Gastroenterology · General Medicine
Viral Hepatitis (Hepatitis A, B, C, D, E)
Also known as Viral hepatitis · Hepatitis A · Hepatitis B · Hepatitis C · Hepatitis D · Hepatitis E · HBV · HCV
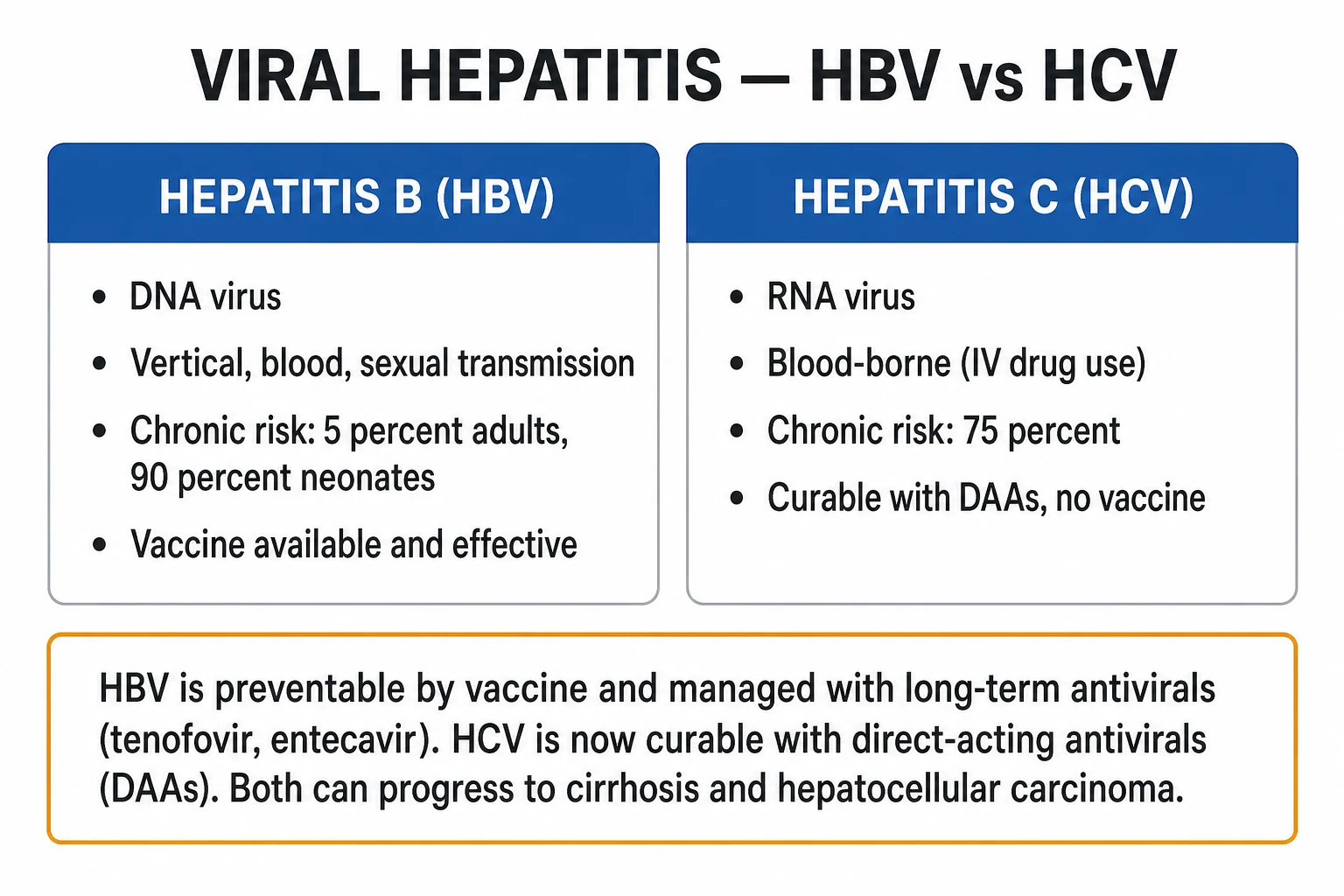
Viral hepatitis is inflammation of the liver caused by the five hepatotropic viruses — A (RNA, faecal-oral, never chronic), B (DNA, parenteral/vertical/sexual, chronic in 90 percent neonates), C (RNA, blood-borne, chronic in 75 percent, curable with DAAs), D (defective RNA requiring HBsAg), E (RNA, faecal-oral, fulminant in pregnancy). Acute infection presents with prodrome (anorexia, nausea, fatigue, arthralgia) then jaundice, dark urine, tender hepatomegaly; chronic infection is often asymptomatic until cirrhosis or HCC. Diagnosis by serology: HBsAg/anti-HBc (HBV), anti-HCV/HCV RNA (HCV), IgM anti-HAV/anti-HEV. Management: supportive for HAV/HEV; long-term nucleos(t)ide analogues (tenofovir, entecavir) for HBV; direct-acting antivirals (DAAs) 8-12 weeks with cure over 95 percent for HCV. Prevention: HAV and HBV vaccines, birth-dose HBV vaccine plus HBIG, harm reduction, blood screening. Both chronic HBV and HCV drive cirrhosis and hepatocellular carcinoma requiring 6-monthly surveillance.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Exam tags
Red flags

Overview & Definition
Viral hepatitis is inflammation of the liver caused by hepatotropic viruses — classically the five named A, B, C, D and E. They share the ability to infect and injure hepatocytes, but differ profoundly in genome, transmission route, chronicity, treatment and prevention.[1][2]
The clinical skill in viral hepatitis is not merely recognising jaundice. It is (1) identifying the responsible virus by serology, (2) distinguishing acute from chronic infection (which changes everything), (3) staging fibrosis non-invasively, (4) treating — curative DAAs for HCV, lifelong suppression for HBV — and (5) preventing transmission by vaccination, birth-dose immunisation, post-exposure prophylaxis and harm reduction. The single most common and most dangerous process error is missing HBV reactivation when an HBsAg-positive (or even anti-HBc-positive) patient starts rituximab, chemotherapy or high-dose steroids — it can cause fatal fulminant hepatitis that is entirely preventable with prophylactic tenofovir or entecavir.[1]
The hepatotropic viruses are distinct from the non-hepatotropic viruses that incidentally cause hepatitis — Epstein-Barr virus (infectious mononucleosis), cytomegalovirus, herpes simplex (rare but fulminant in pregnancy and immunosuppressed), and yellow fever virus. These cause a milder, diffuse hepatitis as part of systemic illness and are not the focus of this chapter.[8]
Classification
The hepatotropic viruses are classified by transmission route (the practical division) and by genome (the virological division). Memory aid: the vowels (A and E) are Enteric (faecal-oral); the consonants (B, C, D) are Blood-borne (parenteral).[1]
| Virus | Genome / Family | Transmission | Chronicity | Vaccine | Treatment |
|---|---|---|---|---|---|
| HAV | RNA / Picornaviridae | Faecal-oral | Never chronic | Yes | Supportive |
| HBV | DNA / Hepadnaviridae | Parenteral, sexual, vertical | 90 percent neonate, under 5 percent adult | Yes | Tenofovir / entecavir |
| HCV | RNA / Flaviviridae | Blood (IVDU) | 75 percent | No | DAAs (cure) |
| HDV | RNA / Deltavirus (defective) | As for HBV (needs HBsAg) | Superinfection 80-90 percent chronic | HBV vaccine protects | Peg-IFN, bulevirtide |
| HEV | RNA / Hepeviridae | Faecal-oral, water-borne | Immunocompetent: no; transplant: yes | China only | Supportive; ribavirin if chronic |
Enteric hepatitis (A and E)
- **RNA viruses**, faecal-oral / water-borne
- Incubation 2-6 weeks (HAV) and 2-9 weeks (HEV)
- **Never chronic** in immunocompetent (HEV can be chronic in transplant/HIV)
- Vaccine exists for HAV (and HEV in China only)
- Fulminant: HAV rare in young; **HEV dangerous in pregnancy (20 percent mortality)**
Parenteral hepatitis (B, C, D)
- **B is DNA; C and D are RNA**
- Transmission by blood, sexual, vertical (HBV)
- All three can become chronic
- **HBV = vaccine-preventable, lifelong antiviral suppression**
- **HCV = no vaccine, curable with DAAs in 8-12 weeks**
- **HDV = defective, needs HBsAg envelope**
Acute versus chronic — the most important clinical division. Acute hepatitis is infection under 6 months' duration; chronic hepatitis is persistence of HBsAg (HBV) or HCV RNA beyond 6 months. HAV and HEV never cause chronic infection in the immunocompetent host.[1][4]
The four immunological phases of chronic HBV (a recurring exam favourite) classify the dynamic state of the host-virus interaction:[1][4]
- HBeAg-positive immune-tolerant phase — high HBV DNA, normal ALT, minimal fibrosis. Typical of perinatally acquired infection; lasts decades. No treatment.
- HBeAg-positive immune-active phase — high DNA (typically over 20,000 IU/mL), ALT elevated, active hepatitis, HBeAg positive. Treat.
- Inactive carrier phase (after HBeAg seroconversion) — HBsAg positive, DNA under 2000 IU/mL, normal ALT. Monitor; low HCC risk but not zero.
- HBeAg-negative immune-active phase — fluctuating DNA over 2000 IU/mL, ALT elevated, anti-HBe positive (precore/basal core promoter mutants). Treat; relapsing course. [1]
Epidemiology & Risk Factors
Viral hepatitis is among the commonest infectious diseases globally and a leading cause of infectious death. The WHO estimates that approximately 296 million people are chronically infected with HBV and approximately 58 million with HCV; viral hepatitis kills over 1 million people per year (mostly from HBV-related cirrhosis and HCC), rivalling tuberculosis and HIV.[7]
Hepatitis A — worldwide; incidence correlates with sanitation. Endemic in India, Africa, South-East Asia (most children infected asymptomatically before age 10). In high-income countries it occurs in travellers, men who have sex with men, food-borne outbreaks, and people who inject drugs. [1]
Hepatitis B — commonest cause of chronic viral hepatitis in India, China, South-East Asia and sub-Saharan Africa, where 5 to 10 percent of adults are HBsAg positive and transmission is predominantly vertical (perinatal). In low-endemicity regions (USA, Western Europe) transmission is sexual and parenteral (IVDU). The HBV vaccine (since 1982) has dramatically reduced prevalence in countries with universal programmes.[1][4]
Hepatitis C — commonest in Egypt (historically from parenteral antischistosomal therapy, genotype 4), Pakistan (reuse of needles, genotype 3), and among people who inject drugs globally (genotype 1 and 3). The global genotype distribution is approximately genotype 1 (46 percent), genotype 3 (22 percent), genotypes 2 and 4 (about 10 percent each).[7]
Hepatitis D — requires HBsAg; prevalence roughly 5 percent of HBsAg-positive persons worldwide (12 to 48 million). Highest in the Amazon basin, Central Africa, Mongolia, Eastern Europe. Prevalence is falling with HBV vaccination.[1]
Hepatitis E — the commonest cause of acute viral hepatitis in adults in India and much of the developing world. Large water-borne outbreaks ( Kashmir 1978, refugee camps). Unique risk of fulminant failure in pregnancy (20 to 25 percent mortality).[8]
- Perinatal exposure to an HBeAg-positive mother (90 percent transmission without immunoprophylaxis — the commonest global route)
- Sexual contact with an infected person (HBV 10 to 30 percent transmission per partner-year)
- Injection drug use (shared equipment)
- Blood transfusion (now rare in countries with screening; still relevant in low-resource settings)
- Haemodialysis, occupational needlestick (surgeons, dentists, laboratory workers)
- Tattooing, piercing, acupuncture with unsterile equipment
- Horizontal transmission in young children in endemic areas (via open cuts, shared toothbrushes) [1]
- Injection drug use (commonest in the West — past or current, even once)
- Blood transfusion or organ transplant before 1992 (before anti-HCV screening)
- Unsafe therapeutic injections (commonest globally — reuse of syringes)
- Haemodialysis (annual incidence 1 to 3 percent even with screening)
- Being born to an HCV-positive mother (vertical transmission approximately 5 percent, higher with HIV co-infection)
- Occupational needlestick (around 2 percent transmission)
- HIV co-infection (accelerates fibrosis)
- Being born between 1945 and 1965 (US "baby boomers" — five times higher HCV prevalence; CDC recommends one-time screening)
- Intranasal cocaine use, unsterile tattooing/piercing [1]
Viral hepatitis — the numbers examiners test
Pathophysiology
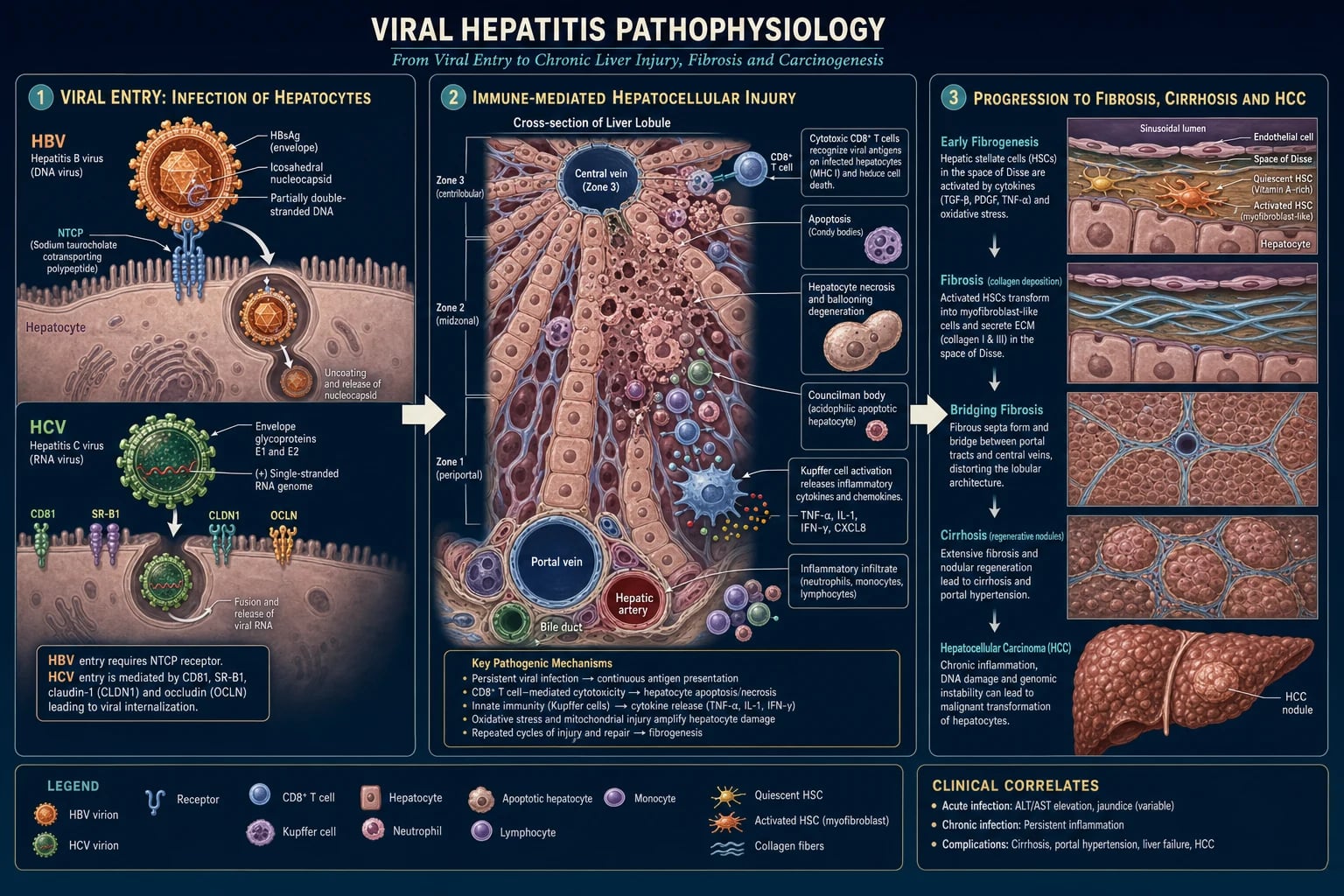
The hepatotropic viruses injure the liver by interlocking mechanisms: direct cytopathic effect (minor for HBV, more relevant for HCV), and — overwhelmingly — immune-mediated hepatocyte apoptosis driven by cytotoxic CD8-positive T-cells. The severity of liver injury reflects the vigour of the host immune response more than the viral load.[1][4]
HBV life cycle — the DNA minichromosome that defies cure
HBV is a small (42 nm) enveloped DNA virus (the Dane particle). Its genome is a partially double-stranded circular DNA of approximately 3200 base pairs. The life cycle:[1][5]
- Binding and entry — HBV binds the sodium taurocholate cotransporting polypeptide (NTCP, SLC10A1) receptor on the hepatocyte basolateral membrane, enabling receptor-mediated endocytosis.
- Nuclear delivery and cccDNA formation — the partially double-stranded DNA is delivered to the nucleus and repaired by host enzymes into covalently closed circular DNA (cccDNA), a stable minichromosome that serves as the transcription template for all viral RNAs.
- Transcription — host RNA polymerase II transcribes the four major RNAs (precore, polymerase, surface, X) from cccDNA.
- Replication via reverse transcription — the pregenomic RNA is packaged with the viral polymerase into core particles; the polymerase's reverse-transcriptase domain reverse-transcribes the RNA into the new partially double-stranded DNA.
- Assembly and secretion — core particles bud into the endoplasmic reticulum, acquire the surface antigen (HBsAg) envelope, and are secreted as complete Dane particles (and as subviral HBsAg filaments/spheres that vastly outnumber complete virions). [1]
Why HBV is not curable with current drugs: the cccDNA minichromosome persists in the hepatocyte nucleus, is not eliminated by nucleos(t)ide analogues (which only block new reverse transcription), and serves as the reservoir for viral rebound when therapy stops. Functional cure (HBsAg loss) occurs in only 1 to 3 percent of patients per year on treatment. True sterilising cure would require cccDNA elimination or removal of all infected hepatocytes — the goal of pipeline therapies (capsid inhibitors, RNA interference, entry inhibitors, therapeutic vaccines).[1]
HCV life cycle — no DNA stage, hence curable
HCV is a small (55 to 65 nm) enveloped positive-sense single-stranded RNA virus of the family Flaviviridae, genome approximately 9600 bases. The life cycle:[2][6]
- Binding and entry — HCV binds a complex of receptors including CD81, scavenger receptor class B type 1 (SR-B1), claudin-1 (CLDN1) and occludin, enabling endocytosis.
- Release and translation — the viral RNA is uncoated and the positive-sense genome is directly translated on the rough endoplasmic reticulum into a single polyprotein, cleaved by viral and host proteases into 10 proteins (core, E1, E2, p7, NS2, NS3/4A protease, NS4B, NS5A, NS5B RNA-dependent RNA polymerase).
- Replication — NS5B copies the positive-sense RNA into a negative-sense intermediate, then into new positive-sense genomes, on a virus-induced membranous web derived from the ER.
- Assembly and secretion — new virions assemble on lipid droplets and are secreted via the very-low-density lipoprotein pathway (HCV is a "lipo-viro-particle"). [1]
Why HCV is curable: there is no DNA intermediate, no integration into the host genome, and no long-lived viral reservoir. DAAs target essential viral enzymes (NS3/4A protease, NS5A, NS5B polymerase) and, given 8 to 12 weeks, eliminate the virus entirely. Sustained virological response at 12 weeks (SVR12) — HCV RNA undetectable 12 weeks after therapy ends — is virologically equivalent to cure.[2][3][6]
Immune pathogenesis and the cytokine cascade
In both HBV and HCV, hepatocyte death is immune-mediated. Infected hepatocytes present viral peptide on MHC class I to CD8-positive cytotoxic T lymphocytes, which release perforin and granzyme and trigger apoptosis. Kupffer cells (resident macrophages) release tumour necrosis factor-alpha, interleukin-1, interferon-gamma, recruiting neutrophils and additional mononuclear cells. The inflammatory infiltrate produces the histological hallmarks: portal inflammation, interface hepatitis ("piecemeal necrosis"), lobular disarray, ballooning degeneration, Councilman bodies (apoptotic hepatocytes), and regenerative double-nucleated hepatocytes.[1]
The outcome — clearance versus chronicity — hinges on the T-cell response. Acute resolving HBV mounts a broad, polyclonal, multi-epitope CD4 and CD8 T-cell response that is sustained for weeks; chronic HBV is associated with a narrow, oligoclonal, exhausted T-cell response (high PD-1 expression) that fails to clear infected hepatocytes. This immunological difference is why neonates (immature immune system) develop chronic infection in 90 percent of cases, while immunocompetent adults clear the virus in over 95 percent.[1][4]
Fibrogenesis and the road to cirrhosis
Persistent hepatocyte injury activates hepatic stellate cells (Ito cells, vitamin-A storing perisinusoidal cells), which transdifferentiate into myofibroblast-like cells, proliferate, and deposit type I and type III collagen in the space of Disse. This produces sinusoidal capillarisation (loss of fenestrated endothelium), impaired hepatocyte-blood exchange, and rising sinusoidal pressure — the substrate of portal hypertension.[10]
Fibrosis accumulates over decades: chronic HBV reaches cirrhosis in 8 to 20 percent over 5 years untreated; chronic HCV in 15 to 30 percent over 20 years. Progression is accelerated by alcohol, obesity and diabetes (MASLD overlap), HIV co-infection, male sex, older age at infection, and HBV-HCV co-infection.[2][10]
Why HBV (and not HCV) is directly oncogenic
HBV causes hepatocellular carcinoma even without cirrhosis. Mechanisms: (1) HBV DNA integration into the host genome (a near-universal event in HBV-related HCC) causes chromosomal instability and insertional mutagenesis; (2) the HBx protein transactivates cellular growth and apoptotic pathways and sequesters p53; (3) chronic inflammation and oxidative damage from immune-mediated injury. By contrast, HCV causes HCC almost exclusively in the context of established cirrhosis, by chronic regenerative injury and oxidative stress — there is no DNA intermediate and no integration. This is why HCC surveillance is offered to all chronic HBV patients with risk factors even in the absence of cirrhosis, whereas for HCV it is offered to those with cirrhosis.[1]
HEV in pregnancy — the special catastrophe
HEV infection in pregnancy (second and third trimester) carries a uniquely high risk of acute liver failure, with maternal mortality of 20 to 25 percent. Mechanisms include immune modulation by pregnancy hormones, suppression of T-helper-1 responses, high viral replication, and increased hepatocyte susceptibility. The fetus suffers high rates of prematurity, stillbirth and vertical transmission. Management is supportive (and urgent delivery if fulminant); ribavirin is contra-indicated in pregnancy (teratogenic).[8]
Clinical Presentation
Acute viral hepatitis — the four classical phases
Acute hepatitis (any of the five viruses) runs a stereotyped course divided into four phases:[8]
- Incubation phase (asymptomatic, viraemic) — HAV 15 to 50 days (median 28); HBV 30 to 180 days (median 75); HCV 2 weeks to 6 months (median 50); HDV as for HBV; HEV 2 to 9 weeks (median 40).
- Prodromal (pre-icteric) phase (1 to 2 weeks) — anorexia (often the first symptom), nausea, vomiting, fatigue, malaise, low-grade fever, right-upper-quadrant discomfort, arthralgia, myalgia, headache, coryza, pharyngitis, and a characteristic distaste for cigarettes (a classic exam pearl). A serum-sickness-like prodrome (arthralgia, urticarial rash, fever) occurs in HBV (immune-complex mediated).
- Icteric phase (1 to 6 weeks) — onset of dark urine (bilirubinuria, the first objective sign), then pale/clay-coloured stools (intrahepatic cholestasis), then jaundice (scleral, then cutaneous), with pruritus. Examination shows tender hepatomegaly, mild splenomegaly (especially in EBV and HBV in children), and cervical lymphadenopathy.
- Convalescent phase (weeks to months) — symptoms resolve; fatigue may persist for months (post-hepatitis fatigue syndrome). ALT and bilirubin normalise. [1]
Atypical acute presentations include: anicteric hepatitis (most common in children, easily missed); relapsing hepatitis (biphasic, common with HAV); cholestatic hepatitis (deep prolonged jaundice and pruritus, especially HAV in older adults); fulminant hepatitis (acute liver failure — see Complications); and aplastic anaemia (rare, classically after HAV in young men). [1]
Chronic viral hepatitis — the silent disease
Chronic HBV and HCV are usually asymptomatic for decades, detected incidentally on LFTs, on screening (blood donation, antenatal, pre-immunosuppression), or at presentation with complications of cirrhosis or HCC. Vague fatigue is the commonest reported symptom. Once cirrhosis develops, patients present with jaundice, ascites, variceal bleed, hepatic encephalopathy, or hepatomegaly/nodule (HCC).[1][2]
Extrahepatic manifestations
These are favourite exam topics and may precede the liver diagnosis. [1]
HBV extrahepatic (immune-complex mediated):[1][4]
- Serum-sickness-like prodrome — arthralgia/arthritis, urticarial rash, fever (10 to 30 percent of acute HBV)
- Polyarteritis nodosa (PAN) — medium-vessel vasculitis, mononeuritis multiplex, mesenteric ischaemia, hypertension; HBV accounts for 10 to 30 percent of PAN
- Membranous glomerulonephritis and membranoproliferative GN (especially in children)
- Gianotti-Crosti syndrome (papular acrodermatitis of childhood) — symmetrical papular rash on cheeks, buttocks and extensor surfaces
- Mixed cryoglobulinaemia (less common than HCV) [1]
HCV extrahepatic (the extrahepatic HCV syndrome):[2][3]
- Mixed cryoglobulinaemia (Type II, with rheumatoid factor monoclonal IgM against polyclonal IgG) — palpable purpura, arthralgia, weakness, glomerulonephritis, peripheral neuropathy
- Membranoproliferative glomerulonephritis
- Porphyria cutanea tarda — photosensitive bullous rash on sun-exposed skin
- Lichen planus — oral Wickham striae, pruritic polygonal purple papules on wrists
- Non-Hodgkin lymphoma (especially marginal-zone and diffuse large B-cell)
- Type 2 diabetes mellitus (HCV-induced insulin resistance)
- Sicca syndrome (dry eyes/dry mouth resembling Sjogren)
- Autoimmune thyroiditis (especially with interferon therapy) [1]
Atypical and high-risk presentations
- Pregnancy with HEV — fulminant hepatitis, coagulopathy, encephalopathy, 20 to 25 percent mortality; test HEV urgently in any pregnant woman with acute hepatitis.
- Immunosuppressed patient — HBV reactivation (rise in HBV DNA and ALT on rituximab, anti-CD20, high-dose steroids, anthracyclines, TNF inhibitors) can be fulminant and fatal; chronic HEV in solid-organ transplant and HIV; fulminant HSV hepatitis in pregnancy and immunosuppressed (aciclovir empirically).
- Children — usually asymptomatic or mild; perinatally acquired HBV is the silent driver of chronic infection.
- Elderly — atypical, more often cholestatic, more severe HAV (fulminant rate rises with age), lower vaccine response.
- Acquired in adulthood — acute HBV may present with frank jaundice and serositis; acute HCV is rarely symptomatic (only 20 to 30 percent symptomatic). [1]
Differential Diagnosis
An acute hepatitis picture (ALT over 10 times the upper limit of normal, with or without jaundice) is not always viral. The complete differential:[8][9]
- Drug-induced liver injury (DILI) — paracetamol (zone-3 necrosis, very high ALT over 1000, history of overdose); anti-tuberculous drugs (isoniazid, rifampicin, pyrazinamide); statins; amoxicillin-clavulanate; NSAIDs; antiepileptics (phenytoin, valproate); herbal and ayurvedic supplements; anabolic steroids. Distinguished by detailed drug history, AST/ALT pattern, eosinophilia, and viral serology negative.
- Ischaemic hepatitis ("shock liver") — AST and ALT both over 1000 with a clear episode of hypotension/shock/sepsis, rapid rise and fall (halves in 24 to 48 hours). Cardiac, respiratory or hypovolaemic cause.
- Alcoholic hepatitis — AST and ALT typically under 300 IU/L, with AST greater than ALT (ratio over 2), GGT markedly elevated, macrocytosis, thrombocytopenia, history of heavy alcohol use.
- Autoimmune hepatitis — high IgG, positive antinuclear antibody / anti-smooth muscle antibody / anti-LKM1, female predominance, other autoimmune disease.
- Wilson disease — young patient, Coombs-negative haemolytic anaemia, low caeruloplasmin, Kayser-Fleischer rings, ALP markedly low, ALP-to-bilirubin ratio under 2.
- Biliary obstruction (choledocholithiasis, pancreatic cancer, cholangiocarcinoma) — ALP and GGT disproportionately high, pain, fever (cholangitis), dilated ducts on ultrasound; magnetic resonance cholangiopancreatography to confirm.
- Budd-Chiari syndrome — painful hepatomegaly, ascites, hypercoagulable state.
- Other infections — EBV, CMV, leptospirosis, Q fever, yellow fever, malaria.
- Haemolysis — unconjugated hyperbilirubinaemia, normal ALT, high LDH, low haptoglobin.
- Hereditary hyperbilirubinaemias — Gilbert (unconjugated, normal ALT, mild, stress-triggered), Dubin-Johnson and Rotor (conjugated, benign). [1]
Distinguishing the five hepatotropic viruses from one another is done entirely by serology (see Investigations) — the clinical picture alone is rarely sufficient, although the exposure history, incubation period, and chronicity risk provide strong clues.[1]
In a patient with chronic HBV and rising ALT, distinguish: immune-active chronic HBV (rising DNA), acute HBV superinfection on chronic HBV (rare), HDV superinfection (test anti-HDV), HCV co-infection (test anti-HCV/HCV RNA), alcoholic or drug injury, hepatotoxic herbal/Ayurvedic, and acute viral superinfection (HAV, HEV) causing decompensation of underlying chronic HBV.[1]
Clinical & Bedside Assessment
A focused bedside examination in suspected viral hepatitis documents (1) the hepatic syndrome, (2) severity, (3) extrahepatic clues, and (4) aetiological risk factors. [1]
Acute hepatitis signs: jaundice (scleral first), tender hepatomegaly (liver edge 1 to 3 cm, soft, tender), mild splenomegaly (especially in children and EBV), cervical lymphadenopathy, low-grade fever. [1]
Severity / decompensation signs (seek these aggressively — they signal acute liver failure): asterixis (flapping tremor of hepatic encephalopathy), fetor hepaticus, constructional apraxia (Reitan trail test, clock drawing), reduced conscious level, bruising/bleeding (coagulopathy), shrinking liver on percussion (a grave sign in fulminant hepatitis).[8][9]
Chronic liver disease stigmata (signal established cirrhosis): palmar erythema, spider naevi, gynaecomastia, testicular atrophy, parotid enlargement, Dupuytren contracture, leuconychia, clubbing, caput medusae, ascites, splenomegaly, peripheral oedema. [1]
Extrahepatic and aetiological clues: palpable purpura on the lower limbs (HCV cryoglobulinaemia), papular acrodermatitis on the extensor surfaces of a child (Gianotti-Crosti, HBV), arthritis and urticarial prodrome (HBV), mononeuritis multiplex and mesenteric ischaemia (HBV-PAN), photosensitive bullae on the dorsum of the hands (porphyria cutanea tarda, HCV), oral Wickham striae and pruritic purple polygonal papules on the wrists (lichen planus, HCV), tattoos, IVDU track marks, piercing, Kayser-Fleischer rings (Wilson), cushion goid habitus (alcohol). [1]
A focused exposure history — every hepatitis patient: blood transfusion (year, country), surgery, dental work, tattoos and piercings (year, sterile?), occupation (healthcare worker?), sexual contacts and partners, IVDU (ever, even once, with shared equipment), intranasal cocaine, occupational needlestick, maternal HBV status, travel to endemic regions, food and water exposure, household or sexual contact with known hepatitis, alcohol, medications (prescribed, OTC, herbal, Ayurvedic), and recent chemotherapy or immunosuppression.[1]
Investigations
Investigations in viral hepatitis serve four questions: (1) is this hepatitis? (2) which virus? (3) acute or chronic, and how severe? (4) what is the fibrosis stage? [1]
First-line bloods in any suspected viral hepatitis
- ALT and AST — markedly elevated (often 10 to 50 times upper limit) in acute viral hepatitis; AST/ALT ratio greater than 2 suggests alcoholic hepatitis.
- Bilirubin (total and fractionated) — both conjugated and unconjuginated rise; dark urine confirms conjugated hyperbilirubinaemia.
- ALP and GGT — mildly elevated; disproportionately high suggests cholestasis or biliary obstruction.
- Albumin and INR — markers of hepatic synthetic function; INR over 1.5 with encephalopathy = acute liver failure.[8][9]
- Full blood count — atypical lymphocytes (EBV, CMV), thrombocytopenia (chronic liver disease/hypersplenism, aplastic anaemia after HAV).
- Urea and electrolytes, glucose — renal function (hepatorenal syndrome), glucose (hypoglycaemia in ALF).
Serology — the key to identifying the virus
HBV serology panel — the most-tested table in hepatology
The HBV serology panel uses six markers to define the phase and natural history of infection. Understanding these patterns is a guaranteed NEET-PG/INICET question.[1][4]
| Marker | Significance | Appears / disappears |
|---|---|---|
| HBsAg | Surface antigen — first marker; presence = infection | 1 to 10 weeks post-exposure; persists beyond 6 months = chronic |
| anti-HBs | Antibody to surface — protective immunity | After recovery or vaccination; anti-HBs over 10 mIU/mL = immune |
| IgM anti-HBc | IgM antibody to core — most sensitive marker of acute HBV | Appears in window period (when HBsAg wanes, anti-HBs not yet) |
| total anti-HBc (IgG + IgM) | Past or current infection (never from vaccine) | Persists for life |
| HBeAg | Soluble antigen — marker of high infectivity and active replication | Present in immune-tolerant and HBeAg-positive immune-active phases |
| anti-HBe | Antibody to e — appears after HBeAg seroconversion | Marks transition to inactive carrier |
| HBV DNA | Direct measure of viral load — guides treatment | Over 20,000 IU/mL HBeAg-positive active; over 2000 IU/mL HBeAg-negative active |
The classic serological patterns (memorise verbatim): [1]
| Clinical state | HBsAg | anti-HBs | IgM anti-HBc | total anti-HBc | HBeAg | anti-HBe | HBV DNA |
|---|---|---|---|---|---|---|---|
| Acute HBV | positive | negative | positive | positive | positive | negative | high |
| Window period | negative | negative | positive | positive | variable | variable | lower |
| Recovered (immune) | negative | positive | negative | positive | negative | positive | undetectable |
| Chronic HBV (HBeAg-pos) | positive | negative | negative | positive | positive | negative | very high |
| Chronic HBV (HBeAg-neg) | positive | negative | negative | positive | negative | positive | high |
| Vaccinated (immune) | negative | positive | negative | negative | negative | negative | undetectable |
| Occult HBV | negative | negative/pos | negative | positive | negative | variable | low/undetectable |
The "window period" — when HBsAg has disappeared (typically 4 to 6 months after acute infection) but anti-HBs has not yet appeared — is bridged by IgM anti-HBc, the most sensitive marker of acute HBV and the single most-tested serological fact.[1][4]
HCV testing — the two-step algorithm
- anti-HCV antibody — screening test (enzyme immunoassay); positive indicates past or current infection (cannot distinguish). Becomes positive 4 to 10 weeks after exposure; never declines.
- HCV RNA (PCR) — confirmatory; positive = active infection; quantifies viral load for treatment and SVR monitoring. [1]
A positive antibody with negative HCV RNA = resolved (spontaneously cleared or treated) infection. There is no reliable IgM anti-HCV; acute HCV is diagnosed on the basis of a documented seroconversion (negative antibody then positive) or HCV RNA positivity in the first 6 months, with a defined exposure.[2][3]
Genotyping (1 through 6) was previously essential for selecting DAA regimen and duration; with pan-genotypic regimens (sofosbuvir/velpatasvir, glecaprevir/pibrentasvir) it is now less critical but still recommended for difficult cases (genotype 3 with cirrhosis).[2]
HAV and HEV serology
- IgM anti-HAV — acute HAV; IgG anti-HAV = past infection or vaccine.
- IgM anti-HEV — acute HEV; IgG anti-HEV = past infection. HEV RNA for immunocompromised/chronic cases. [1]
HDV serology
- Total anti-HDV as a screen in every HBsAg-positive patient (underdiagnosed); IgM anti-HDV for acute coinfection; HDV RNA for active replication. [1]
Non-invasive fibrosis assessment
Staging fibrosis decides who to treat (any bridging fibrosis or active hepatitis), the urgency of HCC surveillance, and prognosis. Non-invasive methods have largely replaced liver biopsy.[1][10]
- Transient elastography (FibroScan) — liver stiffness measurement (LSM): under 7 kPa = no/mild fibrosis (F0-F1); 8 to 12 kPa = significant fibrosis (F2-F3); over 12 to 15 kPa = advanced fibrosis/cirrhosis (F4). A high score in chronic HBV or HCV mandates HCC surveillance and variceal screening.
- FIB-4 score = (age times AST) divided by (platelet count times square root of ALT) — under 1.45 low, over 3.25 significant fibrosis; useful primary-care triage.
- APRI (AST-to-platelet ratio index) — over 1.5 significant; under 0.5 low.
- AST-to-ALT ratio over 1 — suggests advanced fibrosis (or alcoholic hepatitis).
- Liver biopsy — reserved for uncertain aetiology (suspected autoimmune overlap, DILI, Wilson), grading before Peg-IFN, suspected HDV with atypical features, or when elastography is unreliable (ascites, obesity, narrow intercostal space). [1]
Severity scoring — refer to acute liver failure
When acute viral hepatitis progresses to coagulopathy and encephalopathy, apply the King's College Criteria for liver transplantation (paracetamol and non-paracetamol versions — see the related acute-liver-failure topic for the verbatim criteria). Any INR over 1.5 with encephalopathy, under 26 weeks, without pre-existing cirrhosis = acute liver failure — escalate to ICU and transplant centre.[8][9]
Pre-treatment work-up
Before starting antiviral therapy:[1][2][3]
- HBV: HBV DNA viral load, HBeAg/anti-HBe, HBV genotype (if considering Peg-IFN), FibroScan, ALT, INR, platelets, creatinine, HDV (anti-HDV), HIV, HCV serology, TDF renal monitoring (eGFR, phosphate, urine glucose).
- HCV: HCV genotype (where available), viral load, FibroScan, renal function, HBV serology (HBsAg, anti-HBc, anti-HBs — to prevent HBV reactivation, a boxed warning), HIV, pregnancy test, drug-interaction check (amiodarone contra-indicated with sofosbuvir), vaccination status (HAV, HBV). [1]
Management — Resuscitation
Most acute viral hepatitis is mild and self-limiting and needs no resuscitation. The resuscitation priorities apply to the severe and fulminant presentations and to the public-health exposures.[8][9]
Fulminant viral hepatitis (acute liver failure)
ABCDE assessment. Airway — intubate early for grade III to IV encephalopathy (airway protection and intracranial pressure control). Breathing — high-flow oxygen. Circulation — two large-bore cannulae, group and save, IV access; treat hypotension with albumin and noradrenaline. Disability — check and correct hypoglycaemia (10 percent or 50 percent dextrose); treat rising intracranial pressure (head of bed 30 degrees, hypertonic saline to sodium 145 to 155, mannitol 0.5 g/kg). Exposure — full history including drug ingestion, exposure, transplantation history, and pregnancy. [1]
Apply the King's College Criteria immediately (see acute-liver-failure topic). The fulminant viral hepatitis causes are: HBV (1 percent of acute cases), HDV coinfection or superinfection, HAV (rare, more in older adults or underlying liver disease), HEV in pregnancy, and herpes simplex hepatitis (in pregnancy/immunosuppressed — give empirical aciclovir IV 10 mg/kg every 8 hours). HCV almost never causes fulminant failure. [1]
Hepatology referral and transfer to a transplant centre the moment King's College Criteria are met. Do not wait for multi-organ failure. Do not prophylactically correct the INR with fresh frozen plasma — it erases the key prognostic and transplant trigger; correct only if bleeding or before a procedure.[8][9]
Needlestick and occupational exposure to HBV
Immediate management of a needlestick from a known HBsAg-positive (or unknown-status) source:[1][4]
- Source HBsAg positive, exposed person unvaccinated: HBIG 0.06 mL/kg intramuscular within 24 hours (best within 12 hours) plus initiate the HBV vaccine series (dose 1 now, 2 at 1 month, 3 at 6 months).
- Source HBsAg positive, exposed person vaccinated but known non-responder (anti-HBs under 10): HBIG x 2 doses (1 month apart) plus a vaccine booster.
- Source HBsAg positive, exposed person vaccinated and responder (anti-HBs over 10): no treatment; test anti-HBs and booster if low.
- Source unknown or HBsAg negative: vaccinate the unvaccinated; no HBIG. [1]
For HCV needlestick there is no post-exposure prophylaxis — test source anti-HCV and HCV RNA; test the exposed person's HCV RNA at 3 to 6 weeks and antibody at 4 to 6 months; treat with DAAs immediately if seroconversion is documented (this is acute HCV and is highly curable).[2]
HBV reactivation on immunosuppression — prevent it
The most preventable fatal event in viral hepatitis. Every patient starting immunosuppression (chemotherapy, biological therapy, solid-organ or haematopoietic stem cell transplant) must be screened for HBV (HBsAg, anti-HBc, anti-HBs).[1]
- HBsAg-positive patient starting a high-risk regimen (rituximab, ofatumumab, anti-CD20, high-dose corticosteroids over 4 weeks, anthracyclines, haematopoietic stem cell transplant): prophylactic tenofovir or entecavir for the entire duration of immunosuppression plus 12 to 18 months afterwards. Do not wait for reactivation.
- HBsAg-negative, anti-HBc-positive (past infection) on high-risk regimen: prophylactic antiviral (per AASLD for rituximab); monitor closely otherwise.
- If reactivation occurs (rise in DNA over 2 log, or ALT flare): start tenofovir or entecavir immediately; stop or reduce immunosuppression if possible. [1]
Management — Definitive & Stepwise
The definitive therapies for chronic HBV and HCV are entirely different in goal: HBV = lifelong viral suppression (functional cure is rare); HCV = short-course cure (SVR over 95 percent). HAV and HEV are managed supportively. Vaccination is the cornerstone of prevention for HAV and HBV. [1]
Hepatitis A and E — supportive
No specific antiviral alters the course of HAV or HEV in the immunocompetent host. Management is supportive:[8]
- Rest as needed (no evidence that bedrest changes outcome), adequate hydration, antiemetics for nausea, analgesia (avoid paracetamol in full hepatotoxic doses — keep under 2 g/day in acute hepatitis; avoid alcohol).
- Monitor for severity — INR, encephalopathy, bilirubin trend, renal function. Admit if INR over 1.5, encephalopathy, severe vomiting/dehydration, pregnancy with HEV, age over 45, or underlying chronic liver disease (HAV superinfection on chronic liver disease has high mortality).
- Pregnancy with HEV — supportive care, monitor for fulminant failure, deliver if severe; ribavirin contra-indicated (teratogenic).
- Chronic HEV in solid-organ transplant or HIV — ribavirin 600 to 1000 mg orally daily for 12 weeks achieves SVR in most; reduce immunosuppression where possible. [1]
Chronic hepatitis B — long-term viral suppression
The two classes of therapy are oral nucleos(t)ide analogues (NAs) (first-line, the modern standard) and pegylated interferon-alpha (Peg-IFN) (selected patients only).[1][5]
First-line oral nucleos(t)ide analogues (high barrier to resistance, indefinite duration): [1]
- Tenofovir disoproxil fumarate (TDF) 300 mg orally once daily — nucleotide analogue reverse-transcriptase inhibitor; potent, resistance under 1 percent at 5 years; monitor renal function (Fanconi syndrome, eGFR decline) and bone density. Marcellin 2008 (NEJM) demonstrated sustained HBV DNA suppression and histological improvement versus adefovir.[5]
- Tenofovir alafenamide (TAF) 25 mg orally once daily — prodrug with improved renal and bone safety; preferred in renal impairment and osteoporosis.
- Entecavir 0.5 mg orally once daily (1.0 mg daily if lamivudine-resistant) — nucleoside analogue; potent, resistance under 1 percent at 5 years in treatment-naive; dose-reduce in renal impairment.
Pegylated interferon-alpha-2a (Peg-IFN-alpha-2a) 180 microgram subcutaneously once weekly for 48 weeks — for selected young patients with HBeAg-positive chronic HBV, high ALT, low HBV DNA, genotype A or B, who want finite therapy. Achieves HBeAg seroconversion in 30 percent and HBsAg loss in 3 to 7 percent. Contra-indicated in decompensated cirrhosis, pregnancy, autoimmune disease, severe depression, neutropenia/thrombocytopenia. Side effects: flu-like symptoms, depression, bone-marrow suppression, autoimmune thyroiditis.[1][4]
AASLD treatment indications (Terrault 2018):[1]
- HBeAg-positive chronic HBV — ALT over 2 times ULN plus HBV DNA over 20,000 IU/mL (treat if persistent for 3 to 6 months; consider liver biopsy if age over 40 or ALT marginal).
- HBeAg-negative chronic HBV — ALT over 2 times ULN plus HBV DNA over 2000 IU/mL.
- Cirrhosis — treat any detectable HBV DNA (compensated) and treat all decompensated cirrhosis with HBsAg positivity (refer for transplant).
- Always treat: immunosuppression (prophylaxis), prevention of vertical transmission (tenofovir in third trimester if DNA over 200,000 IU/mL), HDV co-infection, HCC, acute liver failure from HBV. [1]
Monitoring on therapy: HBV DNA every 3 to 6 months, ALT, HBeAg/anti-HBe annually (seroconversion may permit stopping in selected HBeAg-positive patients — but cirrhosis = indefinite), creatinine and phosphate (TDF). Adherence is critical — lapses cause viral rebound and resistance (especially with low-genetic-barrier drugs like lamivudine, adefovir, telbivudine, which are now obsolete first-line).[1]
Stopping rules — Asian-Pacific guidance permits finite therapy after HBeAg seroconversion with consolidation for 12 months; AASLD favours indefinite therapy especially for cirrhosis. Never stop in cirrhosis (risk of flare and decompensation).[1]
Chronic hepatitis C — the curative DAA revolution
Modern HCV therapy uses pan-genotypic direct-acting antivirals for 8 to 12 weeks, achieving SVR12 (cure) over 95 percent across genotypes, including in cirrhosis and HIV co-infection. Interferon is obsolete.[2][3][6]
First-line pan-genotypic regimens: [1]
- Sofosbuvir 400 mg plus velpatasvir 100 mg (Epclusa) — one tablet orally once daily for 12 weeks; pan-genotypic, including compensated and decompensated cirrhosis (add ribavirin or extend in decompensated). SVR over 95 percent. Ledipasvir/sofosbuvir (Harvoni) — 12 weeks for genotype 1, 4, 5, 6 (Afdhal 2014, ION-1).[6]
- Glecaprevir 300 mg plus pibrentasvir 120 mg (Mavyret) — three tablets once daily for 8 weeks in treatment-naive, non-cirrhotic, all genotypes; 12 weeks with cirrhosis or prior treatment.
- Sofosbuvir/velpatasvir/voxilaprevir (Vosevi) — salvage regimen for prior DAA failure (12 weeks).
Pre-treatment work-up (essential):[2][3]
- HCV genotype and viral load; FibroScan; renal function; HBV serology (HBsAg, anti-HBc, anti-HBs) — HBV reactivation is a boxed warning (test before DAA; give prophylactic tenofovir/entecavir if HBsAg positive); HIV; pregnancy test (DAAs contra-indicated in pregnancy and with ribavirin, which is teratogenic — 6-month contraception after); drug interaction check (amiodarone contra-indicated with sofosbuvir — bradyarrhythmia; many antiretrovirals, statins, anticonvulsants interact). [1]
After SVR (cure):[2]
- Fibrosis regresses over years; portal hypertension falls.
- HCC surveillance continues if cirrhosis is present at SVR (6-monthly ultrasound plus AFP).
- Re-infection risk remains in people who inject drugs — offer harm reduction. [1]
Hepatitis D — limited therapy
- Pegylated interferon-alpha for 48 weeks — limited efficacy (SVR 25 to 30 percent), considerable side effects.
- Bulevirtide (Hepcludex) — an entry inhibitor (blocks NTCP receptor used by both HBV and HDV); approved in Europe 2020 (EMA); 2 to 10 mg subcutaneously daily for 48 weeks; viral DNA suppression and biochemical response. Not yet FDA-approved.
- Lonafarnib (prenylation inhibitor) with ritonavir boosting, plus Peg-IFN — under study.
- The most effective prevention of HDV is universal HBV vaccination (which prevents HBV and therefore HDV).[1]
Prevention — vaccination is the single most powerful intervention
Hepatitis A vaccine[8]
- Havrix 1440 ELU intramuscularly (adult) at month 0 and 6 to 12 — over 95 percent protective; booster not needed after two doses.
- Indications: travellers to endemic areas, men who have sex with men, IVDU, chronic liver disease (severe HAV on chronic liver disease), haemophilia, occupational exposure, food handlers in outbreaks.
- Post-exposure: HAV vaccine within 2 weeks for contacts aged 1 to 40 years; human normal immunoglobulin 0.02 mL/kg IM for infants under 1, immunocompromised, chronic liver disease, and adults over 40. [1]
- Engerix-B 20 microgram intramuscularly (adults and children over 10) at 0, 1, and 6 months; paediatric dose 10 microgram for under 10.
- Schedule — three doses: 0, 1, 6 months (or 0, 1, 2 with a 12-month booster for rapid protection post-exposure / for haemodialysis).
- Universal birth-dose — within 24 hours of birth (the WHO/AASLD cornerstone of vertical-transmission prevention), followed by doses at 6 weeks, 10 weeks, 14 weeks (or at 1 and 6 months for the 3-dose schedule).
- Combined HAV/HBV (Twinrix) — for those needing both.
- Post-vaccination serology — anti-HBs over 10 mIU/mL = responder; 5 to 10 percent of immunocompetent adults are non-responders (age over 40, obesity, smoking, immunosuppression, renal failure — risk factors). Non-responders: repeat the series (3 further doses); double-dose for haemodialysis.
- No HCV or HEV vaccine is universally available (HEV vaccine licensed in China only). [1]
Post-exposure prophylaxis and prevention of vertical transmission
Vertical HBV transmission — the package:[1]
- Maternal tenofovir 300 mg orally once daily from 28 to 32 weeks of gestation if HBV DNA over 200,000 IU/mL (or HBeAg positive) — halves vertical transmission.
- Birth-dose HBV vaccine within 24 hours (best within 12 hours) for ALL newborns.
- HBIG 0.5 mL intramuscularly within 12 hours of birth for newborns of HBsAg-positive mothers.
- Complete the vaccine series at 1 and 6 months.
- Caesarean section is not routinely indicated solely for HBV; breastfeeding is safe once the baby is immunised.
- Test the infant for HBsAg and anti-HBs at 9 to 12 months. [1]
Harm reduction (people who inject drugs): needle and syringe programmes, opioid substitution therapy (methadone, buprenorphine), vaccination (HAV and HBV), testing and treatment — central to the WHO elimination strategy.[2][3]
Blood safety and universal precautions — screening of all blood products for HBV, HCV, HIV (and HAV/HEV in some settings); single-use needles and syringes; sterile medical and dental equipment; safe tattooing and piercing.[1]
Specific Subtypes & Scenarios
- Acute hepatitis A — incubation 15 to 50 days, faecal-oral; prodrome then jaundice; never chronic; fulminant in 0.1 to 0.5 percent (higher in older adults and underlying liver disease). Treatment supportive. Vaccine preventable.[8]
- Acute hepatitis B — incubation 30 to 180 days; 95 percent of immunocompetent adults resolve spontaneously (with anti-HBs immunity); 1 percent fulminant; 90 percent of neonates and 25 to 50 percent of children under 5 become chronic. Treatment of severe acute HBV is supportive; nucleos(t)ide analogues for fulminant or protracted/severe acute HBV.[1][4]
- Chronic hepatitis B — managed as above; the four immunological phases guide treatment.
- Hepatitis C, acute — 75 to 85 percent become chronic; curative DAA therapy is recommended for acute HCV (treat from 8 to 12 weeks after onset, no longer wait for spontaneous clearance).[2]
- Hepatitis C, chronic — DAA cure in 8 to 12 weeks; HCC surveillance if cirrhotic.
- Hepatitis D — coinfection versus superinfection — a recurring exam distinction:
- Coinfection (acute HBV and HDV together) — more severe acute hepatitis; lower chronicity (2 to 5 percent); fulminant rate higher than HBV alone.
- Superinfection (HDV on chronic HBV) — 80 to 90 percent become chronic; accelerated fibrosis (cirrhosis in 15 years versus 25 to 30 for HBV alone); high HCC risk. Treat per HDV guidance above.[1]
- Hepatitis E in pregnancy — fulminant in 20 to 25 percent; supportive; urgent delivery if severe; ribavirin contra-indicated.[8]
- Chronic HEV in immunosuppressed (solid-organ transplant, HIV) — ribavirin 600 to 1000 mg daily for 12 weeks.
- Acute-on-chronic liver failure from HBV reactivation — identify the precipitant (immunosuppression withdrawal or start, superinfection, alcohol), start tenofovir/entecavir immediately, transplant evaluation.[8][10]
Complications & Pitfalls
Complications of chronic viral hepatitis
- Cirrhosis — over 8 to 20 percent of untreated chronic HBV at 5 years; over 15 to 30 percent of untreated chronic HCV at 20 years. Once cirrhotic, the patient is at risk of variceal bleed, ascites, SBP, encephalopathy, hepatorenal syndrome.[10]
- Hepatocellular carcinoma — annual risk 2 to 5 percent in HBV cirrhosis; HBV causes HCC even without cirrhosis (direct oncogenesis). HCV causes HCC mainly through cirrhosis.[1]
- Acute liver failure — fulminant HBV (1 percent), HDV coinfection/superinfection, HEV in pregnancy, HAV in older adults/chronic liver disease. HCV almost never.[8][9]
- Extrahepatic syndromes — HCV cryoglobulinaemia, MPGN, porphyria cutanea tarda, NHL, lichen planus, diabetes; HBV PAN, membranous GN, Gianotti-Crosti.
- Aplastic anaemia (rare, after HAV in young men).
Classic pitfalls — the high-yield errors
- Missing HBV reactivation on rituximab/chemotherapy/high-dose steroids — screen every patient (HBsAg, anti-HBc, anti-HBs) before immunosuppression; give prophylactic tenofovir or entecavir for high-risk regimens. Can be fatal; entirely preventable.[1]
- Not screening for HDV in HBsAg-positive patients — HDV is underdiagnosed; test total anti-HDV in every HBsAg-positive patient, especially if from endemic regions (Amazon, Central Africa, Mongolia, Eastern Europe).[1]
- Assuming a positive anti-HCV antibody means active infection — confirm with HCV RNA. A positive antibody with negative RNA = resolved infection.[2]
- Not checking HBV status before DAA therapy for HCV — HBV reactivation is a boxed warning; can cause fulminant hepatitis.[2][3]
- Confusing IgM anti-HBc with HBsAg in acute hepatitis — IgM anti-HBc is positive in the window period when HBsAg has waned; it is the most sensitive marker of acute HBV.
- Stopping nucleos(t)ide therapy in HBV cirrhosis — risk of fatal flare and decompensation; therapy is indefinite in cirrhosis.
- Assuming "hepatitis B vaccine failure" without checking anti-HBs response — 5 to 10 percent non-responders need a repeat series or double dose.
- Forgetting that HBV causes HCC without cirrhosis — HCC surveillance applies to high-risk chronic HBV patients even without cirrhosis (Asian men over 40, Asian women over 50, Africans over 20, family history of HCC, HDV, cirrhosis of any cause).[1]
- Prophylactically correcting the INR with fresh frozen plasma in acute liver failure — erases the prognostic and transplant marker; correct only if bleeding or before procedures.[8][9]
- Overlooking HEV in a pregnant woman with acute hepatitis — high fulminant rate; test IgM anti-HEV urgently.[8]
Factors accelerating fibrosis progression
In chronic HBV or HCV, progression to cirrhosis is accelerated by: alcohol, obesity and diabetes (MASLD overlap), HIV co-infection, male sex, older age at infection, HBV/HCV co-infection, iron overload, schistosomal co-infection.[2][10]
Prognosis & Disposition
- Acute hepatitis A — full recovery in over 99 percent; fulminant in 0.1 to 0.5 percent (higher in older adults and underlying chronic liver disease); prolonged cholestasis in a few. Immune for life.[8]
- Acute hepatitis B — over 95 percent resolution in immunocompetent adults; 1 percent fulminant; chronicity 90 percent perinatal, 25 to 50 percent under 5, under 5 percent adults. Recovery confers anti-HBs immunity for life.[1][4]
- Chronic hepatitis B — untreated: 8 to 20 percent develop cirrhosis over 5 years; annual HCC risk 2 to 5 percent in cirrhotics. Treated (TDF/ETV): viral suppression halts fibrosis, reduces HCC risk by approximately 50 percent, but does not eliminate it — surveillance continues.
- Chronic hepatitis C — untreated: 15 to 30 percent develop cirrhosis over 20 years; of cirrhotics, 1 to 4 percent per year develop HCC. Treated with DAAs: SVR (cure) over 95 percent; fibrosis regresses; HCC risk falls but persists if cirrhosis was present — continue surveillance.[2][3]
- Hepatitis D superinfection — 70 to 80 percent chronic; accelerated cirrhosis (15 years versus 25 to 30 for HBV alone).
- Hepatitis E — self-limiting in immunocompetent; maternal mortality up to 20 to 25 percent in pregnancy; chronic in transplant/HIV.
- Admit if INR over 1.5, encephalopathy, severe vomiting/dehydration, age over 45, comorbidity, pregnancy with HEV. Discharge when asymptomatic, INR normal, ALT falling, and a safe social situation with safety-net advice.
Special Populations
- Pregnancy — screen all pregnancies for HBsAg (universal antenatal). HBV: give maternal tenofovir 300 mg daily from 28 to 32 weeks if HBV DNA over 200,000 IU/mL; birth-dose vaccine plus HBIG within 12 hours; complete vaccine series; caesarean not routinely indicated; breastfeeding safe once baby immunised. HCV: screen all pregnant women (CDC/AASLD 2020 universal screening); DAAs postpartum (avoid in pregnancy and breastfeeding; ribavirin is teratogenic — 6-month contraception after); vertical transmission approximately 5 percent (no prophylaxis available). HEV: high fulminant risk — supportive, urgent delivery if severe.[1][2][3]
- Neonates and children — perinatal HBV transmission is the commonest global route; 90 percent become chronic without immunoprophylaxis — prevented by the universal birth-dose vaccine plus HBIG; paediatric HBV vaccine dose is half the adult; HCV vertical transmission 5 percent (no prophylaxis; treat later, DAAs approved from age 3).
- Immunosuppressed (HIV, transplant, chemotherapy) — high risk of HBV reactivation: screen all and give prophylactic tenofovir/entecavir for high-risk regimens (rituximab, ofatumumab, high-dose steroids over 4 weeks, anthracyclines, HSCT). HCV-HIV co-infection accelerates fibrosis — treat both, with careful DAA-antiretroviral interaction checks.[1]
- End-stage renal disease / dialysis — prefer entecavir (dose-adjusted to eGFR) or TAF over TDF (nephrotoxicity); double the HBV vaccine dose and schedule extra doses; screen regularly for HBV and HCV (outbreak risk in dialysis units).[1]
- Healthcare workers — HBV vaccination mandatory; check anti-HBs response (booster if under 10); HBsAg-positive surgeons may be restricted from exposure-prone procedures depending on national policy (UK: restricted if HBeAg-positive and DNA over 200 IU/mL).
- Elderly — atypical, cholestatic, more severe HAV (fulminant rate rises with age); lower vaccine response; multiple comorbidities complicate DAAs (drug interactions); polypharmacy risk.
- Decompensated cirrhosis — refer for transplant evaluation; HBV: continue tenofovir or entecavir (avoid Peg-IFN — contra-indicated); HCV: sofosbuvir-based regimens with ribavirin (cautious, specialist); HCC surveillance mandatory.[8][10]
Evidence, Guidelines & Regional Differences
AASLD 2018 HBV guidance (Terrault 2018)[1] — treat HBeAg-positive and HBeAg-negative active chronic hepatitis, cirrhosis with detectable DNA, immunosuppression, pregnancy with high DNA; first-line tenofovir/entecavir/TAF; Peg-IFN for selected young patients with high ALT and low DNA; HCC surveillance in chronic HBV with risk factors regardless of cirrhosis.
Lok and McMahon 2007 (AASLD) Hepatology[4] — the foundational AASLD HBV guideline that codified the four immunological phases and the treatment indications; superseded by 2018 update but still the standard reference for serology interpretation.
AASLD-IDSA HCV Guidance 2018 and 2023 Update[2][3] — universal one-time HCV screening of all adults aged 18 years and over plus all pregnant women; pan-genotypic sofosbuvir/velpasvir (Epclusa) or glecaprevir/pibrentasvir (Mavyret) first-line; SVR over 95 percent across genotypes; simplified regimens; check HBV before starting DAAs.
- Marcellin 2008 (NEJM) — tenofovir disoproxil fumarate versus adefovir for chronic HBV: superior HBV DNA suppression and histological improvement at 48 weeks, establishing tenofovir as first-line.
- Afdhal 2014 ION-1 (NEJM) — ledipasvir/sofosbuvir for untreated HCV genotype 1: SVR over 95 percent at 12 weeks, the trial that began the DAA era for genotype 1. [1]
Global epidemiology[7] — Polaris Observatory 2017 (Lancet Gastroenterol Hepatol): HCV global prevalence 1.0 percent in 2015 (approximately 71 million); genotype 1 commonest (46 percent), genotype 3 second (22 percent); informed WHO elimination targets.
Baveno VII (2022)[10] — renewed consensus on portal hypertension; defines cirrhosis by liver stiffness over 12 to 15 kPa (or over 25 kPa with platelets under 150 with varices), the criteria that determine HCC and variceal surveillance in chronic viral hepatitis with cirrhosis.
- India (NACO/INASL) — HBV commonest chronic viral hepatitis; high HAV and HEV burden (HEV is the commonest cause of acute viral hepatitis in Indian adults); universal HBV birth-dose vaccine now part of the national immunisation schedule; cost still limits DAA and transplant access; the National Viral Hepatitis Control Programme (NVHCP) launched 2018 to deliver free HBV/HCV diagnosis and treatment.
- USA (AASLD/CDC) — HCV commonest chronic (baby-boomer cohort); universal one-time adult and pregnancy screening; pan-genotypic DAAs first-line.
- Europe (EASL) — similar to AASLD; bulevirtide approved for HDV (EMA 2020); harm reduction central in PWID.
- WHO 2030 elimination targets — 90 percent of HBV/HCV diagnosed, 80 percent of eligible treated, 65 percent reduction in mortality; 90 percent HBV birth-dose coverage and three-dose infant coverage; needle-syringe programmes and opioid substitution therapy for people who inject drugs. [1]
Controversies:[1]
- Stopping HBV nucleos(t)ide analogues — Asian-Pacific guidance permits finite therapy after HBeAg seroconversion with consolidation; AASLD favours indefinite therapy especially for cirrhosis.
- HDV prevalence is underestimated — many HBsAg-positive patients are never tested for anti-HDV.
- HCC surveillance after HCV SVR in non-cirrhotics — not recommended (insufficient yield) but controversial in high-risk ethnic groups.
- DAA access in low-income settings — generic manufacturing has lowered cost dramatically but barriers remain. [1]
Exam Pearls
The five hepatotropic viruses — chronicity, transmission, vaccine
ABCDE
Incubation 2-6 weeks; never chronic; vaccine; fulminant rare in young
Chronic in 90% neonates, under 5% adults; vaccine-preventable; tenofovir/entecavir; causes HCC without cirrhosis
Chronic in 75%; no vaccine; curable with DAAs in 8-12 weeks (>95% SVR); HCC via cirrhosis only
Requires HBsAg; coinfection (severe acute, low chronicity) vs superinfection (80-90% chronic, accelerated cirrhosis); prevent by HBV vaccine
Commonest acute viral hepatitis in Indian adults; 20-25% maternal mortality in pregnancy; ribavirin for chronic in transplant
HBV serology — the markers that decide every MCQ
MASTERS
Positive in the window period (HBsAg gone, anti-HBs not yet); most sensitive marker of acute HBV
First to appear (1-10 weeks); persists beyond 6 months = chronic
Protective; over 10 mIU/mL = responder to vaccine; also appears after recovery
Past OR current infection (never from vaccine); persists for life
Marker of active replication and high viral load; seroconversion to anti-HBe marks inactive carrier
Over 20,000 IU/mL in HBeAg-positive active; over 2000 IU/mL in HBeAg-negative active; guides treatment
Loss of HBeAg and gain of anti-HBe; rarely achievable for HBsAg (functional cure 1-3% per year)
Exam application bank (NEET-PG / INICET)
One-line answer
Viral hepatitis is inflammation of the liver caused by the five hepatotropic viruses — A (RNA, faecal-oral, never chronic), B (DNA, parenteral/vertical/sexual, chronic in 90 percent neonates), C (RNA, blood-borne, chronic in 75 percent, curable with DAAs), D (defective RNA requiring HBsAg), E (RNA, faecal-oral, fulminant in pregnancy). Acute infection presents with prodrome (anorexia, nausea, fatigue, arthralgia) then jaundice, dark urine, tender hepatomegaly; chronic infection is often asymptomatic until cirrhosis or HCC. Diagnosis by serology: HBsAg/anti-HBc (HBV), anti-HCV/HCV RNA (HCV), IgM anti-HAV/anti-HEV. Management: supportive for HAV/HEV; long-term nucleos(t)ide analogues (tenofovir, entecavir) for HBV; direct-acting antivirals (DAAs) 8-12 weeks with cure over 95 percent for HCV. Prevention: HAV and HBV vaccines, birth-dose HBV vaccine plus HBIG, harm reduction, blood screening
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard. [1]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes. [1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change. [1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each. [1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory. [1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Viral Hepatitis (Hepatitis A, B, C, D, E).
References
- [1]Terrault NA, Lok ASF, McMahon BJ, et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance Hepatology, 2018.PMID 29405329
- [2]AASLD-IDSA HCV Guidance Panel. Hepatitis C Guidance 2018 Update: AASLD-IDSA Recommendations for Testing, Managing, and Treating Hepatitis C Virus Infection Clin Infect Dis, 2018.PMID 30215672
- [3]Ghany MG, Marks KM, Morgan TR, et al. Hepatitis C Guidance 2023 Update: AASLD-IDSA Recommendations for Testing, Managing, and Treating Hepatitis C Virus Infection Clin Infect Dis, 2023.PMID 37229695
- [4]Lok ASF, McMahon BJ. Chronic hepatitis B Hepatology, 2007.PMID 17256718
- [5]Marcellin P, Heathcote EJ, Buti M, et al. Tenofovir disoproxil fumarate versus adefovir dipivoxil for chronic hepatitis B N Engl J Med, 2008.PMID 19052126
- [6]Afdhal N, Zeuzem S, Kwo P, et al. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection N Engl J Med, 2014.PMID 24725239
- [7]Polaris Observatory HCV Collaborators. Global prevalence and genotype distribution of hepatitis C virus infection in 2015: a modelling study Lancet Gastroenterol Hepatol, 2017.PMID 28404132
- [8]Stravitz RT, Lee WM. Acute liver failure Lancet, 2019.PMID 31498101
- [9]Bernal W, Wendon J. Acute liver failure N Engl J Med, 2013.PMID 24369077
- [10]de Franchis R, Bosch J, Garcia-Tsao G, et al. Baveno VII - Renewing consensus in portal hypertension J Hepatol, 2022.PMID 35120736