 (Bissler 2013, Lancet) — everolimus reduced renal AML volume by ≥50% in 42% of patients vs 0% on placebo; long-term extension showed durable response and preserved renal function.[7][17][19]
- EXIST-3 (French 2016, Lancet) — adjunctive everolimus reduced refractory focal seizure frequency by ≥50% in 40% of patients (high-exposure arm) vs 15% on placebo; an important adjunct where seizures persist despite multiple ASMs.[10]
Sirolimus (rapamycin itself, the parent compound) is used orally for TSC-LAM at 2 mg daily, titrated to trough 5–15 ng/mL, based on the MILES trial (McCormack 2011, NEJM), which showed stabilisation of FEV1 and improvement in quality of life and forced vital capacity over 12 months compared with placebo.[8][14][15]
Topical sirolimus at 0.1% ointment twice daily is the modern first-line treatment for facial angiofibromas, based on the TREATMENT trial (Koenig 2018, JAMA Dermatol), which showed significant improvement in lesion size and erythema compared with placebo.[9]
Antiseizure management
Vigabatrin is first-line for TSC-associated infantile spasms at 50 mg/kg/day, escalating to 150 mg/kg/day over days to weeks; sustained response rates around 95% are characteristic.[1][2][13] The two notable adverse effects are irreversible visual-field constriction (monitor visual fields every 6 months in older children and adults; not possible in infants) and T2/FLAIR MRI signal changes in the basal ganglia and thalami that are typically reversible.[1]
For older children and adults with focal seizures, the choice of antiseizure medication (ASM) follows standard epilepsy guidelines (levetiracetam, valproate, lamotrigine, lacosamide, carbamazepine), with everolimus as adjunctive therapy for refractory cases.[10]
Organ-specific ladders
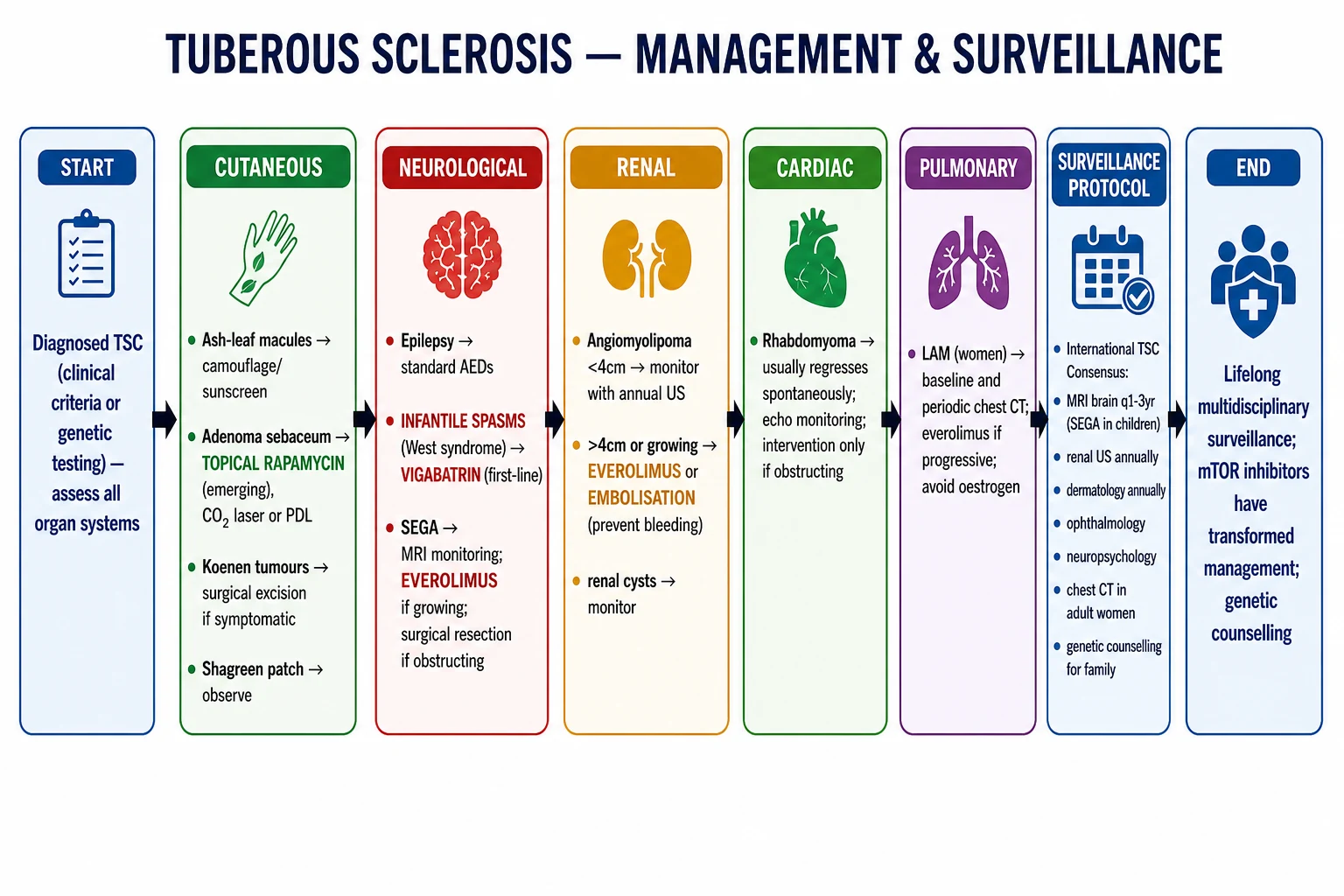
| Organ | Observation | Pharmacologic | Procedural / Surgical |
|---|---|---|---|
| Facial angiofibromas | Cosmetic camouflage | Topical sirolimus 0.1% BD[9] | Pulsed dye laser, CO2 ablation |
| Subependymal giant cell astrocytoma | Serial MRI | Everolimus[5][6] | Neurosurgical resection |
| Renal angiomyolipoma | MRI / CT surveillance | Everolimus ≥3 cm[7][17] | Selective arterial embolisation, nephron-sparing surgery |
| Lymphangioleiomyomatosis | PFTs, oxygen | Sirolimus[8][14] | Pleurodesis, lung transplantation |
| Refractory focal epilepsy | Standard ASMs | Everolimus adjunct[10] | Vagal nerve stimulator, epilepsy surgery |
| Cardiac rhabdomyoma | Echocardiography | mTOR inhibitor rarely | Resection only if obstructive |
Surveillance schedule
Adapted from the 2012/2021 International TSC Consensus recommendations and updated with mTOR-inhibitor monitoring.[2][4]
- Brain MRI: every 1–3 years in children with SEGA or subependymal nodules; every 5–10 years in adults if previously normal.[2]
- EEG: at diagnosis in infants; then as clinically indicated; consider serial EEG in infants with normal imaging to identify pre-symptomatic epileptiform activity.[13]
- Renal MRI (or CT/MR angiography): every 1–3 years to track AML growth and aneurysm formation.[11]
- Echocardiogram: at diagnosis in infants; repeat if symptomatic or to follow-up an obstructing lesion.[2]
- Chest CT (HRCT): in adult women at diagnosis and every 5–10 years; not routine in men.[8]
- Serum VEGF-D: in adult women with suspected LAM.[15]
- Everolimus / sirolimus trough level: every 1–3 months.[5][7][8]
- TAND screening: at diagnosis, then annually — formal neuropsychology assessment.[1][2]
- Dental examination: at diagnosis and as needed.[2]
- Ophthalmology: at diagnosis and as needed.[2]
Specific Subtypes and Scenarios
TSC1 vs TSC2 genotype-phenotype
TSC2 mutations are associated with earlier seizure onset, more cortical tubers, larger SEGA volume, greater seizure burden, more autism spectrum disorder, and more renal AML than TSC1 mutations.[1][16] The two genes do not differ in mTOR-inhibitor responsiveness — response to everolimus is independent of the mutated gene and of the specific mutation site within the gene.[16]
TSC2-PKD1 contiguous gene syndrome
Approximately 2% of patients with TSC2 mutations carry a large deletion on chromosome 16p13 that removes both TSC2 and the adjacent PKD1 gene, producing a contiguous gene syndrome combining TSC with autosomal dominant polycystic kidney disease (ADPKD).[1][11][18] These patients present earlier with hypertension and progressive cystic kidney disease, and require blood pressure control, tolvaptan consideration in selected adults, and combined nephrology-genetics care.[11]
Mosaic / segmental TSC
A subset of TSC patients have post-zygotic somatic mosaicism, in which the TSC1 or TSC2 mutation is present only in a subset of cells. The clinical phenotype is correspondingly segmental or attenuated: cutaneous features may be limited to a unilateral band (along Blaschko lines), and systemic involvement may be minimal.[1] Genetic testing on blood may be negative — testing of affected tissue (skin biopsy of the involved segment) is required. Surveillance is modulated by the extent of disease.
TSC and pregnancy
Pregnancy in TSC is increasingly common and requires pre-conception multidisciplinary review.[2]
- mTOR inhibitors are teratogenic (FDA category D / EMA contraindicated in pregnancy); they should be stopped 12 weeks before conception where feasible, with cardiology and neurology review of alternative seizure control during the transition.
- Fetal surveillance with mid-trimester cardiac ultrasound (20–24 weeks) for cardiac rhabdomyoma, and detailed CNS ultrasound.
- Mode of delivery — vaginal delivery is safe unless there is a large symptomatic SEGA, AML or cardiac lesion; an anaesthetic plan should anticipate difficult airway if facial angiofibromas distort the lips and gums.
- Post-partum — resumption of mTOR inhibitors requires weighing seizure/AML/LAM control against lactation and breastfeeding (mTOR inhibitors pass into breast milk).[2]
TSC-LAM in men
LAM is approximately 10-fold more common in women, but is reported in men. Diagnostic delay is common because LAM is not considered in male patients; any adult man with TSC who develops progressive dyspnoea, pneumothorax, chylous effusion or abdominal lymphangioleiomyomas should have HRCT.[8]
Forehead plaque as a presenting feature
A congenital forehead fibrous plaque can be the only cutaneous feature at birth, may be mistaken for a congenital melanocytic naevus or aplasia cutis, and should prompt Wood's lamp examination, brain MRI, and echocardiography if any other feature is suspicious.[12]
Complications and Pitfalls
The complications of TSC are the complications of its organ-specific hamartomas, plus a small but distinctive set of treatment-related pitfalls.[1][5][7][8]
Prognosis and Disposition
Prognosis in TSC has improved substantially since the introduction of mTOR inhibitors, but remains organ-dependent.[1][5][7][8]
- Brain — vigabatrin and everolimus substantially reduce seizure burden and SEGA growth; modern cohorts report seizure freedom in about 60% of children and stable SEGA in 75–85% of treated adults.[5][6][10]
- Kidneys — everolimus slows AML growth and preserves renal function over at least 5 years of follow-up; bleeding episodes have declined where surveillance is structured.[7][17][19]
- Lungs — sirolimus stabilises FEV1 and improves quality of life in TSC-LAM; lung transplantation is reserved for advanced disease.[8][14]
- Skin — topical sirolimus improves facial angiofibromas; other cutaneous lesions are stable.[9]
The principal prognostic determinants are:[14]
- Genotype — TSC2 generally more severe than TSC1.
- Seizure control — freedom from spasms and good early seizure control are the strongest predictors of cognitive outcome.[13]
- Renal function — preserved eGFR, controlled blood pressure, no Wunderlich event.
- LAM status — sirolimus-responsive disease has a markedly better outlook than transplant-listed LAM.[8][14]
Disposition. Most patients with TSC are managed as outpatients in a multidisciplinary TSC clinic combining paediatrics/adult medicine, neurology, dermatology, nephrology, pulmonology (women), cardiology, ophthalmology, clinical genetics, and neuropsychology.[2] Admission is reserved for status epilepticus, acute SEGA, ruptured AML, pneumothorax, drug toxicity, or pre-/peri-operative mTOR-inhibitor management.[5][7][8]
Survival. Reported median life expectancy in modern cohorts extends well into adulthood; historical mortality curves from the pre-mTOR era reflected premature deaths in childhood from SEGA and status epilepticus, and in adult life from AML and LAM. Modern mTOR-based management is improving these curves.[1][2]
Special Populations
Neonates and infants
The leading presenting features are prenatal or neonatal cardiac rhabdomyoma (detected on fetal echocardiography from 20 weeks), ash-leaf macules visible at birth, and infantile spasms at 4–9 months. A structured neonatal work-up includes brain MRI with gadolinium, echocardiogram, Wood's lamp skin examination, abdominal ultrasound, ophthalmology review, and EEG.[1][2][12] Genetic testing is recommended because early diagnosis opens the door to preventive epilepsy management.
Children and adolescents
Childhood is when TSC is most active diagnostically (facial angiofibromas, ash-leaf macules on dark skin, subependymal nodules visible on MRI) and therapeutically. The major issues are seizure control (vigabatrin first-line for infantile spasms; everolimus for refractory disease), TAND management (formal neuropsychology, behavioural therapy, school accommodations), renal AML surveillance and treatment once lesions exceed 3 cm, and family genetic counselling.[1][2][10][13]
Adults with newly diagnosed LAM
Adult women newly diagnosed with TSC should have baseline HRCT, serum VEGF-D, pulmonary function testing, and consideration of sirolimus. Pneumothorax prevention includes avoiding scuba diving, avoiding abrupt barometric changes, and prompt treatment of chest infections.[8][15]
Pregnancy
Pre-conception review of mTOR inhibitors (teratogenic), fetal cardiac ultrasound, and a multidisciplinary obstetric / anaesthetic plan are mandatory. Vigabatrin is generally continued during pregnancy where seizure control requires it, with monitoring of vision in the infant.[2]
Elderly patients
In elderly patients, renal function, cardiovascular comorbidity, drug–drug interactions (CYP3A4), and the higher risk of sirolimus/everolimus pneumonitis become management priorities. Everolimus dose reduction is often necessary.[1]
Darker skin phototypes (Fitzpatrick IV–VI)
Ash-leaf macules can be subtle on dark skin without Wood's lamp; the macule is detected much more reliably under a UV lamp in pigmented skin. Genetic testing plays a greater proportionate role. Facial angiofibromas are more clinically prominent and more cosmetically distressing. Skin-of-colour counselling should address post-inflammatory hyperpigmentation from procedures or irritant topicals.[1][12]
Founder mutations and consanguinity
In populations with high consanguinity, autosomal recessive phenocopies of TSC are exceedingly rare; TSC remains autosomal dominant. Founder mutations (e.g., specific TSC2 mutations reported in particular populations) may cluster locally and inform cascade screening.[1]
Evidence, Guidelines and Regional Differences
Landmark trials
| Trial | Drug | Indication | Year / Journal | Primary result |
|---|---|---|---|---|
| EXIST-1[5] | Everolimus | TSC-SEGA | 2013 Lancet | 35% SEGA response vs 0% placebo |
| EXIST-1 extension[6] | Everolimus | TSC-SEGA | 2014 Lancet Oncol | Sustained response at 2 years |
| EXIST-2[7] | Everolimus | TSC-AML | 2013 Lancet | 42% AML response vs 0% placebo |
| EXIST-2 extension[19] | Everolimus | TSC-AML | 2016 NDT | Long-term AML control |
| MILES[8] | Sirolimus | TSC-LAM | 2011 NEJM | Stabilisation of FEV1 |
| TREATMENT[9] | Topical rapamycin | Facial angiofibromas | 2018 JAMA Dermatol | Significant lesion and erythema improvement |
| EXIST-3[10] | Everolimus | Refractory seizures | 2016 Lancet | 40% seizure responder-rate (high-exposure) |
International consensus
The 2012 International Tuberous Sclerosis Complex Consensus Conference published two landmark papers in Pediatric Neurology in October 2013 — diagnostic criteria update (Northrup, Krueger)[3] and surveillance and management recommendations (Krueger, Northrup)[4] — which together underpin the modern diagnostic and surveillance framework. The 2021 update (Northrup et al., Pediatr Neurol)[2] refined wording, added fibrous cephalic plaque as a separate major criterion, and integrated mTOR-inhibitor evidence. The 2015 TSC strategic planning conference (Sahin, Henske, Manning et al., Pediatr Neurol 2016)[20] set research priorities, including clinical trials of preventive vigabatrin.
Regional differences
Global — International TSC Consensus. The 2012/2021 criteria and surveillance recommendations are the global reference. The Tuberous Sclerosis Association (TSA) and Tuberous Sclerosis Alliance (TS Alliance) maintain patient registries (TOSCA) and educational resources.[2]
Australia and New Zealand — RANZCD and TSC support organisations. Everolimus for SEGA and AML, and topical sirolimus, are accessible through specialised centres; sirolimus for LAM is funded under defined criteria.
Controversies
- Systematic surveillance vs targeted screening — the 2012/2021 Consensus recommends systematic surveillance MRI / renal imaging / EEG, but cost-effectiveness in low-resource settings is debated.
- mTOR inhibitors in mild disease — whether to start everolimus in minimally symptomatic AML or in a slowly growing SEGA, or to defer until a clinical trigger. Current practice favours early treatment where the natural history predicts complications.
- Preventive vigabatrin — the EPISTOP findings support vigabatrin in infants with subclinical epileptiform activity; pending confirmatory trials, this is not yet standard care everywhere.
- Topical sirolimus vehicle — pharmacy-compounded formulations differ in stability and skin penetration; commercial products are emerging.[13]
Exam Pearls and High-Yield Minutiae
HAMARTOMAS — major criteria mnemonic
HAMARTOMAS
Ash-leaf spots, ≥5 mm, 2012/2021 major criterion
Bilateral facial; part of forehead-plaque criterion family
Cortical tubers + cerebral white-matter radial migration lines
Distinct from angiofibromas above
Often regresses spontaneously
Pathogenic TSC1/TSC2 variant
≥2 subependymal nodules; multiple retinal hamartomas
Minor criterion set for oral findings
Major; mTOR responsive
Three organ-specific major features
Self-test: 7-month-old boy with flexion-spasms clusters, hypsarrhythmia on EEG, 4 hypomelanotic macules on Wood's lamp, MRI showing 3 cortical tubers. What is the most likely diagnosis, the most likely causative gene, and the first-line treatment?
Diagnosis: Tuberous sclerosis complex (TSC) — five major criteria (≥3 hypomelanotic macules, cortical dysplasias on MRI, infantile spasms as a clinical correlate, plus presumed others to come). Most likely causative gene: TSC2 (more common, more severe neuro phenotype, earlier seizure onset). First-line treatment for the infantile spasms: vigabatrin (50 mg/kg/day, escalating to 150 mg/kg/day), with subsequent transition to a standard ASM regimen, EEG monitoring, and multidisciplinary staging for SEGA, AML, cardiac and renal involvement. (AI reasoning by examiner.)
Self-test: 28-year-old woman with TSC on everolimus for SEGA presents with acute left flank pain, hypotension and falling haemoglobin. CT angiogram shows a 6 cm left renal angiomyolipoma with active extravasation. Outline your management.
This is a Wunderlich haemorrhage from a ruptured renal AML. Management: (1) Resuscitate with two large-bore cannulae, cross-match 4–6 units, tranexamic acid, maintain haemodynamics. (2) Urgent CT angiography to localise active bleeding (already done). (3) Selective arterial embolisation of the bleeding arterial branch — the haemostatic procedure of choice because it is nephron-sparing. (4) Continue everolimus for the contralateral kidney and residual lesions to prevent rebleeding; consider titration of trough to 5–15 ng/mL. (5) Total nephrectomy is a last resort and reserved for uncontrolled life-threatening haemorrhage. (6) Multidisciplinary TSC clinic review for ongoing AML surveillance and (in women) LAM screening with HRCT and serum VEGF-D.
Red Flags
Exam application bank (NEET-PG / INICET)
One-line answer
Tuberous sclerosis complex (TSC) is an autosomal dominant, multi-organ, hamartomatous neurocutaneous disorder caused by loss-of-function mutations in TSC1 (hamartin) or TSC2 (tuberin) leading to constitutive mTORC1 activation. Fellowship-level assessment requires the 2012/2021 International TSC Diagnostic Criteria reproduced verbatim, the full cutaneous tetrad (ash-leaf macule, adenoma sebaceum, shagreen patch, Koenen tumour), multi-organ surveillance (SEGA, cardiac rhabdomyoma, renal angiomyolipoma, pulmonary LAM, retinal hamartomas), infantile-spasm management with vigabatrin, mTOR-inhibitor pharmacology (everolimus systemic, sirolimus/rapamycin topical for facial angiofibromas and oral for LAM), TAND, and the TSC2-PKD1 contiguous gene syndrome.
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard.[10]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes.[1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change.[1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each.[1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory.[5]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Tuberous sclerosis complex.
References
- [1]Henske EP, Jóźwiak S, Kingswood JC, et al. Tuberous sclerosis complex. Nature Reviews Disease Primers, 2016.PMID 27226234
- [2]Northrup H, Aronow ME, Bebin EM, et al. Updated International Tuberous Sclerosis Complex Diagnostic Criteria and Surveillance and Management Recommendations. Pediatric Neurology, 2021.PMID 34399110
- [3]Northrup H, Krueger DA, International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatric Neurology, 2013.PMID 24053982
- [4]Krueger DA, Northrup H, International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatric Neurology, 2013.PMID 24053983
- [5]Franz DN, Belousova E, Sparagana S, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. The Lancet, 2013.PMID 23158522
- [6]Franz DN, Belousova E, Sparagana S, et al. Everolimus for subependymal giant cell astrocytoma in patients with tuberous sclerosis complex: 2-year open-label extension of the EXIST-1 study. The Lancet Oncology, 2014.PMID 25456370
- [7]Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. The Lancet, 2013.PMID 23312829
- [8]McCormack FX, Inoue Y, Moss J, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. New England Journal of Medicine, 2011.PMID 21410393
- [9]Koenig MK, Bell CS, Hebert AA, et al. Efficacy and Safety of Topical Rapamycin in Patients With Facial Angiofibromas Secondary to Tuberous Sclerosis Complex: The TREATMENT Randomized Clinical Trial. JAMA Dermatology, 2018.PMID 29800048
- [10]French JA, Lawson JA, Yapici Z, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. The Lancet, 2016.PMID 27613521
- [11]Lam HC, Siroky BJ, Henske EP. Renal disease in tuberous sclerosis complex: pathogenesis and therapy. Nature Reviews Nephrology, 2018.PMID 30232410
- [12]Staley BA, Vail EA, Thiele EA. Tuberous sclerosis complex: diagnostic challenges, presenting symptoms, and commonly missed signs. Pediatrics, 2011.PMID 21173003
- [13]De Ridder J, Verhelle B, Vervisch J, et al. Early epileptiform EEG activity in infants with tuberous sclerosis complex predicts epilepsy and neurodevelopmental outcomes. Epilepsia, 2021.PMID 33778971
- [14]Gupta N, Lee HS, Young LR, et al. Analysis of the MILES cohort reveals determinants of disease progression and treatment response in lymphangioleiomyomatosis. European Respiratory Journal, 2019.PMID 30846465
- [15]Young LR, Lee HS, Inoue Y, et al. Serum VEGF-D a concentration as a biomarker of lymphangioleiomyomatosis severity and treatment response: a prospective analysis of the Multicenter International Lymphangioleiomyomatosis Efficacy of Sirolimus (MILES) Trial. The Lancet Respiratory Medicine, 2013.PMID 24159565
- [16]Kwiatkowski DJ, Palmer MR, Jozwiak S, et al. Response to everolimus is seen in TSC-associated SEGAs and angiomyolipomas independent of mutation type and site in TSC1 and TSC2. European Journal of Human Genetics, 2015.PMID 25782670
- [17]Bissler JJ, Budde K, Sauter M, et al. Effect of everolimus on renal function in patients with tuberous sclerosis complex: evidence from EXIST-1 and EXIST-2. Nephrology Dialysis Transplantation, 2019.PMID 30053159
- [18]European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell, 1993.PMID 8269512
- [19]Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus for renal angiomyolipoma in patients with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis: extension of a randomized controlled trial. Nephrology Dialysis Transplantation, 2016.PMID 26156073
- [20]Sahin M, Henske EP, Manning BD, et al. Advances and Future Directions for Tuberous Sclerosis Complex Research: Recommendations From the 2015 Strategic Planning Conference. Pediatric Neurology, 2016.PMID 27267556