Endocrinology · General Medicine
Phaeochromocytoma
Also known as Phaeochromocytoma · Pheochromocytoma · Paraganglioma · Catecholamine-secreting tumour
Phaeochromocytoma is a catecholamine-secreting tumour arising from chromaffin cells of the adrenal medulla; an identical tumour outside the adrenal (organ of Zuckerkandl, sympathetic chain, bladder) is a paraganglioma. About 80 to 85 percent are intra-adrenal and 15 to 20 percent extra-adrenal. It presents with the classic triad of episodic headache, sweating and palpitations with paroxysmal or sustained hypertension and striking pallor. The historical rule of 10 (about 10 percent each bilateral, malignant, extra-adrenal, familial and paediatric) is outdated for heritability: modern genotyping shows 30 to 40 percent carry a germline mutation (RET/MEN 2, VHL, NF1, SDHx, MAX, TMEM127). Diagnosis rests on plasma free metanephrines or 24-hour urine fractionated metanephrines, with CT/MRI for localisation and MIBG or 68Ga-DOTATATE PET for extra-adrenal and metastatic disease. The cardinal management rule is ALPHA-block before beta-block — phenoxybenzamine for 10 to 14 days, then a beta-blocker for tachycardia, then laparoscopic adrenalectomy. A beta-blocker given first leaves alpha-1 vasoconstriction unopposed and triggers a fatal hypertensive crisis; intra-operative crisis is treated with IV phentolamine or sodium nitroprusside.
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags
Overview & Definition
Phaeochromocytoma is one of the few genuinely curable causes of secondary hypertension, yet it is also one of the most dangerous to miss: an unrecognised tumour can unleash a catastrophic catecholamine crisis during anaesthesia, surgery, radiological contrast or beta-blockade, with a high mortality. It arises from chromaffin cells of neural-crest origin. Tumours within the adrenal medulla are termed phaeochromocytomas; histologically identical tumours in extra-adrenal sympathetic paraganglia (the organ of Zuckerkandl, sympathetic chain, urinary bladder, thorax) are paragangliomas. The two are increasingly grouped as PPGL (phaeochromocytoma and paraganglioma), because they share genetics, biochemistry and management.[1][3]
The disease is biochemically distinctive: the tumour continuously leaks metanephrine and normetanephrine into the circulation, regardless of the episodic catecholamine surges that produce the symptoms. This single insight underpins modern diagnosis — metanephrines, not catecholamines, are the sensitive first-line test. Management is built on one non-negotiable sequence: alpha-blockade before beta-blockade, then surgical resection. A high index of suspicion is essential, because the presentation overlaps with commoner and more benign complaints — anxiety, menopausal flushes, migraine and simple palpitations — and because the tumour may be silent until a crisis.[1][2]
Classification
PPGL is classified by anatomical site, sympathetic versus parasympathetic lineage, and secretory profile, and — increasingly — by underlying genotype. About 80 to 85 percent of catecholamine-producing tumours arise in the adrenal medulla and 15 to 20 percent are extra-adrenal paragangliomas. Sympathetic paragangliomas (abdominal, thoracic, pelvic, bladder) usually secrete noradrenaline and produce the classic pressor phenotype; parasympathetic paragangliomas of the head and neck (carotid body, glomus jugulare, vagal) are usually non-functional and present as a mass, hearing loss or cranial-nerve palsies rather than hypertension.[1][3]
The secretory profile itself is informative. Tumours producing adrenaline and metanephrine (an "adrenergic" phenotype) are typically adrenal, intra-tumoural, and often MEN 2 or NF1-related; tumours producing predominantly noradrenaline and normetanephrine (a "noradrenergic" phenotype) point to extra-adrenal disease, VHL or SDHx mutations. This biochemistry-to-genotype link guides which genes to test first.[3]
The traditional teaching rule of 10 — that roughly 10 percent each are bilateral, malignant, extra-adrenal, familial and paediatric — remains a useful memory device but is historically outdated on heritability and malignancy. Modern series show 30 to 40 percent carry a germline mutation, and the risk of malignancy is far higher than 10 percent for extra-adrenal tumours — up to 35 to 40 percent for SDHB-mutated paragangliomas. The rule of 10 should be remembered for the examination, but a candidate must also state that it has been superseded for genetics.[1][3]

Phaeochromocytoma — key numbers
Epidemiology & Risk Factors
Phaeochromocytoma is rare. The annual incidence is about 2 to 8 per million population per year, and it accounts for only about 0.1 to 0.6 percent of all patients with hypertension. It is found in roughly 5 to 7 percent of adrenal incidentalomas, which is why every adrenal incidentaloma must be biochemically screened for catecholamine excess before any intervention. Sporadic disease peaks in the 4th to 5th decade with no sex predilection; bilateral, multifocal or paediatric disease should immediately raise suspicion for a hereditary syndrome.[1][5]
The case-finding yield is highest in patients with one or more of: resistant or paroxysmal hypertension; hypertension with the classic triad of headache, sweating and palpitations; an adrenal incidentaloma; a family history or known carrier status for a susceptibility gene (RET, VHL, NF1, SDHx, MAX, TMEM127); prior resected phaeochromocytoma (surveillance); paradoxical hypertension on a beta-blocker; hypertension in pregnancy; and spell-like episodes (anxiety, flushing, tremor, pallor). Hereditary tumours tend to present younger, are more often bilateral and multifocal, and carry a higher risk of recurrence and malignancy (especially SDHB).[1][3]
Pathophysiology
The tumour is composed of chromaffin cells — neuroendocrine cells of neural-crest origin — arranged in nested clusters called "Zellballen" (German, "cell balls"), separated by a rich vascular network and supported by sustentacular (S100-positive) cells. This architecture is the histological signature and is shared with normal adrenal medulla and other paragangliomas.[3]
Catecholamine biosynthesis follows a fixed cascade: tyrosine is converted by tyrosine hydroxylase (the rate-limiting enzyme) to L-DOPA, then to dopamine, to noradrenaline, and finally — only in the adrenal medulla — to adrenaline by phenylethanolamine N-methyltransferase (PNMT). PNMT is induced by the high local cortisol delivered by the adrenal cortex via the portal system, which is why extra-adrenal tumours, lacking this cortisol bath, usually cannot synthesise adrenaline and secrete predominantly noradrenaline. This explains the noradrenergic biochemistry of paragangliomas and is a useful diagnostic clue.[1][3]
The single most important biochemical principle for the examination is this: metanephrine and normetanephrine are produced continuously inside the tumour by the enzyme catechol-O-methyltransferase (COMT), independently of the episodic catecholamine release that produces symptoms. They leak steadily into the bloodstream. Random plasma or urine catecholamines, by contrast, surge and fall with each spell and are easily missed between attacks. This is why plasma free metanephrines and 24-hour urine fractionated metanephrines are far more sensitive than catecholamines or vanillylmandelic acid (VMA), and are the recommended first-line tests.[1]
The excess catecholamines act on adrenergic receptors to produce the clinical picture: alpha-1 activation causes vasoconstriction (hypertension, pallor, cold extremities); beta-1 activation drives tachycardia, palpitations and increased myocardial contractility; beta-2 activation produces tremor and a vasodilatory counter-regulation. Orthostatic hypotension occurs paradoxically in chronic disease, through chronic vasoconstriction causing volume depletion and impaired baroreflex buffering — its presence in a hypertensive patient is a useful clue. Chronic catecholamine excess is cardiotoxic and can produce catecholamine cardiomyopathy, Takotsubo (stress) cardiomyopathy, myocarditis and arrhythmia — sometimes presenting as cardiogenic shock or acute coronary syndrome with normal coronaries.[2]
The cardiotoxicity is worth emphasising. Sustained catecholamine exposure causes coronary vasospasm, microvascular injury, focal myocyte necrosis and interstitial fibrosis, and may precipitate Takotsubo cardiomyopathy (apical ballooning) even in the absence of epicardial coronary disease. The result is a patient who may present not with hypertension at all but with acute heart failure, pulmonary oedema, cardiogenic shock or ventricular arrhythmia — the so-called "normotensive phaeochromocytoma" in crisis. Recognising this is critical, because administering a beta-blocker to such a patient without alpha-blockade can precipitate an unopposed-alpha catastrophe, and standard heart-failure therapy (beta-blockers, ACE inhibitors) may worsen the underlying catecholamine excess until the tumour is controlled.[2]
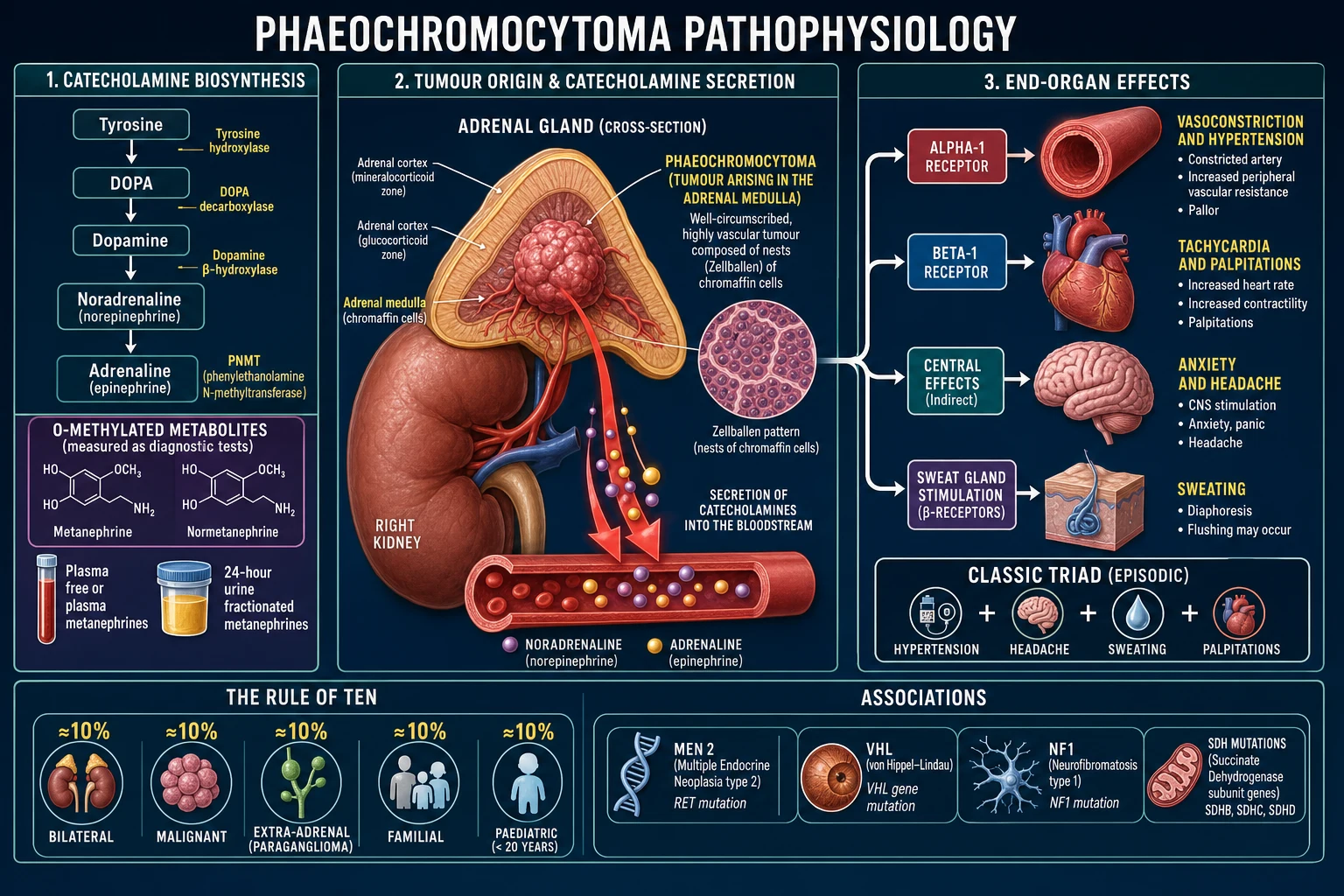
Clinical Presentation
The clinical hallmark is the paroxysm (a "spell"): an abrupt, dramatic episode lasting a few minutes to an hour, dominated by the classic triad. [1]
- The classic triad — episodic hypertension with headache, sweating and palpitations/tachycardia. The combination is highly specific; in a hypertensive patient it carries a likelihood ratio that should prompt immediate biochemical screening. The headache is severe, throbbing and bilateral; sweating is profuse and generalised; palpitations are felt as a pounding, racing heart.[1][5]
- Pallor, not flushing — spells are accompanied by marked pallor from alpha-1-mediated vasoconstriction. This is a key discriminator: flushing suggests carcinoid syndrome, not phaeochromocytoma. Other spell features include anxiety, terror, a sense of impending doom, tremor, nausea and abdominal pain.[2]
- Triggers — spells may be precipitated by straining, bending, micturition, defaecation, palpation of the abdomen, exertion, anaesthesia, radiological contrast, tyramine-rich foods (cheese, red wine, smoked meats) and certain drugs (tricyclics, MAOIs, decongestants, cocaine, opioids). Micturition-triggered spells specifically suggest a bladder paraganglioma.[1]
- Between spells — the patient may be normotensive, but more often has sustained hypertension. Other interparoxysmal features include orthostatic hypotension and dizziness, weight loss, hyperglycaemia or new-onset diabetes, constipation, anxiety, and pallor.[1]
- Atypical or silent presentations — many are found as an adrenal incidentaloma on cross-sectional imaging done for another reason. The tumour may also declare itself through catecholamine cardiomyopathy, Takotsubo, cardiogenic shock, acute coronary syndrome with normal coronaries, arrhythmia, or sudden death.[2]
- Crisis presentation — a hypertensive emergency precipitated by induction of anaesthesia, tumour handling at surgery, biopsy, or administration of a beta-blocker, metoclopramide, glucocorticoids or contrast in an unblocked patient. This may be the first manifestation of the disease and carries a high mortality if not recognised.[2]
At a molecular level, PPGL is now understood through transcriptional clusters that predict behaviour and biochemistry, and that link directly to the germline susceptibility genes. The pseudo-hypoxia cluster (driven by VHL, SDHx, and EPAS1/HIF2A) acts through dysregulated hypoxia-signalling pathways; these tumours are typically noradrenergic, often extra-adrenal, and carry the highest metastatic risk (notably SDHB). The kinase-signalling cluster (driven by RET/MEN 2, NF1, RAS, MAX and TMEM127) activates the MAPK and mTOR pathways; these tumours are usually adrenergic and adrenal with a lower metastatic risk. A smaller WNT-signalling cluster (MAML3 fusions) is associated with metastatic, mixed adrenergic-noradrenergic tumours. This molecular taxonomy is why the biochemical phenotype predicts the genotype: a noradrenergic extra-adrenal tumour prioritises testing for SDHx and VHL, while a bilateral adrenergic adrenal tumour prioritises RET and NF1.[3]
Differential Diagnosis
The differential of episodic hypertension or "spells" is broad. The decisive discriminator is always biochemistry (raised metanephrines) combined with anatomical localisation. The most important mimics to distinguish are: [1]
Phaeochromocytoma
- Episodic HTN + headache + sweating + palpitations with PALLOR
- Raised plasma or urine METANEPHRINES
- Adrenal mass on CT/MRI over 10 HU; treat alpha before beta
Panic / anxiety disorder
- Episodic fear, palpitations, trembling
- Normal metanephrines; no orthostatic drop; flush rather than pale
- No adrenal mass; a diagnosis of exclusion after biochemistry is normal
Carcinoid syndrome
- FLUSHING (not pallor), secretory diarrhoea, bronchospasm, right-heart valvular disease
- Raised 24-hour urine 5-HIAA
- Usually a metastatic midgut neuroendocrine tumour with hepatic deposits
Menopause / hyperthyroidism
- Hot flushes, sweating, palpitations, heat intolerance
- Normal metanephrines; menopausal age or raised free T4 with suppressed TSH
- No adrenal mass; thyroid function and FSH settle it
Drug / toxin
- Cocaine, amphetamine, MAOI-tyramine interaction, clonidine withdrawal, liquorice
- History and urine drug screen; transient
- Normal metanephrines after drug clearance
Mast cell / histamine disorders
- Flushing, hypotension or hypertension, GI symptoms
- Raised serum tryptase; normal metanephrines
- Urticaria pigmentosum; triggers include opioids and NSAIDs
For the adrenal mass itself, phaeochromocytoma (high CT attenuation over 10 HU, raised metanephrines) must be distinguished from a cortical adenoma (lipid-rich, under 10 HU on unenhanced CT, over 60 percent washout at 10 minutes), a cortisol-producing adenoma (Cushing phenotype, failure to suppress cortisol on 1 mg overnight dexamethasone), an aldosteronoma (hypokalaemic hypertension, raised aldosterone-to-renin ratio), myelolipoma (macroscopic fat density), adrenal metastasis (known primary, often bilateral, non-functional), and adrenocortical carcinoma (large over 4 cm, irregular, heterogeneous, often mixed steroid excess). Every adrenal incidentaloma therefore deserves a "triple screen" — metanephrines, dexamethasone suppression and an aldosterone-renin ratio (with potassium corrected).[1][5]
Clinical & Bedside Assessment
Bedside assessment is built around the blood pressure pattern, spell characteristics, syndromic stigmata and end-organ damage. [1]
- Blood pressure — measure both lying and standing. Look for paroxysmal or sustained hypertension with an orthostatic (postural) drop greater than 20/10 mmHg. An orthostatic drop in a hypertensive patient is a valuable clue to chronic catecholamine excess with volume depletion.[1]
- Syndromic stigmata — examine for the cutaneous and somatic markers of hereditary disease: a marfanoid habitus with mucosal neuromas and a thyroid nodule (MEN 2); café-au-lait patches, axillary freckling, cutaneous neurofibromas and Lisch nodules (NF1); retinal and CNS haemangioblastomas, renal cysts and renal cell carcinoma (VHL); and multiple paragangliomas (SDHx, especially SDHD with paternal transmission).[1][3]
- Micturition spells — a history of headache, sweating and hypertension during or immediately after micturition strongly suggests a bladder paraganglioma; ask specifically, as patients rarely volunteer it.[1]
- End-organ assessment — look for hypertensive retinopathy, signs of heart failure or cardiomyopathy, and evidence of prior stroke. Examine the abdomen carefully but avoid deep palpation of a known mass, which can precipitate a crisis.[1]
Investigations
Investigation proceeds in a fixed sequence: biochemistry first, then anatomical localisation, then functional imaging and genetic testing. The cardinal rule is biochemistry before any biopsy or intervention.[1]
First-line biochemistry is plasma free metanephrines (highest sensitivity, 96 to 100 percent) or 24-hour urine fractionated metanephrines and catecholamines (slightly lower sensitivity but higher specificity, and useful when plasma results are borderline). Before testing, stop interfering drugs for at least 48 hours — tricyclic antidepressants, phenoxybenzamine, sympathomimetic decongestants (pseudoephedrine), caffeine, MAOIs, and many antidepressants — and avoid paracetamol for 24 hours (it interferes with the plasma assay). Withdrawal of antihypertensives is generally not required, but calcium-channel blockers and alpha-blockers can raise levels modestly. Use the clonidine suppression test for equivocal biochemistry: clonidine normally suppresses sympathetic noradrenaline release, but a phaeochromocytoma fails to suppress plasma normetanephrine/noradrenaline.[1][5]
Interpretation turns on the pre-test probability. The Endocrine Society considers raised plasma free normetanephrine (over 0.8 nmol/L) or metanephrine (over 0.5 nmol/L) highly suggestive, and levels more than four times the upper limit of normal are essentially diagnostic. Because specificity is imperfect, false positives are common and arise from interfering drugs, physiological stress (severe pain, surgery, myocardial infarction, stroke, obstructive sleep apnoea), and renal impairment. A recognised mimic is pseudopheochromocytoma — episodic hypertension with normal metanephrines driven by baroreflex failure or anxiety. To suppress false positives, repeat the test with the patient supine and rested for at least 30 minutes, withdraw all interfering agents, and confirm with 24-hour urine fractionated metanephrines or the clonidine suppression test before committing the patient to imaging and blockade.[1][5]
Anatomical localisation — CT or MRI of the abdomen and pelvis (with contrast) is first-line. Phaeochromocytoma is typically over 10 HU on unenhanced CT (often 40 to 50 HU, reflecting vascularity), shows intense heterogeneous enhancement, and has slow washout (under 60 percent absolute washout at 10 minutes — the opposite of an adenoma). MRI shows high signal on T2-weighted images (the "light-bulb" sign, though not always present). Localise before surgery; the tumour is vascular and well-defined.[1]
Functional imaging is reserved for extra-adrenal disease, metastatic or malignant disease, hereditary syndromes, large tumours (over 5 cm), biochemical-radiological mismatch, and recurrence. Options are 123I-meta-iodobenzylguanidine (MIBG) scintigraphy (taken up by noradrenaline transporters; traditional whole-body tracer), 18F-DOPA PET (excellent for SDHx-related disease), 18F-FDG PET (high uptake in SDHB-mutated and aggressive tumours), and increasingly 68Ga-DOTATATE PET (somatostatin-receptor imaging, the most sensitive tracer for metastatic and SDHx disease). The choice is tailored to genotype and intent.[1][3]
From suspicion to surgery — the diagnostic pathway
Genetic testing should be offered to all patients, because over 30 percent carry a germline mutation. A tiered approach is used: the biochemical profile and presentation direct testing — adrenergic/adrenal tumours to RET, NF1, MAX, TMEM127; noradrenergic/extra-adrenal to VHL and SDHx (especially SDHB, SDHD). Identifying a mutation changes surveillance, predicts malignancy risk, enables cascade testing of relatives, and informs cortical-sparing surgery decisions.[1][3]
Exam application bank (NEET-PG / INICET)
One-line answer
Phaeochromocytoma is a catecholamine-secreting tumour arising from chromaffin cells of the adrenal medulla; an identical tumour outside the adrenal (organ of Zuckerkandl, sympathetic chain, bladder) is a paraganglioma. About 80 to 85 percent are intra-adrenal and 15 to 20 percent extra-adrenal. It presents with the classic triad of episodic headache, sweating and palpitations with paroxysmal or sustained hypertension and striking pallor. The historical rule of 10 (about 10 percent each bilateral, malignant, extra-adrenal, familial and paediatric) is outdated for heritability: modern genotyping shows 30 to 40 percent carry a germline mutation (RET/MEN 2, VHL, NF1, SDHx, MAX, TMEM127). Diagnosis rests on plasma free metanephrines or 24-hour urine fractionated metanephrines, with CT/MRI for localisation and MIBG or 68Ga-DOTATATE PET for extra-adrenal and metastatic disease. The cardinal man
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard. [1]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes. [1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change. [1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each. [1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory. [1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Phaeochromocytoma.
Management — Resuscitation

A catecholamine crisis is a hypertensive emergency — typically precipitated by anaesthesia induction, tumour handling at surgery, biopsy, contrast, or a beta-blocker, metoclopramide or glucocorticoid in an unblocked patient. Immediate management is: [1]
- IV phentolamine (a short-acting, non-selective alpha-blocker) — 2.5 to 5 mg IV bolus, repeated or as an infusion titrated to blood pressure. This is the first-line vasodilator because it directly antagonises the alpha-mediated vasoconstriction driving the crisis.[2]
- Alternatives are a sodium nitroprusside infusion (0.3 to 2 mcg/kg/min, titrated; useful for rapid, titratable control) or a nicardipine infusion (5 to 15 mg/hour), a clevidipine infusion, or urapidil.[2]
- Never give a beta-blocker alone first — it leaves alpha-mediated vasoconstriction unopposed and worsens the crisis dramatically. Treat arrhythmia with a short-acting beta-blocker such as esmolol (loading 500 mcg/kg over 1 minute, then 50 to 200 mcg/kg/min) only after alpha-blockade is established.[2]
- Intravenous fluids to restore intravascular volume, and management in a high-dependency or intensive-care setting with invasive arterial monitoring. Magnesium sulphate (2 to 4 g IV) is a useful adjunct for both blood pressure and arrhythmia control.[2]
- For an undiagnosed phaeochromocytoma precipitated on induction of anaesthesia, the priority is to recognise it, abort the procedure if possible, give IV phentolamine, fluids and vasopressors as needed, and arrange definitive workup once stabilised.[2]
Phentolamine (crisis)
Short-acting IV alpha-blocker for catecholamine crisis / intra-operative hypertensive surges
Dose
2.5 to 5 mg IV bolus, repeat; or infusion 0.1 to 2 mg/min titrated to BP
Management — Definitive & Stepwise
The definitive treatment is complete surgical resection of the tumour, but only after meticulous preoperative preparation. The goal of preparation is to convert a patient who might die on the table into one who tolerates the catecholamine swings of induction, tumour handling and tumour-vein clamping. The non-negotiable sequence is: alpha-blockade first, then volume expansion, then a beta-blocker for tachycardia, then surgery.[1]
Preoperative preparation — the 10 to 14 day sequence
Alpha-blockade first — phenoxybenzamine 10 mg BD
Start phenoxybenzamine (a non-competitive, irreversible alpha-blocker) 10 mg twice daily; titrate up by 10 to 20 mg every 2 to 3 days over 10 to 14 days to about 1 mg/kg/day (e.g. 40 to 100 mg/day in divided doses), until seated blood pressure is about 130/80 with a mild orthostatic drop and nasal congestion appears.
Volume expansion
Encourage a high-salt diet and give IV normal saline — the patient is volume-depleted from chronic vasoconstriction. Aim for a haematocrit drop as volume is restored.
Add a beta-blocker — only after alpha-blockade
Add propranolol (e.g. 20 to 40 mg three times daily), bisoprolol (2.5 to 5 mg daily) or atenolol, for reflex tachycardia. NEVER give a beta-blocker first — unopposed alpha vasoconstriction triggers a fatal crisis.
Optimise — 10 to 14 days
Endpoints: seated BP under 130/80, mild orthostatic drop but no syncope, controlled tachycardia (resting under 80), no spells for a week, nasal congestion (a clinical sign of complete alpha-blockade).
Laparoscopic adrenalectomy
Standard of care for most tumours; open for large, invasive or clearly malignant disease. Cortical-sparing (partial) adrenalectomy in bilateral/hereditary disease to avoid permanent steroid dependence.
Postoperative care and surveillance
Anticipate hypotension (give fluids and vasopressors) and hypoglycaemia (monitor glucose). Lifelong biochemical surveillance — plasma metanephrines at 6 weeks then 6 to 12 monthly — for recurrence, metastasis and new primaries.
Selectively, alternatives to phenoxybenzamine are used. Selective alpha-1 blockers such as doxazosin (2 to 8 mg/day), terazosin (1 to 10 mg/day) or prazosin (1 to 5 mg two to three times daily) are competitive and shorter-acting; some centres prefer them on the grounds of less postoperative hypotension and fewer side-effects, and a retrospective comparison found comparable intraoperative stability between phenoxybenzamine and doxazosin.[7] A landmark multicentre randomised controlled trial (Buitenwerf et al, 2020), however, questioned whether routine preoperative phenoxybenzamine meaningfully improves intraoperative haemodynamic control versus no phenoxybenzamine — a finding that has fuelled ongoing debate but has not displaced alpha-blockade from standard practice, which the Endocrine Society guideline continues to recommend.[4] Alpha-methyl-p-tyrosine (metyrosine), a tyrosine hydroxylase inhibitor that cuts catecholamine synthesis at source (250 mg every 8 to 12 hours, up to 4 g/day), is added for refractory, large or high-output tumours and to reduce intraoperative surges.[1]
Calcium-channel blockers (e.g. nicardipine, amlodipine, nifedipine) are used either as adjuncts to alpha-blockade or as sole agents in some centres for mild disease, controlling blood pressure without the full vasodilatory consequences of phenoxybenzamine.[1]
Phenoxybenzamine (preparation)
Non-competitive, irreversible alpha-blocker — first step in preoperative preparation
Dose
10 mg twice daily, titrate by 10 to 20 mg every 2 to 3 days to 1 mg/kg/day over 10 to 14 days
Surgery. Laparoscopic adrenalectomy is the standard of care for most phaeochromocytomas, offering shorter stay and quicker recovery with comparable safety, provided the tumour is not too large or invasive. Open adrenalectomy is reserved for large tumours (over 6 to 8 cm), locally invasive disease, suspected malignancy, or when laparoscopic control is difficult. In bilateral or hereditary disease (especially MEN 2 and VHL), cortical-sparing (partial) adrenalectomy is preferred to preserve cortisol and aldosterone production and avoid lifelong steroid dependence, accepting a slightly higher recurrence risk. Anaesthesia demands an experienced team, invasive arterial monitoring, rapid access to phentolamine/nitroprusside and vasopressors, and anticipation of the swings: hypertension on tumour handling, then hypotension after the adrenal vein is clamped (treat with fluids and noradrenaline/phenylephrine) and hypoglycaemia (treat with dextrose; monitor glucose for 24 to 48 hours).[1][5]
Anaesthetic and intraoperative considerations. Surgery demands an experienced anaesthetic team and invasive arterial-line monitoring from induction. Two predictable haemodynamic swings bracket tumour devascularisation: on tumour handling, catecholamine surges cause severe hypertension (treat with boluses of IV phentolamine or magnesium, or a nitroprusside/nicardipine infusion); and immediately after the adrenal vein is ligated, the abrupt loss of catecholamine tone on a fully alpha-blockaded, vasodilated patient causes hypotension (treat with IV fluids and noradrenaline or phenylephrine, weaning as vascular tone returns). Avoid drugs that release histamine or catecholamines (morphine, atracurium, metoclopramide, droperidol); prefer remifentanil, propofol, fentanyl, rocuronium or vecuronium, and volatile agents. Postoperatively, watch for hypoglycaemia (the catecholamine-driven hyperglycaemia is gone while insulin action persists) and for transient adrenal insufficiency after bilateral resection (give stress-dose hydrocortisone). Most patients need 24 to 48 hours in a high-dependency or intensive-care setting for haemodynamic and glucose monitoring.[2][5]
Specific Subtypes & Scenarios
Hereditary phaeochromocytoma and paraganglioma now account for 30 to 40 percent of all cases and demand a different surveillance strategy. The major syndromes, each with a distinctive phenotype, are: [1]
MEN 2 (RET)
- Bilateral adrenal phaeochromocytomas, adrenergic phenotype
- Medullary thyroid carcinoma (the dominant cause of death), hyperparathyroidism
- Marfanoid habitus, mucosal neuromas (MEN 2B); rarely malignant
von Hippel-Lindau (VHL)
- Adrenal phaeochromocytomas, noradrenergic phenotype, often bilateral
- Renal cell carcinoma, CNS and retinal haemangioblastomas, pancreatic neuroendocrine tumours, endolymphatic sac tumours
- Low metastatic risk
NF1
- Café-au-lait patches, axillary/inguinal freckling, cutaneous neurofibromas, Lisch nodules
- Phaeochromocytoma in a minority; adrenergic phenotype
- Optic glioma, skeletal and learning features
SDHx (SDHB, SDHD, SDHC)
- Extra-adrenal, multifocal, recurrent paraganglioma
- SDHB carries the HIGHEST risk of malignancy and metastasis
- SDHD shows parental imprinting (paternal transmission); head-and-neck paragangliomas
MAX, TMEM127
- Less common susceptibility genes
- Often bilateral adrenal tumours
- Intermediate metastatic risk
Pregnancy. Phaeochromocytoma in pregnancy is rare but, untreated, carries a high maternal and fetal mortality (historically up to 40 to 50 percent, now much lower with good care). The presentation may mimic pre-eclampsia; a key clue is hypertensive spells that are paroxysmal rather than sustained, and orthostatic hypotension (unusual in pre-eclampsia). Alpha-blockade with phenoxybenzamine is safe in pregnancy; add a beta-blocker cautiously for tachycardia. Delivery is by planned caesarean section with a multidisciplinary team (endocrinology, obstetrics, anaesthesia, surgery); avoid vaginal pushing as straining can trigger a crisis, and avoid certain oxytocic and anaesthetic agents that release catecholamines. The tumour is resected either at caesarean or soon after.[1][5]
Adrenal incidentaloma. Every adrenal incidentaloma must be biochemically screened with the triple panel — metanephrines, overnight dexamethasone suppression, and aldosterone-renin ratio (with potassium corrected). A phaeochromocytoma identified this way must never be biopsied; prepare with alpha-blockade and resect.[1]
Malignant and metastatic phaeochromocytoma. Malignancy is defined by the presence of metastases at non-chromaffin sites (bone, lung, liver, lymph nodes distant from the primary) — histology alone cannot reliably predict it, although scoring systems such as the GAPP (Grading of Adrenal Phaeochromocytoma and Paraganglioma) system and PASS (Phaeochromocytoma of the Adrenal Gland Scaled Score) attempt to stratify risk. Metastatic disease is most often associated with SDHB mutations and with extra-adrenal primary site. Management is multidisciplinary: surgical debulking to reduce catecholamine load; radionuclide therapy with 131I-MIBG (for MIBG-avid disease) and 177Lu-DOTATATE (for somatostatin-receptor-positive disease); chemotherapy with the CVD regimen (cyclophosphamide 750 mg/m² day 1, vincristine 1.4 mg/m² day 1, dacarbazine 600 mg/m² days 1 and 2, every 21 days); tyrosine-kinase inhibition; radiotherapy for painful bone metastases; and metyrosine to reduce catecholamine synthesis.[3][6]
FIRSTMAPPP (Baudin et al, 2024)
Lancet
Academic, multicentre, international, randomised, placebo-controlled, double-blind phase 2 trial of sunitinib in progressive, metastatic phaeochromocytoma and paraganglioma.
Key finding
Sunitinib improved progression-free survival at 12 months versus placebo in patients with progressive metastatic PPGL — the first randomised evidence for a systemic therapy in this rare disease.
Practice change
Establishes sunitinib as a reference systemic therapy for progressive metastatic PPGL, though overall response remains modest and follow-up therapies (CVD chemotherapy, 131I-MIBG, 177Lu-DOTATATE) are still needed.
Bladder paraganglioma presents with micturition spells (headache, sweating, palpitations and hypertension during or after voiding), sometimes with haematuria; it is often small and requires functional imaging to localise. Head-and-neck paragangliomas (carotid body, glomus jugulare, glomus tympanicum, vagal) are usually parasympathetic and non-functional, presenting as a pulsatile neck mass, hearing loss, tinnitus or cranial-nerve palsies rather than hypertension, and are frequently SDHD/SDHC-related.[1][3]
Who to screen, and the silent phaeochromocytoma. Because the disease is rare but dangerous, targeted screening of high-yield groups is far more cost-effective than population screening. The Endocrine Society recommends testing in: any patient with paroxysmal hypertension or the classic triad; an adrenal incidentaloma (5 to 7 percent of which are phaeochromocytomas); resistant or early-onset hypertension; a family history or known carrier of a susceptibility gene; any patient with a pressor response to anaesthesia, contrast or beta-blockade; and anyone with spell-like episodes. Importantly, a substantial minority of phaeochromocytomas — up to 15 to 30 percent — are normotensive at presentation, and many are entirely silent, found only on incidental imaging or declared by a crisis. This silent subset is precisely why biochemical screening of every adrenal incidentaloma is non-negotiable: a deceptively bland mass on CT may still be a phaeo, and biopsying it unblocked is fatal. The combination of plasma free metanephrines (sensitivity 96 to 100 percent) and a dedicated CT or MRI will catch virtually all tumours; a negative plasma metanephrine in a low-pre-test-probability patient effectively excludes the disease, while a positive result mandates imaging and blockade. The clonidine suppression test resolves the difficult middle ground of borderline levels in a stressed patient.[1][5]
Complications & Pitfalls
Disease-related complications arise from chronic and acute catecholamine excess: hypertensive crisis (stroke, intracerebral haemorrhage, hypertensive encephalopathy, myocardial infarction, arrhythmia), catecholamine cardiomyopathy and Takotsubo, acute pulmonary oedema, acute kidney injury, lactic acidosis, and multi-organ failure. Metabolic complications include hyperglycaemia, diabetes and weight loss.[2]
Classic pitfalls that cost marks — and lives — include: giving a beta-blocker before an alpha-blocker (precipitates an unopposed-alpha crisis); biopsying an unblocked phaeochromocytoma; inadequate preoperative alpha-blockade or volume expansion (intra-operative haemodynamic instability and post-resection hypotension); missing the diagnosis in pregnancy by attributing the spells to pre-eclampsia; relying on random catecholamines or VMA instead of metanephrines; not stopping interfering drugs before biochemical testing (false positives); failing to test genetics in a young, bilateral or recurrent patient; and omitting lifelong surveillance after resection.[1][2]
Post-resection, two predictable physiological shifts must be anticipated: hypotension from the abrupt loss of catecholamine tone on a fully alpha-blockaded, vasodilated patient (treat with IV fluids and vasopressors such as noradrenaline or phenylephrine), and hypoglycaemia from the loss of catecholamine-driven hyperglycaemia and residual insulin action (monitor glucose for 24 to 48 hours and treat with dextrose). Transient adrenal insufficiency may occur after bilateral surgery and requires glucocorticoid replacement.[2]
Prognosis & Disposition
A completely resected benign phaeochromocytoma is curative, and blood pressure usually normalises, though some patients remain hypertensive from chronic vascular remodelling. Lifelong biochemical surveillance is mandatory — plasma metanephrines at 6 weeks postoperatively (to confirm biochemical cure), then every 6 to 12 months indefinitely — for local recurrence, metastasis, and new primaries (especially in hereditary disease, where metachronous tumours are common). Patients with a germline mutation and their relatives enter a surveillance programme from childhood.[1][3]
Malignant or metastatic disease has a variable but often guarded prognosis; 5-year survival is roughly 50 to 80 percent depending on tumour burden, site and SDHB status, with some indolent metastatic courses lasting many years. Catecholamine-crisis mortality is high when unrecognised — reinforcing the absolute necessity of meticulous preoperative alpha-blockade and a high index of suspicion in any patient with an adrenal mass or refractory hypertension. After surgery, most patients are admitted to a high-dependency or intensive-care unit for 24 to 48 hours for haemodynamic and glucose monitoring.[1][2][6]
Special Populations
- Pregnancy — phaeochromocytoma is rare but dangerous; untreated it threatens both mother and fetus. Alpha-blockade (phenoxybenzamine) is safe; a beta-blocker is added cautiously. Plan a multidisciplinary, scheduled caesarean delivery; avoid vaginal pushing (straining triggers crisis) and catecholamine-releasing agents. Resect at caesarean or soon after. The key is to think of it — spells in pregnancy are easily misattributed to pre-eclampsia.[1][5]
- Children — paediatric phaeochromocytoma is more often bilateral, extra-adrenal and hereditary; genetic testing is mandatory and frequently reveals SDHx, VHL or MEN 2. Cortical-sparing surgery is preferred in bilateral disease.[3]
- Hereditary carriers — surveillance begins in childhood (age and modality gene-specific) and continues lifelong; cortical-sparing surgery is offered to avoid permanent steroid dependence while preserving adrenal cortical function.[3]
- Elderly with an incidentaloma — balance the surgical risk of adrenalectomy against the risk of catecholamine crisis from an unblocked tumour; biochemical screening is mandatory, and alpha-blockade is required before any intervention (including non-adrenal surgery) if metanephrines are raised.[1]
- Patients needing non-adrenal surgery — a known or occult phaeochromocytoma must be blocked before any operation; an undiagnosed tumour is a classic cause of unexplained haemodynamic collapse on induction.[2]
Evidence, Guidelines & Regional Differences
The Endocrine Society 2014 clinical practice guideline (Lenders et al) sets the international standard and is the most frequently cited reference in this field: it recommends first-line plasma free metanephrines or 24-hour urine fractionated metanephrines, CT/MRI for localisation, preoperative alpha-blockade, surgical resection, and genetic testing for all patients.[1] The 2024 scoping review by Saavedra et al consolidates contemporary practice from presentation to management.[5]
A live controversy is the choice of preoperative alpha-blockade. Traditional teaching (and the Endocrine Society guideline) favours non-competitive phenoxybenzamine for its complete, irreversible blockade. The Buitenwerf 2020 multicentre randomised controlled trial found that phenoxybenzamine did not significantly improve intraoperative haemodynamic control compared with no phenoxybenzamine — a finding that has stimulated debate but has not yet overturned standard practice pending larger trials.[4] A large single-centre comparison (Zhu et al, 2022) found comparable intraoperative stability between phenoxybenzamine and doxazosin-based regimens, with less postoperative hypotension in some selective alpha-blocker groups.[7] Most centres continue to use phenoxybenzamine for large, symptomatic or high-output tumours and selective alpha-blockers or calcium-channel blockers for smaller, less secretory lesions.[1]
For metastatic disease, the FIRSTMAPPP trial (Baudin et al, 2024) is a landmark: the first randomised, placebo-controlled, phase 2 trial to show a progression-free survival benefit for sunitinib in progressive metastatic PPGL, establishing it as a reference systemic therapy.[6] 131I-MIBG radionuclide therapy, 177Lu-DOTATATE peptide radioligand therapy, and CVD chemotherapy remain important for selected patients, and metyrosine reduces catecholamine synthesis. The personalised-management framework of Nölting et al (2022) ties genotype to imaging tracer and therapy choice — 18F-FDG for SDHB, 68Ga-DOTATATE for somatostatin-receptor-positive disease, 18F-DOPA for sporadic and SDHx disease.[3]
Regional practice varies mainly in functional imaging (MIBG versus PET tracers, with European centres using 68Ga-DOTATATE earlier), blockade drug preference (phenoxybenzamine in the UK and much of Europe; doxazosin or calcium-channel blockers in some North American centres), and surveillance intensity. The modern understanding has retired the rule of 10 for heritability — over 30 percent (commonly quoted 30 to 40 percent) carry a germline mutation, and genetic testing is now offered to all patients.[1][3]
Regional and resource considerations. In the UK, NICE hypertension guidance directs screening for secondary causes in patients with resistant or early-onset hypertension, and most endocrine units follow the Endocrine Society pathway with phenoxybenzamine-based blockade and laparoscopic adrenalectomy in specialist centres. In North America, the AACE/ATA adrenal-incidentaloma guidance endorses the same biochemical triple screen, with a growing preference for selective alpha-1 blockers or calcium-channel blockers in lower-risk lesions and earlier use of 68Ga-DOTATATE PET. In India and similar resource-limited settings, the 24-hour urine fractionated metanephrines and VMA remain widely used because plasma free metanephrine assays are not universally available; MIBG is more accessible than PET tracers, and genetic testing — now standard in high-income settings — is often prioritised to bilateral, extra-adrenal, recurrent or young-onset cases rather than offered universally. The principles do not change: biochemistry before any biopsy, alpha before beta, and complete surgical resection after adequate preparation.[1][5]
Exam Pearls
- Triad: episodic or sustained hypertension plus headache plus sweating plus palpitations; pallor (not flushing — flushing points to carcinoid).[1]
- Rule of 10: bilateral, malignant, extra-adrenal, familial, paediatric — historical; 30 to 40 percent now carry a germline mutation.[1][3]
- Associations: MEN 2 (RET), VHL, NF1, SDHx (especially SDHB = highest malignant risk).[3]
- Diagnosis: plasma free metanephrines (highest sensitivity) or 24-hour urine fractionated metanephrines; localise with CT/MRI (over 10 HU, slow washout, T2 light-bulb); MIBG / 68Ga-DOTATATE / 18F-FDG PET for extra-adrenal, metastatic, hereditary or recurrent disease.[1]
- ALPHA before BETA: phenoxybenzamine 10 mg BD titrated over 10 to 14 days to about 1 mg/kg/day, then add a beta-blocker (propranolol), then operate. Beta-first = fatal unopposed-alpha crisis.[1]
- Crisis: IV phentolamine (2.5 to 5 mg bolus) or sodium nitroprusside infusion; never a beta-blocker first; treat arrhythmia with esmolol only after alpha-blockade.[2]
- Histology: chromaffin cells in Zellballen nests; never biopsy an unblocked phaeo.[1]
- Metanephrines are produced continuously intra-tumourally by COMT — more sensitive than catecholamines or VMA.[1]
- Screen metanephrines in EVERY adrenal incidentaloma (with dexamethasone suppression and aldosterone-renin ratio) before any intervention.[1]
- Postoperative pitfalls: anticipate hypotension (give fluids and vasopressors) and hypoglycaemia (monitor and treat with dextrose).[2]
- Metyrosine = tyrosine hydroxylase inhibitor (cuts catecholamine synthesis); sunitinib = first-line TKI for metastatic disease (FIRSTMAPPP).[6]
- Pregnancy: spells mislabelled as pre-eclampsia; alpha-blockade safe; elective caesarean, avoid pushing.[1]
Phaeochromocytoma associations — ORGAN
ORGAN
classic site of extra-adrenal paraganglioma (secretes noradrenaline)
bilateral adrenal phaeo + medullary thyroid carcinoma + hyperparathyroidism; adrenergic
SDHB carries the highest risk of malignant, extra-adrenal, metastatic paraganglioma
phenoxybenzamine first for 10 to 14 days, then beta-blocker, then surgery
neurofibromatosis 1 (café-au-lait, neurofibromas) and von Hippel-Lindau (RCC, haemangioblastomas)
The classic triad and crisis drug — HSP
HSP
with episodic hypertension and sweating (and pallor, not flushing)
complete the classic triad; screen plasma or urine metanephrines
IV drug for catecholamine crisis; never give a beta-blocker first
Rule of 10 — the historical teaching device
10s
more in MEN 2 and VHL
higher for extra-adrenal and SDHB (up to 40 percent)
historical; now superseded — 30 to 40 percent carry a germline mutation
Self-test — what three clinical signs tell you alpha-blockade is adequate?
A seated blood pressure of about 130/80 with a mild orthostatic drop (but no syncope), the appearance of nasal congestion, and resolution of spells for about a week. Add the beta-blocker only once these are achieved.[1]
Self-test — why does a beta-blocker given first kill the patient?
A beta-blocker antagonises beta-2-mediated vasodilation while leaving alpha-1-mediated vasoconstriction unopposed, causing catastrophic vasoconstriction and a hypertensive crisis. Always establish alpha-blockade first.[2]
References
- [1]Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline J Clin Endocrinol Metab, 2014.PMID 24893135
- [2]Nazari MA, Hasan R, Haigney M, et al. Catecholamine-induced hypertensive crises: current insights and management Lancet Diabetes Endocrinol, 2023.PMID 37944546
- [3]Nölting S, Bechmann N, Taieb D, et al. Personalized Management of Pheochromocytoma and Paraganglioma Endocr Rev, 2022.PMID 34147030
- [4]Buitenwerf E, Osinga TE, Timmers HJLM, et al. Efficacy of α-Blockers on Hemodynamic Control during Pheochromocytoma Resection: A Randomized Controlled Trial J Clin Endocrinol Metab, 2020.PMID 31714582
- [5]Saavedra TJS, Nati-Castillo HA, Valderrama Cometa LA, et al. Pheochromocytoma: an updated scoping review from clinical presentation to management and treatment Front Endocrinol (Lausanne), 2024.PMID 39735644
- [6]Baudin E, Goichot B, Berruti A, et al. Sunitinib for metastatic progressive phaeochromocytomas and paragangliomas: results from FIRSTMAPPP, an academic, multicentre, international, randomised, placebo-controlled, double-blind, phase 2 trial Lancet, 2024.PMID 38402886
- [7]Zhu CY, Hong JC, Kamdar NV, et al. Comparison of Preoperative Alpha-blockade for Resection of Paraganglioma and Pheochromocytoma Endocr Pract, 2022.PMID 35809774