General Surgery · General Surgery
Pancreatic Carcinoma
Also known as Pancreatic cancer · Pancreatic adenocarcinoma · Carcinoma head of pancreas · Periampullary carcinoma
Pancreatic carcinoma (ductal adenocarcinoma) is an aggressive malignancy with a 5-year survival under 10% — the worst of any solid organ cancer. Risk factors: smoking, chronic pancreatitis, diabetes, obesity, family history, BRCA2. Head of pancreas (70%): presents with painless obstructive jaundice, weight loss, Courvoisier's sign (palpable non-tender gallbladder). Body/tail: presents late with pain radiating to the back. Diagnosis: CT pancreas protocol + CA 19-9 + EUS-FNA biopsy. Only 15 to 20% are resectable at presentation (Whipple / pancreatoduodenectomy for head tumours). FOLFIRINOX or gemcitabine/nab-paclitaxel for metastatic. Jaundice relief by ERCP stenting. Almost uniformly fatal.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Exam tags
Red flags
Overview & Definition[1]
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive epithelial malignancy arising from the exocrine pancreatic ducts (the small intercalated and intralobular ducts that line the glandular parenchyma). It is the fourth leading cause of cancer death in the United States and most developed countries (after lung, colorectal, and breast/prostate) and has the worst 5-year survival of any major malignancy — approximately 10% overall, a figure that has barely budged in four decades despite extraordinary efforts in surgery, chemotherapy, radiation, targeted therapy, and immunotherapy.[1][2][5]
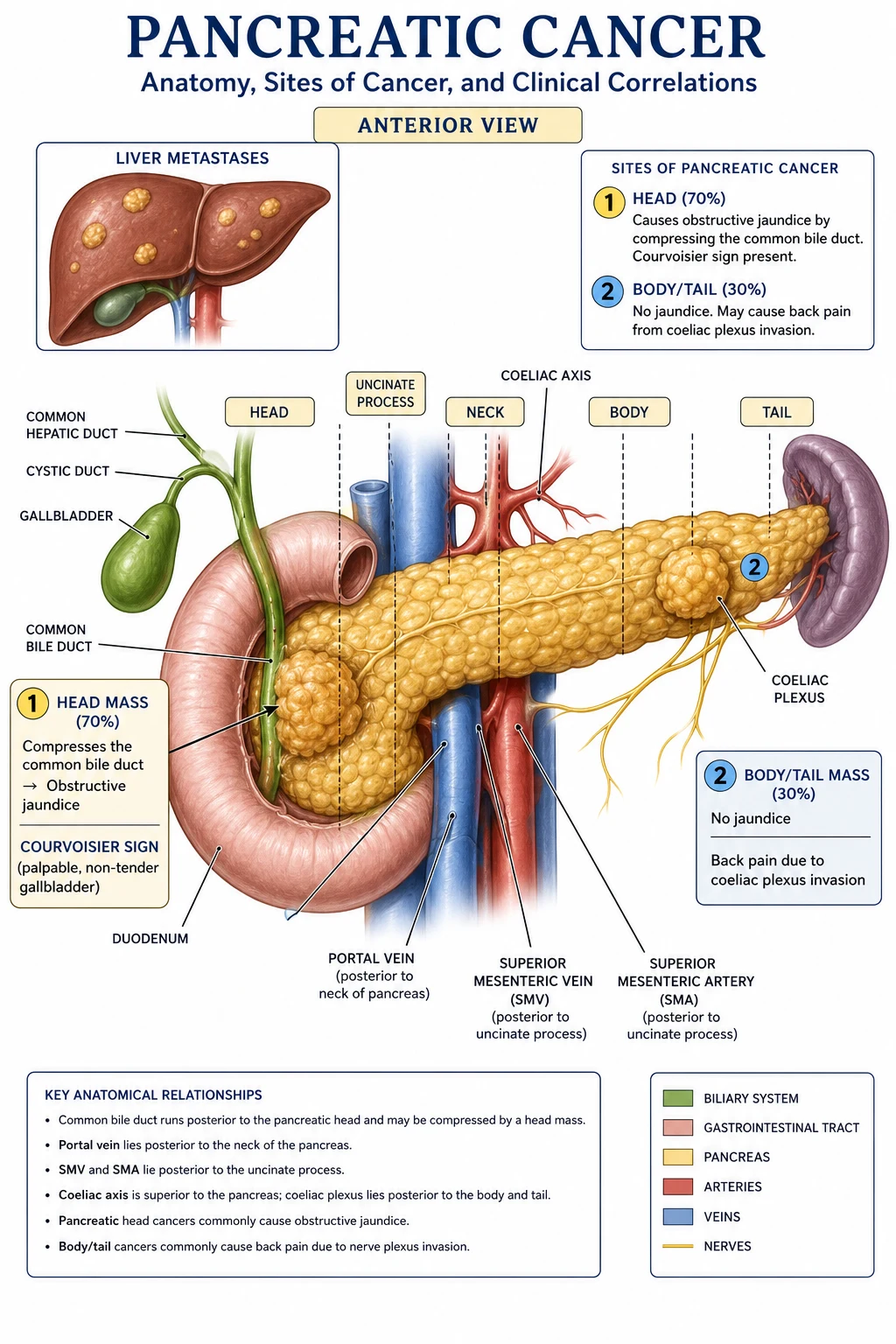
Approximately 70% arise in the head of the pancreas, 20% in the body, and 10% in the tail. Head tumours present earlier because they obstruct the intrapancreatic common bile duct and produce painless obstructive jaundice. Body and tail tumours are clinically silent until they invade the retroperitoneal coeliac/superior mesenteric plexus (causing back pain) or metastasise to liver or peritoneum, at which point they are almost never resectable.[2][5]
The clinical challenge is early detection. There is no effective screening test for the general population; symptoms are non-specific until biliary obstruction or back pain develop; the pancreas is retroperitoneal and hidden from palpation; and tumour biology is uniquely aggressive, with rapid local invasion, perineural spread, early lymphatic and haematogenous dissemination, and a dense desmoplastic stroma that excludes chemotherapy and immune cells. The result is that over 80% of patients present at an advanced, unresectable stage, and even those who reach the operating table often recur within 24 months.[1][6]
PDAC must be distinguished from a wider family of pancreatic neoplasms. The pancreatic ductal adenocarcinoma is the dominant lesion (over 90% of all pancreatic malignancies), but the same anatomical region gives rise to ampullary carcinoma, cholangiocarcinoma of the distal bile duct, pancreatic neuroendocrine tumours (pNETs), cystic neoplasms (IPMN, MCN, serous cystadenoma, solid pseudopapillary), acinar cell carcinoma, and the rare pancreatoblastoma. Each has its own biology, staging system, and treatment pathway; the focus of this chapter is pancreatic ductal adenocarcinoma, with periampullary and pNET touched on where they are the commonest source of confusion at the bedside and in the exam hall.[2][5]
Classification[1]
By anatomical site (determines presentation and operability)[1]
The anatomical location of a PDAC within the gland dictates both how it presents and whether it is resectable:[2][5]
- Head of pancreas (60 to 70%) — the commonest location. The head sits within the C-loop of the duodenum and contains the intrapancreatic distal common bile duct and the main pancreatic duct (duct of Wirsung) as it joins the ampulla of Vater. A tumour in this region compresses the distal CBD early, producing painless obstructive jaundice (the textbook presentation), and may also obstruct the duodenum (gastric outlet obstruction late in the disease). Head tumours present earlier than body/tail tumours and have a higher resection rate (around 20 to 25%) because symptoms develop before distant spread.[2]
- Uncinate process — the inferior/medial hook of the pancreatic head. Uncinate tumours characteristically invade the superior mesenteric vein (SMV)/portal vein (PV) axis and the superior mesenteric artery (SMA), and they often present with duodenal obstruction rather than jaundice (because the uncinate lies inferior to the CBD). Uncinate tumours have a higher R1 margin rate and a worse prognosis than other head tumours.[5]
- Body (15 to 20%) — sits anterior to the SMA, coeliac axis, and aorta. Body tumours are clinically silent until they invade the coeliac plexus and the superior mesenteric plexus, producing the characteristic epigastric pain radiating through to the back, worse at night, partially relieved by leaning forward. By the time this pain develops, vascular encasement is usually present and resection is rarely possible.[2]
- Tail (5 to 10%) — the most lateral portion, abutting the splenic hilum. Tail tumours present even later than body tumours, often with left upper quadrant pain, splenomegaly, splenic-vein thrombosis with isolated gastric varices (sinistral/left-sided portal hypertension), and early haematogenous metastases to the liver and peritoneum. Almost never resectable at diagnosis.[5]
- Diffuse/multifocal (5 to 10%) — extensive glandular involvement at diagnosis. Behaviour is that of an advanced, unresectable lesion.[2]
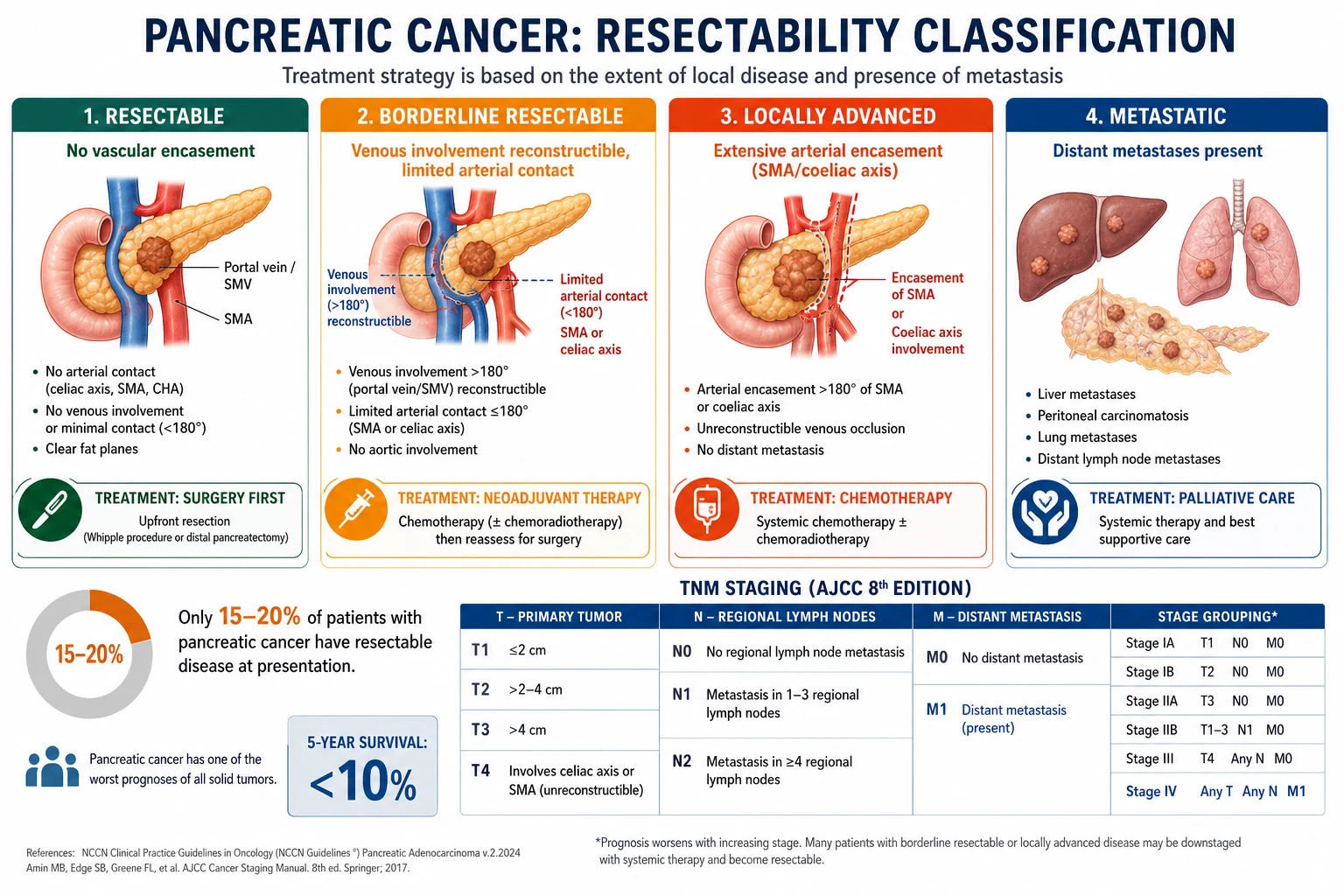
By resectability (the most important clinical classification — drives treatment)[1]
This four-tier NCCN/AHPBA/SSO consensus classification is the cornerstone of treatment selection and is now the standard for MDT discussion, trial entry, and exam answers:[1][5][4]
| Category | Vascular criteria | Treatment | Approx share |
|---|---|---|---|
| Resectable | No tumour contact with SMA, coeliac axis, or common hepatic artery. ≤180° contact with SMV/PV without vein deformity. | Upfront surgery (Whipple/distal/total pancreatectomy) followed by adjuvant mFOLFIRINOX. | 15 to 20% |
| Borderline resectable | ≤180° contact with SMA; ≤180° contact with coeliac axis (body/tail); >180° contact or reconstructible occlusion of SMV/PV. | Neoadjuvant chemotherapy (FOLFIRINOX or gemcitabine/nab-paclitaxel) for 4 to 6 months, restage, then surgery if response. | ~10% |
| Locally advanced (unresectable) | >180° encasement of SMA or coeliac axis. Non-reconstructible SMV/PV occlusion. No distant metastases. | Induction chemotherapy (FOLFIRINOX or gem/nab) ± chemoradiotherapy. Median OS 15 to 20 months. | ~30% |
| Metastatic | Any. Plus distant metastases (liver, peritoneum, lung, distant nodes). | Systemic chemotherapy ± targeted therapy; palliative care. Median OS 8 to 11 months (treated), 3 to 6 months (untreated). | ~50% |
Periampullary carcinoma[1]
Periampullary carcinoma is an umbrella term for tumours arising from the ampulla of Vater, the intrapancreatic distal common bile duct, or the second part of the duodenum (not the pancreas itself). They are grouped with pancreatic head cancer because they present in the same way — progressive painless jaundice — and are treated with the same operation (pancreatoduodenectomy). However, periampullary tumours have a substantially better prognosis than PDAC (5-year survival 40 to 50% after Whipple vs 20 to 25% for PDAC), partly because they are biologically less aggressive and partly because they declare themselves with jaundice while still small. The differential within the periampullary group is:[2][5]
- Ampullary adenocarcinoma (true ampullary, intestinal type) — best prognosis; 5-year survival 50 to 70% after Whipple.
- Distal cholangiocarcinoma (intrapancreatic CBD) — intermediate prognosis; 5-year survival 30 to 40%.
- Duodenal adenocarcinoma — 5-year survival 40 to 50%.
- PDAC of the head — worst; 5-year survival 20 to 25% (with modern adjuvant FOLFIRINOX).[2]
Histological variants of pancreatic cancer[1]
Although "pancreatic cancer" is shorthand for PDAC, the differential is worth knowing:[2][5]
- Ductal adenocarcinoma (PDAC) — over 90% of pancreatic malignancies. Driven by KRAS (over 90%), TP53 (50 to 75%), CDKN2A/p16 (over 90%), SMAD4/DPC4 (about 55%). Arises from pancreatic intraepithelial neoplasia (PanIN) precursor lesions.
- Pancreatic neuroendocrine tumour (pNET) — 3 to 5% of pancreatic neoplasms. Subdivided into functional (insulinoma, gastrinoma/Zollinger-Ellison, glucagonoma, VIPoma, somatostatinoma) and non-functional. Slower growing, hypervascular on CT (a useful discriminator), better prognosis (5-year survival 50 to 60% for localised disease). Associated with MEN1 and von Hippel-Lindau syndromes.
- Acinar cell carcinoma — rare; presents with the Schmid triad of subcutaneous fat necrosis, polyarthralgia, and eosinophilia from lipase hypersecretion.
- Mucinous cystic neoplasm (MCN) — almost exclusively in women in their 40s, body/tail, ovarian-type stroma, premalignant.
- Intraductal papillary mucinous neoplasm (IPMN) — mucin-producing cystic tumour of the pancreatic duct. Main-duct IPMN has a 40 to 70% lifetime risk of malignancy (resect); branch-duct IPMN is lower risk (observe unless cyst >3 cm, mural nodule, or main pancreatic duct dilated >5 mm — the Fukuoka/Kyoto criteria).
- Solid pseudopapillary neoplasm — young women; low-grade malignant; excellent prognosis after resection.
- Serous cystadenoma — benign; "honeycomb" microcystic appearance; observe unless symptomatic.
- Pancreatoblastoma — paediatric tumour; responds to chemotherapy.
- Secondary tumours — renal cell carcinoma, breast, lung, melanoma; rare; usually multifocal.[1][5]
Epidemiology & Risk Factors[1]
Burden of disease[1]
Pancreatic cancer sits in the unenviable position of being the twelfth most common cancer globally but the fourth leading cause of cancer death (after lung, colorectal, and breast/prostate in most registries) — incidence and mortality are almost equal because the disease is so lethal.[1][2][13] In 2020, the GLOBOCAN estimate was 495,773 new cases and 466,003 deaths worldwide. In the United States, 64,050 new cases and 50,550 deaths were projected for 2023, making PDAC the 4th cause of cancer death. By 2030, PDAC is projected to become the 2nd leading cause of cancer death in the United States (surpassing colorectal and breast/prostate) on current incidence trends. The median age at diagnosis is 70 years; the male:female ratio is approximately 1.3:1; and incidence is highest in populations of European ancestry and lowest in those of African or Asian ancestry within multi-ethnic countries.[13][1]
Non-modifiable risk factors[1]
Pancreatic cancer — the worst prognosis in oncology
- Older age — peak incidence in the 7th to 8th decade; rare under 40; over 90% of cases occur in those over 55.
- Male sex — modest 1.3:1 male predominance.
- Non-O blood group — modestly increased risk (A, B, AB), possibly because of cross-reactivity with bacterial surface antigens.
- Family history — present in 5 to 10% of patients. A single first-degree relative confers a 2-fold increased risk; two or more first-degree relatives confer a 6 to 10-fold increased risk ("familial pancreatic cancer", defined by the consensus criteria as ≥2 first-degree relatives with PDAC, none of whom carry an identifiable germline mutation).[3][14]
- Inherited cancer syndromes — a major focus of the modern high-risk clinic.[3][14][2]
- BRCA2 mutation (and to a lesser extent BRCA1) — confers a 3 to 10-fold lifetime risk of pancreatic cancer (cumulative risk 5 to 10%). Same gene implicated in hereditary breast and ovarian cancer. PARP inhibitors (olaparib) are the matched targeted therapy in metastatic disease.
- PALB2 — partner and localiser of BRCA2; lifetime pancreatic cancer risk ~5%.
- ATM — heterozygous ATM carriers have a 2 to 3-fold increased risk.
- Peutz-Jeghers syndrome (STK11/LKB1) — mucocutaneous pigmentation, hamartomatous GI polyps, and a cumulative lifetime pancreatic cancer risk of 11 to 36% (the highest of any hereditary syndrome for PDAC). Surveillance recommended from age 30 to 35.
- Lynch syndrome (mismatch-repair mutations MLH1, MSH2, MSH6, PMS2) — modest 4 to 8-fold increased pancreatic cancer risk; MSI-H/dMMR tumours respond to pembrolizumab.
- Familial atypical multiple mole melanoma (FAMMM) syndrome, CDKN2A mutation — lifetime pancreatic cancer risk 15 to 20% in carriers.
- Hereditary pancreatitis (PRSS1, SPINK1 mutations) — chronic pancreatitis from childhood, with a cumulative lifetime pancreatic cancer risk of 40 to 55%, predominantly in the second half of life. The combination of chronic inflammation and a germline cancer-predisposition mutation is uniquely carcinogenic.
- Von Hippel-Lindau — serous cystadenomas, pNETs; modest PDAC risk.
- MEN1 — pNETs (gastrinoma, insulinoma), not classical PDAC.
- Li-Fraumeni (TP53) — modestly increased PDAC risk; many other cancers.
- Cystic fibrosis — modestly increased PDAC risk.
Modifiable risk factors[1]
- Smoking — the single most important modifiable risk factor. Current smokers have a 2- to 3-fold increased risk of pancreatic cancer, attributable to 20 to 25% of all PDAC cases in population terms. Risk rises with pack-years, and persists for 10 to 20 years after cessation (a long latency). The carcinogens (NNK, PAH) reach the pancreas via refluxed bile and via the bloodstream, where they induce KRAS mutations in the ductal epithelium. The 2020 IARC monograph confirms the causal link.[3][13]
- Chronic pancreatitis — a sustained inflammatory state drives PanIN progression. The relative risk is 5- to 15-fold in cohort studies, and absolute risk is highest in hereditary (PRSS1) pancreatitis and tropical (nutritional) pancreatitis, where 40 to 55% of patients develop PDAC by age 70.[2][3]
- Diabetes mellitus — a bidirectional relationship with PDAC. Long-standing type 2 diabetes confers a 1.5- to 2-fold increased risk (likely through hyperinsulinaemia and chronic low-grade inflammation). Conversely, new-onset diabetes in an older, thin patient with weight loss is a paraneoplastic marker of pancreatic cancer: up to 1% of patients over 50 with new diabetes will be diagnosed with PDAC within 3 years. The mechanism is tumour-derived factors (adrenomedullin, islet amyloid polypeptide) that cause insulin resistance and beta-cell dysfunction before the tumour is radiologically detectable.[1][3]
- Obesity — body-mass index (BMI) ≥30 kg/m² confers a 20 to 30% increased risk, mediated through insulin resistance, adipokine imbalance, and chronic inflammation. Central obesity is the more specific risk factor.
- Alcohol — heavy alcohol intake (>30 to 40 g/day) increases PDAC risk primarily through the intermediate step of chronic pancreatitis. Pure alcohol in the absence of chronic pancreatitis is a weak independent risk factor.
- Diet — high intake of red and processed meat, sugar-sweetened beverages, and saturated fat modestly increase risk; diets rich in fruits, vegetables, and the Mediterranean pattern are associated with lower risk. Coffee and tea are not independent risk factors.
- Occupational exposures — chlorinated hydrocarbons, pesticides, certain metalworking fluids, and radiation (atomic-bomb survivors) are recognised but small contributors.
- Chronic H. pylori infection, hepatitis B, and periodontal disease have all been linked in observational data; the effect sizes are modest and the mechanisms uncertain.
Non-modifiable
fixed at birth
- Age 70+ (peak)
- Male sex (1.3:1)
- Non-O blood group
- **Germline mutation** — BRCA2, PALB2, ATM, STK11 (Peutz-Jeghers), CDKN2A (FAMMM), PRSS1 (hereditary pancreatitis), mismatch-repair (Lynch)
- Family history — 1 FDR = 2×; ≥2 FDR = 6 to 10×
Modifiable
target for prevention
- **Smoking** — 2 to 3× risk; 20 to 25% of cases
- **Chronic pancreatitis** — 5 to 15× (40 to 55× in hereditary)
- **Obesity** — BMI ≥30 = 1.2 to 1.3×
- Long-standing **type 2 diabetes** — 1.5 to 2×
- Heavy **alcohol**, red/processed meat, occupational exposures
- New-onset diabetes in older thin patient = paraneoplastic
Pathophysiology[1]
The PanIN-to-carcinoma sequence[1]
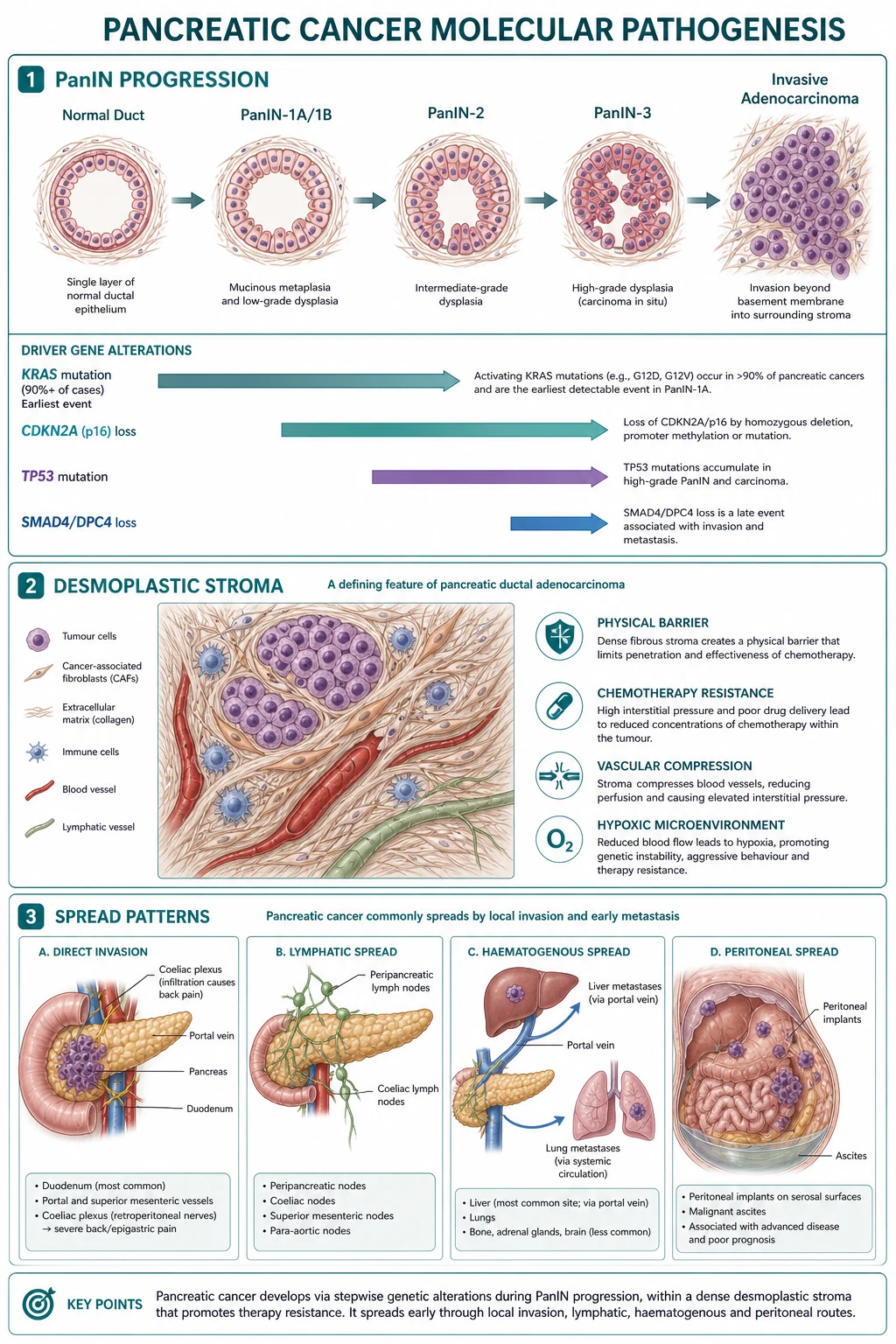
PDAC develops from a stepwise accumulation of genetic and epigenetic changes in the ductal epithelium. The dominant precursor lesion is pancreatic intraepithelial neoplasia (PanIN) — a microscopic, non-invasive lesion graded PanIN-1A, PanIN-1B, PanIN-2, and PanIN-3 (carcinoma in situ). PanIN-1A lesions are flat epithelial lesions with mild atypia; PanIN-1B are papillary without atypia; PanIN-2 show flat or papillary architecture with moderate atypia; PanIN-3 show marked atypia and cribriform architecture, with an intact basement membrane. Most PanIN lesions are clinically silent and are detected incidentally in resected specimens or at autopsy. The progression from low-grade PanIN to invasive PDAC takes 10 to 20 years on average — a window of opportunity for early detection that is, as yet, unrealised at the population level.[1][5][6]
The genetic cascade parallels the histological progression:[5][2][1]
- KRAS mutation — >90% of PDACs carry an activating point mutation in KRAS (most commonly G12D, then G12V, G12C, G12R, Q61H). KRAS mutation is the earliest and most common event, present even in low-grade PanIN-1. The mutation locks KRAS in its GTP-bound active state, driving constitutive RAS/MAPK and PI3K/AKT signalling and uncontrolled proliferation. KRAS G12C is rare in PDAC (about 1%) and is targetable by sotorasib/adagrasib; the more common G12D is the focus of MRTX1133 and other next-generation inhibitors in trials.
- CDKN2A (p16) inactivation — occurs in over 90% of PDACs by mutation, deletion, or promoter methylation. CDKN2A encodes the p16 cell-cycle checkpoint, and its loss removes a critical brake on the G1/S transition.
- TP53 mutation — present in 50 to 75% of PDACs, predominantly in advanced PanIN-3 and invasive disease. Loss of p53 removes the DNA-damage response, allowing accumulation of further mutations and genomic instability.
- SMAD4 (DPC4) loss — present in about 55% of PDACs, predominantly in metastatic disease. SMAD4 is a downstream effector of TGF-beta signalling; its loss abolishes TGF-beta-mediated growth inhibition and is strongly associated with widely metastatic phenotype and poor prognosis.[5]
The roughly 5 to 10% of PDACs that are KRAS-wild-type are enriched for actionable alterations — BRCA1/2, PALB2, ATM, mismatch-repair deficiency (MSI-H), NRG1, ALK, NTRK, BRAF — and are the primary beneficiaries of next-generation sequencing and matched targeted therapy.[5]
The PanIN-to-PDAC genetic cascade
The desmoplastic stroma[1]
A defining histological feature of PDAC is the dense desmoplastic stroma that surrounds the malignant ducts. The stroma is produced by pancreatic stellate cells activated by tumour-derived TGF-beta, PDGF, and sonic hedgehog; it contains dense collagen, fibronectin, and hyaluronic acid, and is populated by myofibroblasts, suppressive immune cells (M2 macrophages, myeloid-derived suppressor cells, regulatory T cells), and a sparse microvasculature.[5][1]
The stroma has three major consequences for tumour biology:[5][1]
- Hypovascularity — the stroma compresses intratumoural vessels, producing a hypoxic, nutrient-poor microenvironment. This explains why PDAC is hypoenhancing on arterial-phase CT (the radiologist's sign) and is poorly penetrated by systemic chemotherapy.
- Chemoresistance — the high interstitial pressure and physical barrier exclude drug molecules from the tumour parenchyma. Even when drug reaches the stroma, the hypoxic environment selects for quiescent, drug-resistant clones.
- Immune exclusion — the stroma is profoundly immunosuppressive; checkpoint inhibitors (anti-PD-1, anti-CTLA-4) have shown essentially no activity in microsatellite-stable PDAC (response rates <5%) outside the rare MSI-H subset.[1]
Stromal depletion (PEGPH20, sonic-hedgehog inhibitors) has so far failed in randomised trials — paradoxically, aggressive stromal depletion in some models accelerated disease — and the field has moved on to combination immunotherapy and KRAS-targeted therapy.[5]
Molecular subtypes[1]
Bulk and single-cell transcriptomic studies (Bailey 2016, Moffitt 2015) have defined two main molecular subtypes of PDAC:[5][1]
- Classical / pancreatic progenitor — expresses adhesion and epithelial genes (GATA6, PDX1); better prognosis; more responsive to 5-FU-based chemotherapy.
- Basal-like / squamous — expresses mesenchymal and inflammatory genes (TP63, S100A2); worse prognosis; more chemoresistant; the dominant subtype in SMAD4-mutated tumours.[5]
A clinically useful genomic classification (Waddell 2015, ICGC whole-genome sequencing) divides PDAC into four structural variants: stable (≤50 structural variants, often aneuploid, KRAS wild-type enriched), locally rearranged (focal events on one or two chromosomes, BRCA-mutated enriched), scattered (moderate number of events, the largest group), and unstable (>200 events, BRCA/PALB2/DNA-repair mutated). The unstable and locally rearranged subtypes are the targets of platinum-based chemotherapy and PARP inhibitors.[5]
Spread[1]
PDAC disseminates by four principal routes:[1][2][5]
- Direct invasion — duodenum, distal stomach, common bile duct, portal vein, splenic vein, superior mesenteric vessels, and the coeliac and superior mesenteric plexuses (producing the characteristic back pain that is often the first symptom of body/tail tumours).
- Lymphatic — to peripancreatic, coeliac, superior mesenteric, para-aortic, and mediastinal nodes. Nodal involvement is present in over 70% of resected specimens and is the most powerful adverse prognostic factor after margin status.
- Haematogenous — to the liver (most common, via portal venous drainage), lungs, bone, and brain. The liver is the dominant first site of distant metastasis.
- Peritoneal — carcinomatosis and malignant ascites; common with tail tumours and after surgical violation of the tumour capsule.
- Perineural — the desmoplastic stroma tracks along the retroperitoneal nerve sheaths, a route considered characteristic of PDAC and the anatomical basis for the intractable back pain that dominates advanced disease.[1]
Clinical Presentation[1]
Head of pancreas (the classic presentation)[1]
Painless obstructive jaundice is the textbook entry point. A tumour in the pancreatic head compresses the intrapancreatic distal common bile duct, and the obstructed bile regurgitates into the systemic circulation. The result is:[2][1]
- Progressive yellowing of skin and sclera — the patient or family notices yellow eyes first, then a sallow complexion. The jaundice is painless (distinguishing it from the painful jaundice of biliary colic and choledocholithiasis) and progressive (worsens over weeks).
- Dark urine (bilirubinuria) — conjugated bilirubin is water-soluble and excreted in the urine; this is often the first symptom, predating scleral icterus by days.
- Pale/clay-coloured (acholic) stools — the absence of stercobilin in the gut produces putty-coloured, smelly, floating stools (steatorrhoea may coexist from pancreatic exocrine insufficiency).
- Pruritus — bile salt deposition in the skin produces intense, generalised itching, often worse at night; excoriations are visible on examination.
- Weight loss — often marked (over 10% of body weight) and rapid (over months), driven by anorexia, early satiety, malabsorption, and the catabolic state of cancer cachexia.
- Anorexia and fatigue — non-specific but almost universal.
- Dull epigastric pain — may be present but is not the dominant feature; pain is more typical of body/tail tumours.[2]
The combination of painless jaundice + weight loss + palpable non-tender gallbladder (Courvoisier's sign) is the diagnostic triad that should trigger urgent cross-sectional imaging within 24 to 48 hours. In real practice, however, the gallbladder is palpable in only 20 to 30% of head-tumour patients — the absence of a palpable gallbladder does not exclude cancer.[1][2]
Body and tail[1]
Body and tail tumours lack the early-warning biliary obstruction of head tumours, so they declare themselves only when they invade the retroperitoneum or metastasise. The classic picture is:[2][5]
- Severe epigastric pain radiating through to the back — gnawing, constant, often worse at night, and partially relieved by leaning forward (the patient may be seen hunched over a chair). The pain is mediated by invasion of the coeliac and superior mesenteric plexuses and is one of the most treatment-resistant cancer pain syndromes known.
- Weight loss and anorexia — the same cachexia syndrome as head tumours, often more severe because diagnosis is delayed.
- Palpable epigastric mass — late; usually represents the tumour or a metastatic liver.
- Splenomegaly, gastric varices, isolated gastric variceal haemorrhage — from splenic-vein occlusion by tail tumours (sinistral/left-sided portal hypertension). This is rare but classic; a patient with bleeding gastric varices and a normal liver should be assumed to have a tail-of-pancreas tumour until proven otherwise.
- Hepatomegaly and ascites — from liver metastases and peritoneal carcinomatosis.[2]
Paraneoplastic and associated features (the exam pearls)[1]
A handful of paraneoplastic phenomena are so characteristic of PDAC that they earn their own line in a viva answer:[1][3][2]
- New-onset diabetes — the bidirectional relationship between PDAC and diabetes makes new-onset diabetes in an older, thin, weight-losing patient a paraneoplastic marker. Up to 1% of patients over 50 with new diabetes will be diagnosed with PDAC within 3 years. The mechanism is tumour-derived factors (adrenomedullin, islet amyloid polypeptide) that produce insulin resistance and beta-cell dysfunction, and the diabetes often resolves after tumour resection. New-onset diabetes in this context is now a recognised indication for cross-sectional imaging.[1][3]
- Trousseau syndrome (migratory thrombophlebitis) — recurrent, sterile, migratory venous thromboses at unusual sites (subclavian, portal, jugular, superficial limb veins), driven by tumour mucins and tissue factor. Trousseau himself described the sign in himself and died of gastric cancer; it is most strongly associated with pancreatic adenocarcinoma. Treatment is therapeutic low-molecular-weight heparin.[1][2]
- Venous thromboembolism — PDAC carries one of the highest VTE risks of any malignancy (8-fold the general population); recurrent VTE despite anticoagulation should raise suspicion of occult PDAC.
- Depression — present in up to 50% of patients in the year preceding diagnosis; new-onset depression in a previously well older patient may be a subtle presenting feature. (Rigorous evidence for this is limited and the association is debated.)
- Subcutaneous fat necrosis and polyarthralgia — the Schmid triad of acinar cell carcinoma (rare; lipase hypersecretion).
- Acute pancreatitis — pancreatic duct obstruction by a small tumour may present as acute pancreatitis; in an older patient with no gallstones or alcohol history, this is an indication for follow-up imaging to exclude a mass.
Atypical presentations[1]
Examiners reward the candidate who recognises the presentations that are easy to miss:[1][2]
- Acute pancreatitis as the first manifestation — ductal obstruction by a small tumour triggers a first episode of pancreatitis in an older patient with no traditional risk factors. Investigate the pancreas 4 to 6 weeks after resolution.
- Upper GI bleed from duodenal invasion or from isolated gastric varices (splenic-vein thrombosis from a tail tumour) — think tail of pancreas in a patient with variceal bleeding and a normal liver.
- Virchow's node (left supraclavicular lymphadenopathy, Troisier sign) — a metastasis from below the diaphragm; classically associated with gastric cancer but seen in advanced PDAC.
- Sister Mary Joseph nodule (periumbilical metastasis) — Sister Mary Joseph Dempsey, the surgical assistant who first noted it.
- Malignant ascites and Sister Mary Joseph nodule — peritoneal carcinomatosis.
- Migratory thrombophlebitis in an apparently well patient — occult PDAC; image the pancreas.
- Melaena or anaemia from occult duodenal invasion — rare first presentation.[2]
Head (70%)
earlier presentation
- **Painless obstructive jaundice** — the hallmark
- **Courvoisier's sign** — palpable non-tender gallbladder
- Pale stools, dark urine, pruritus
- Presents earlier due to bile duct compression
Body/tail (30%)
late presentation
- **Back pain** radiating, worse at night
- **No jaundice** (duct not obstructed)
- **Weight loss**, epigastric mass
- Almost always unresectable at diagnosis
Differential Diagnosis[1]
The differential of obstructive jaundice is broad, and the examiner will test the candidate's ability to discriminate pancreatic cancer from its look-alikes. The key is to combine the clinical picture with the first-line imaging:[2][1]
| Condition | Key distinguishing feature | Test that clinches the diagnosis |
|---|---|---|
| Choledocholithiasis | Painful jaundice (biliary colic); fever (cholangitis); gallbladder usually NOT palpable (chronic cholecystitis → contracted/fibrotic); RUQ tenderness | US shows CBD stone; MRCP confirms |
| Choledochal cyst | Young patient, recurrent jaundice, RUQ mass; cystic dilation of the biliary tree on MRCP | MRCP |
| Cholangiocarcinoma (Klatskin / hilar) | Painless progressive jaundice, hilar stricture on MRCP; CA 19-9 elevated; older patient, PSC/UC risk | MRCP, ERCP brush cytology, biopsy |
| Ampullary carcinoma | Similar presentation to pancreatic head cancer (jaundice) but better prognosis; visible at duodenoscopy | Side-viewing duodenoscopy + biopsy |
| Autoimmune pancreatitis (AIP) / IgG4-related disease | "Sausage-shaped" pancreas, peripancreatic halo, raised serum IgG4, associated with other IgG4 disease (sialadenitis, RPF); steroid-responsive | Serum IgG4; biopsy shows storiform fibrosis, IgG4+ plasma cells; trial of steroids |
| Chronic pancreatitis | Chronic pain, steatorrhoea, diabetes, calcifications on CT, history of alcohol/idiopathic/tropical; pancreatic duct irregularity; can mimic cancer on imaging | EUS-FNA, IgG4 (to exclude AIP), secretin-MRCP |
| Pancreatic neuroendocrine tumour (pNET) | Often hypervascular (vs hypoenhancing PDAC), may have functional syndrome (insulinoma, gastrinoma); slower growing; may be MEN1-associated | EUS-FNA, plasma chromogranin A, functional tests |
| Primary sclerosing cholangitis (PSC) | Younger patient, IBD (ulcerative colitis), multifocal biliary strictures ("beads on a string" on MRCP), p-ANCA positive | MRCP; cholangiogram at ERCP |
| Viral hepatitis (A, B, E) | Acute onset, fever, RUQ pain, dark urine, hepatocellular LFT pattern (ALT/AST >> ALP); viral serology positive | Serology |
| Drug-induced cholestasis | Recent drug exposure (antibiotics — amoxicillin-clavulanate, flucloxacillin, macrolides; OCP; anabolic steroids; azathioprine) | History; resolves on withdrawal |
| Haemolytic jaundice | Unconjugated hyperbilirubinaemia, anaemia, raised LDH, reticulocytosis | Blood film, Coombs, haptoglobin |
| Gilbert syndrome | Mild unconjugated hyperbilirubinaemia, provoked by stress/fasting; benign | Clinical; no treatment needed |
The two commonest exam traps are (a) mistaking ampullary carcinoma for pancreatic head cancer (it presents identically, but prognosis is much better and the operation may be the same) and (b) missing autoimmune pancreatitis in a patient labelled as having inoperable pancreatic cancer (AIP responds to steroids; a missed diagnosis costs the patient a Whipple and a correct diagnosis saves them from one).[1][2]
Clinical & Bedside Assessment[1]
A focused clinical assessment in a patient with suspected pancreatic cancer is rapid and methodical. The history is taken in the context of a patient who is often cachectic, deeply jaundiced, and frightened.[2][1]
General examination[1]
- Nutritional state — temporal wasting, sarcopenia, loss of subcutaneous fat, BMI. Cachexia is a poor prognostic feature independent of stage.
- Jaundice — scleral icterus, cutaneous icterus. Note the depth and distribution.
- Scratch marks — from pruritus.
- Skin — look for Peutz-Jeghers pigmentation (buccal mucosa, lips, fingers — autosomal dominant inheritance of STK11); FAMMM atypical naevi (large, irregular, multiple); jaundice; excoriations.
- Lymphadenopathy — left supraclavicular (Virchow's/Troisier sign), periumbilical (Sister Mary Joseph nodule), cervical, axillary.
- Thrombophlebitis — palpable cord-like superficial veins; check unusual sites (upper arm, chest, neck) for migratory Trousseau thromboses.
- Signs of chronic liver disease or chronic pancreatitis — spider naevi, palmar erythema, Dupuytren's contracture, parotid enlargement (alcoholic), abdominal surgical scars.[2]
Abdominal examination[1]
- Inspection — distension (ascites), surgical scars, caput medusae, prominent superficial veins.
- Palpation — the key findings are:
- Palpable, non-tender, distended gallbladder (Courvoisier's sign) — in the right upper quadrant, smooth, round, moving with respiration, non-tender. Pathognomonic for malignant biliary obstruction (pancreatic head cancer, cholangiocarcinoma, ampullary cancer).
- Hepatomegaly — from metastases or biliary congestion. Hard, irregular, knobbly liver = metastatic disease.
- Epigastric mass — large head tumour, body/tail tumour (late), or a metastatic deposit in the omentum.
- Splenomegaly — tail tumour with splenic-vein thrombosis (look for dilated gastric varices in the LUQ).
- Ascites — peritoneal carcinomatosis; malignant cells on ascitic tap.
- Percussion — shifting dullness for ascites; loss of liver dullness in perforation (not relevant here).
- Auscultation — silent abdomen in ileus (late disease); bruit over the epigastrium (rare, vascular encasement).[1][2]
The Courvoisier law and sign[1]
The examiner is guaranteed to ask about Courvoisier. The sign is the palpable, non-tender, distended gallbladder in a jaundiced patient; the law is the inference.[2][1]
Courvoisier's law (Ludwig Courvoisier, 1890): in the presence of jaundice, a palpable gallbladder is unlikely to be due to gallstones. The reasoning is that gallstones cause chronic recurrent cholecystitis, with mural fibrosis and a contracted, non-distensible gallbladder that cannot enlarge. A palpable, smoothly distended gallbladder implies a progressive, painless, low-grade obstruction that allows the gallbladder to distend gradually — and that pattern is the hallmark of malignant obstruction of the distal common bile duct (pancreatic head cancer, cholangiocarcinoma, ampullary carcinoma). The classic exception is Mirizzi syndrome (an impacted cystic-duct stone that compresses the common hepatic duct) and double-duct stones.[1]
In real practice the gallbladder is palpable in only 20 to 30% of patients with head-of-pancreas cancer; the absence of the sign does not exclude cancer, but the presence of the sign is a near-pathognomonic bedside finding that should trigger urgent cross-sectional imaging.[2]
Investigations[1]
The workup of suspected pancreatic cancer is structured, multimodal, and time-critical. The goal of the first 48 to 72 hours is to (a) confirm the diagnosis, (b) stage the disease, and (c) assess fitness for surgery, chemotherapy, or palliation.[1]
Blood tests[1]
- Liver function tests (LFTs) — an obstructive pattern: elevated bilirubin (predominantly conjugated/direct), elevated alkaline phosphatase (ALP) and gamma-GT out of proportion to transaminases. Transaminases (ALT/AST) may be mildly elevated but the picture is one of cholestasis, not hepatitis.[1][2]
- CA 19-9 (carbohydrate antigen 19-9, sialylated Lewis-a antigen) — the principal tumour marker for PDAC. Elevated in 75 to 85% of patients, with a sensitivity and specificity around 80% at the 37 U/mL cut-off. CA 19-9 is not diagnostic — it is also elevated in benign biliary obstruction, cholangitis, and other GI cancers. 5 to 10% of the population are Lewis-negative (Le(a−b−)) and cannot synthesise CA 19-9, so a normal CA 19-9 in a confirmed PDAC is uninterpretable rather than reassuring. CA 19-9 normalises with biliary decompression independent of tumour response, so always measure after stenting. CA 19-9 is used for monitoring response and detecting recurrence, not for screening or diagnosis.[1][2]
- CEA (carcinoembryonic antigen) — may be elevated in PDAC but is less sensitive (about 50%) and less specific than CA 19-9. Useful as a complementary marker and for monitoring.
- Glucose and HbA1c — to identify new-onset diabetes (a paraneoplastic marker in older, thin patients) and to characterise pre-existing diabetes for perioperative management.
- Coagulation (PT/INR, aPTT) — prolonged PT/INR from vitamin K malabsorption in obstructive jaundice (bile is needed to absorb fat-soluble vitamins). Correct with vitamin K 10 mg IV or PO daily for 3 days before any procedure; FFP only for active bleeding or imminent surgery.
- Renal function, electrolytes, full blood count, group and save — baseline; needed for chemotherapy dosing (cisplatin/gemcitabine in some regimens, irinotecan).
- Nutritional markers — albumin, prealbumin, transferrin. Hypoalbuminaemia is a poor prognostic factor.
- Hepatic and renal function — for chemotherapy dosing and surgical fitness.
Imaging[1]
- Contrast-enhanced CT (pancreatic protocol) — the gold standard for diagnosis and staging. The protocol is triphasic: non-contrast, arterial phase at 25 to 40 seconds (to see the SMA, coeliac axis, common hepatic artery), and portal-venous phase at 60 to 70 seconds (to see the SMV, portal vein, and liver). PDAC is characteristically hypoenhancing on the arterial phase (because of the desmoplastic stroma and hypovascularity), giving it a low-density appearance against the background of normally enhancing parenchyma. CT is evaluated for: (a) tumour size and location, (b) vascular involvement (degrees of circumference of SMA, coeliac, common hepatic artery, SMV/PV), (c) liver metastases, (d) peritoneal disease, (e) lymphadenopathy, and (f) the double-duct sign (dilated CBD + dilated main pancreatic duct) which, in the right clinical context, is highly suggestive of head-of-pancreas PDAC. CT accuracy for resectability is over 90% in expert hands.[1][5][6]
- Endoscopic ultrasound (EUS) with fine-needle aspiration (FNA) or fine-needle biopsy (FNB) — provides high-resolution imaging of the pancreas, direct visualisation of the tumour, and a route to tissue diagnosis (EUS-FNA sensitivity over 90% for PDAC, especially with rapid on-site evaluation, ROSE). EUS also assesses local vascular invasion, identifies small liver metastases missed on CT, and is the preferred method for histological confirmation before chemotherapy. EUS-FNA is safe (bleeding and pancreatitis rate <2%) and is now routine before resection. EUS is the most sensitive modality for small (<2 cm) PDAC and for IPMN-associated lesions.[1][2]
- Magnetic resonance cholangiopancreatography (MRCP) — non-invasive mapping of the biliary tree and pancreatic duct. Useful for distinguishing cancer from chronic pancreatitis (the duct-penetrating sign on secretin-MRCP), for indeterminate liver lesions, and for evaluation of side-branch IPMN. MRCP is increasingly used as a problem-solver in place of diagnostic ERCP.[1]
- ERCP (endoscopic retrograde cholangiopancreatography) — primarily therapeutic in pancreatic cancer: biliary stenting (plastic or self-expanding metal SEMS) to relieve jaundice before surgery or as palliation. ERCP also allows brush cytology of strictures (sensitivity about 30 to 50%) and cholangioscopy with SpyGlass for direct visualisation. ERCP is no longer used purely for diagnosis because of the risk of post-ERCP pancreatitis (3 to 5%).[1][2]
- 18F-FDG PET-CT — selected cases: occult distant metastases in high-risk patients (CA 19-9 >500, large primary, equivocal CT findings), restaging after neoadjuvant therapy, and evaluation of recurrence when CA 19-9 rises and CT is negative. Not routine in resectable disease.[5]
- Staging laparoscopy — performed before planned Whipple in many centres to detect occult peritoneal or liver metastases missed on CT (10 to 20% of apparently resectable cases). Indicated when CA 19-9 is markedly elevated (>500 U/mL), tumour is borderline resectable, or imaging is equivocal. Laparoscopy with peritoneal cytology has been shown to upstage 10 to 15% of patients and avoid non-therapeutic laparotomy.[1][5]
Histology and molecular testing[1]
- Core biopsy is required before any non-surgical therapy (neoadjuvant or palliative chemotherapy). Cytology from FNA is acceptable in the metastatic setting.
- Immunohistochemistry confirms ductal origin (CK7, CK19, CA 19-9) and excludes other primaries (CDX2 for intestinal, TTF-1 for lung, GATA3 for breast).
- Next-generation sequencing (NGS) on the biopsy identifies actionable mutations: KRAS (90%+), TP53, CDKN2A, SMAD4, BRCA1/2, PALB2, ATM, mismatch-repair status (MMR/MSI), NTRK, BRAF. Germline testing (blood) for BRCA1/2, PALB2, ATM, and the Lynch genes is now recommended for all patients with pancreatic cancer by NCCN (2020 onward) because of the implications for relatives and for PARP-inhibitor therapy. MSI-H/dMMR testing is mandatory because of the option of pembrolizumab.[1][3]
- CA 19-9 baseline for monitoring, drawn after biliary decompression (stenting) if performed.
Management — Resuscitation[1]
Pancreatic cancer rarely presents as a surgical emergency. The acute presentations are:[1]
- Biliary obstruction with cholangitis (Charcot triad — fever, jaundice, RUQ pain) — IV fluids, broad-spectrum antibiotics (piperacillin-tazobactam 4.5 g IV 8-hourly, or ceftriaxone 2 g IV daily + metronidazole 500 mg IV 8-hourly), and urgent ERCP with biliary stenting within 24 hours. If ERCP fails or is not available, percutaneous transhepatic biliary drainage (PTBD). Definitive surgery or oncology referral follows.
- Coagulopathy from obstructive jaundice — vitamin K 10 mg IV or PO daily for 3 days; FFP only for active bleeding or imminent surgery. Correct before any procedure.
- Gastric outlet obstruction (duodenal invasion) — NG decompression, IV fluids and electrolytes, PPI. Definitive treatment is palliative gastrojejunostomy (open or laparoscopic) or endoscopic duodenal stent.
- Venous thromboembolism / Trousseau syndrome — therapeutic enoxaparin 1.5 mg/kg SC daily (or 1 mg/kg 12-hourly). PDAC patients hospitalised for any reason should receive prophylactic enoxaparin 40 mg SC daily unless bleeding.
- Severe pain — WHO analgesic ladder (paracetamol → mild opioid → strong opioid); coeliac plexus block or neurolysis for refractory pain (CT-guided or EUS-guided injection of absolute alcohol or phenol into the coeliac plexus).
- Malnutrition and exocrine insufficiency — pancreatic enzyme replacement therapy (PERT) with meals (pancreatin 25,000 to 40,000 units of lipase per main meal, half dose with snacks); dietitian referral; oral nutritional supplements; enteral feeding if oral intake fails.
- New-onset diabetes — metformin first line; insulin if uncontrolled or for perioperative control.[1]
Management — Definitive & Stepwise[1]
Treatment is driven by resectability status, performance status (ECOG/Karnofsky), and patient preference. The MDT (medical oncology, surgical oncology, radiation oncology, hepatobiliary radiology, pathology, palliative care) makes the call.[1]
Resectable disease (15 to 20% of patients)[1]
Pancreatoduodenectomy (Whipple procedure) is the operation for tumours of the head of the pancreas, distal CBD, ampulla, and duodenum. It is one of the largest general surgical operations and should be performed at high-volume centres (mortality <3% vs >10% at low-volume centres).[1][5]
Resection:[1]
- Head of pancreas — divided at the level of the portal vein (over the SMV).
- Duodenum — second, third, and sometimes fourth parts.
- Distal bile duct — divided above the cystic duct; the gallbladder is removed en bloc.
- Distal stomach — classically (Whipple, 1935); the pylorus-preserving modification (Traverso-Longmire) preserves the entire stomach and the first part of the duodenum, with comparable oncological outcomes and better nutritional recovery. Pylorus-preserving is now the default at most centres.
- Regional lymphadenectomy — standard lymphadenectomy includes stations 5, 6, 8a, 12b1, 12b2, 12c, 13a, 13b, 14p, 14d (Japanese classification); an extended template to stations 9 and 16 has not shown survival benefit in the European EXTROP trial.
- Vascular resection — segmental resection of the SMV/PV with primary anastomosis or interposition graft (internal jugular, PTFE) is now standard for venous involvement without arterial encasement, with similar R0 and survival outcomes to standard resection in experienced hands. Arterial resection (SMA, coeliac) remains controversial; most centres reserve it for highly selected fit patients in a trial setting.[1]
Reconstruction (three anastomoses, in order):[1][2]
- Pancreaticojejunostomy — the pancreatic remnant is anastomosed end-to-side to the jejunum. The most prone to leak; the source of postoperative pancreatic fistula (POPF). Variations: duct-to-mucosa, dunking, invagination. The prophylactic octreotide, internal pancreatic stent, and patch coverage of the jejunal staple line are all used to reduce leak rates.
- Hepaticojejunostomy — end-to-side anastomosis of the proximal bile duct to the jejunum, downstream of the pancreatic anastomosis.
- Gastrojejunostomy (or duodenojejunostomy in pylorus-preserving Whipple) — restores gastrointestinal continuity; placed distal to the other anastomoses.[1]
Whipple procedure — the 3 anastomoses
PHG
pancreatic remnant to jejunum (most prone to leak)
bile duct to jejunum
stomach (or duodenum) to jejunum
Distal pancreatectomy ± splenectomy is the operation for body and tail tumours. Often performed laparoscopically or robotically. The splenic artery and vein run in close proximity to the pancreatic body/tail and are typically sacrificed, so splenectomy is routine; spleen-preserving distal pancreatectomy (Warshaw technique) is possible in selected benign or low-grade lesions. The pancreatic remnant is closed (stapler, hand-sewn, or patch) — there is no anastomosis. Spleen loss mandates vaccination against encapsulated organisms (meningococcus, pneumococcus, H. influenzae type b) at least 2 weeks preoperatively or 2 weeks postoperatively.[1][5]
Total pancreatectomy removes the entire gland, the spleen, the gallbladder, and the duodenum, with a single Roux-en-Y reconstruction. Indicated for multifocal disease (main-duct IPMN with high-grade dysplasia), for positive neck margin on frozen section, and for hereditary pancreatitis with extensive neoplasia. Resulting insulin-dependent diabetes is brittle (loss of both alpha and beta cells, no glucagon response to hypoglycaemia).[5]
Post-operative morbidity and mortality. Whipple has a 30 to 40% overall complication rate, but a 1 to 3% mortality rate in high-volume centres. Major complications:[1][2][5]
- Postoperative pancreatic fistula (POPF) — 10 to 20%; the most feared; defined by the International Study Group on Pancreatic Surgery (ISGPS) as drain amylase >3× serum on or after POD 3. Grades B and C are clinically significant. Management: percutaneous drainage, octreotide, antibiotics, nutritional support (enteral or TPN), and rarely completion pancreatectomy. Grade C leaks carry up to 30% mortality.
- Delayed gastric emptying (DGE) — 10 to 30%; the most common; prolonged NG drainage and inability to tolerate oral intake beyond POD 7 to 14. Management: prokinetics (erythromycin, metoclopramide), patience, nutritional support.
- Post-pancreatectomy haemorrhage (PPH) — early (within 24 hours, surgical) or late (after POD 5 to 14, pseudoaneurysm). A sentinel bleed (a small, self-limiting haematemesis or drain bleed) before massive haemorrhage is the classic warning of a pseudoaneurysm from a POPF eroding the gastroduodenal artery stump. Urgent mesenteric angiography and coil embolisation is the treatment of choice; mortality of untreated rupture exceeds 50%.
- Anastomotic leak (hepaticojejunostomy or gastrojejunostomy) — uncommon (<5%); reoperation or percutaneous drainage.
- Intra-abdominal abscess — usually secondary to POPF; percutaneous drainage.
- Wound infection, pneumonia, DVT/PE, urinary tract infection — generic post-op complications.
- Pancreatic exocrine insufficiency — common after Whipple (>50%); PERT is lifelong.
- New-onset diabetes — after Whipple (10 to 25%) or total pancreatectomy (100%).[1]
Adjuvant chemotherapy after resection begins within 4 to 8 weeks of surgery, ideally as soon as the patient is recovered. The current standard of care for fit patients is modified FOLFIRINOX (mFOLFIRINOX) for 12 cycles (about 6 months), as established by the PRODIGE 24/ACCORD 24 trial (Conroy 2018): median OS 54.4 months vs 35.0 months with gemcitabine alone (HR 0.64, p<0.001). For patients not fit for FOLFIRINOX, gemcitabine plus capecitabine (ESPAC-4) is the alternative (median OS 28.0 months vs 25.5 months with gemcitabine alone). Single-agent gemcitabine is reserved for those who cannot tolerate either combination. Adjuvant chemoradiotherapy is no longer routine after ESPAC-1 showed no benefit over chemotherapy alone; reserved for R1 margins in some centres.[1][9][10]
Borderline resectable disease[1]
Borderline resectable PDAC is now treated with neoadjuvant chemotherapy first in nearly all major centres. The intent is to (a) downstage the tumour to a resectable state, (b) treat occult micrometastatic disease early, and (c) identify patients with aggressive biology (who progress on chemotherapy and are spared a non-therapeutic operation).[1][5][12]
- FOLFIRINOX (mFOLFIRINOX) for 4 to 6 months (8 cycles) is the preferred regimen for fit patients. R0 resection rate in contemporary series is 60 to 85%.
- Gemcitabine + nab-paclitaxel for 4 to 6 months is the alternative for patients not fit for FOLFIRINOX.[5]
After neoadjuvant therapy, restaging with pancreatic-protocol CT and CA 19-9 is performed. If the tumour has responded (CA 19-9 down, no progression) and the patient remains operable, proceed to pancreatoduodenectomy (with vascular resection/reconstruction as needed). If progression, convert to palliative chemotherapy. The PREOPANC trial (Dutch, 2018) showed neoadjuvant gemcitabine-based chemoradiotherapy improved R0 and median OS vs upfront surgery in (borderline) resectable PDAC. The Murphy 2019 trial of total neoadjuvant FOLFIRINOX + losartan + chemoradiotherapy achieved a 61% resection rate in locally advanced PDAC, with a 33-month median OS in resected patients — the strongest evidence for total neoadjuvant therapy.[12][5]
Locally advanced unresectable disease[1]
Defined as >180° encasement of SMA or coeliac axis, or non-reconstructible SMV/PV occlusion, with no distant metastases. Treatment is induction chemotherapy (FOLFIRINOX or gemcitabine/nab-paclitaxel) for 4 to 6 months, then restaging. If response — particularly if the tumour drops below 180° SMA contact — consider surgical exploration in high-volume centres. If stable disease, consider consolidation chemoradiotherapy (capecitabine-based 50.4 to 54 Gy in 28 to 30 fractions, or SBRT in selected centres) for local control. If progression, convert to second-line chemotherapy. Median OS 15 to 20 months.[1][5]
Metastatic disease (50% of patients)[1]
The goal is palliation and life extension. For fit patients (ECOG 0 to 1, age <70, normal bilirubin, adequate organ function), the standard first-line is FOLFIRINOX (per PRODIGE 4/ACCORD 11, Conroy 2011): oxaliplatin 85 mg/m², leucovorin 400 mg/m², irinotecan 180 mg/m² on day 1, then 5-FU 400 mg/m² bolus followed by 2,400 mg/m² over 46 hours, every 14 days. Median OS 11.1 months vs 6.8 months with gemcitabine (HR 0.57), but grade 3/4 toxicities are common (neutropenia 45%, fatigue 24%, diarrhoea 13%, sensory neuropathy 9%) and G-CSF prophylaxis is standard. Gemcitabine + nab-paclitaxel (MPACT, Von Hoff 2013) is the alternative for less fit patients or those intolerant of FOLFIRINOX: gemcitabine 1,000 mg/m² + nab-paclitaxel 125 mg/m² on days 1, 8, 15 every 28 days; median OS 8.5 months vs 6.7 months with gemcitabine alone. Gemcitabine monotherapy (1,000 mg/m² weekly × 7 then weekly × 3 of 4) is the option for elderly or frail patients; median OS 6 to 7 months. Best supportive care alone is appropriate for ECOG 3 to 4.[1][7][8]
Second-line options after progression on first-line: nanoliposomal irinotecan + 5-FU/leucovorin (NAPOLI-1, Wang-Gillam 2016) improved median OS 6.2 vs 4.2 months; FOLFOX is widely used off-trial. Pembrolizumab is reserved for the rare MSI-H/dMMR tumour (<1% of PDAC; KEYNOTE-158).[1]
Maintenance therapy for germline BRCA1/2-mutated patients who have not progressed on platinum-based first-line (≥16 weeks of FOLFIRINOX or cisplatin-gemcitabine): olaparib 300 mg orally twice daily (capsule) or 150 mg twice daily (tablet) until progression (POLO trial, Golan 2019). Olaparib improved median PFS from 3.8 to 7.4 months (HR 0.53), with no significant OS gain at first analysis. Olaparib is the first precision-medicine indication in pancreatic cancer and the proof of principle that PARP inhibition is effective in BRCA-mutated PDAC.[11][1]
Palliative care runs in parallel: pain control (coeliac plexus block for back pain, WHO analgesic ladder), biliary decompression (covered metal stent, SEMS), duodenal stenting for gastric outlet obstruction, PERT for exocrine insufficiency, antiemetics, psychological support, early referral to specialist palliative care (improves both quality of life and survival, per Temel 2010 in lung cancer, replicated in pancreatic).[1]
Key evidence in pancreatic cancer systemic therapy
FOLFIRINOX
triplet chemo
- **5-FU 2,400 mg/m² over 46 h + irinotecan 150 mg/m² + oxaliplatin 85 mg/m²** every 14 days
- Survival advantage over gemcitabine in **adjuvant (PRODIGE 24)** and **metastatic (PRODIGE 4)**
- **40% grade 3/4 fatigue, 45% neutropenia** — G-CSF prophylaxis required
- Preferred for fit (ECOG 0–1), age under 70, normal bilirubin
Gemcitabine + nab-paclitaxel
doublet chemo
- **Gemcitabine 1,000 mg/m² + nab-paclitaxel 125 mg/m²** days 1, 8, 15 q28d × 6 cycles
- Median OS 8.5 mo vs 6.7 mo gemcitabine alone (MPACT)
- **Better tolerated than FOLFIRINOX** — preferred for ECOG 1, older, or borderline fitness
- First-line for unfit/frail patients who need more than gemcitabine alone
Staging & Systemic Therapy — Doses, Trials, and NCCN Criteria[1]
AJCC TNM 8th edition[1]
The AJCC 8th edition (Amin 2017) is the current staging system. Resectability is a separate clinical-radiological assessment, not a T-stage.[4]
| Stage | T | N | M | Approx 5-yr OS |
|---|---|---|---|---|
| 0 | Tis (in situ) | N0 | M0 | rare; curable |
| IA | T1a (≤0.5 cm) or T1b (0.5 to <1 cm) or T1c (1 to 2 cm) | N0 | M0 | ~40 to 50% |
| IB | T2 (>2 to ≤4 cm) | N0 | M0 | ~30 to 35% |
| IIA | T3 (>4 cm) | N0 | M0 | ~20 to 25% |
| IIB | T1–T3 | N1 (1–3 nodes) | M0 | ~10 to 15% |
| III | T1–T3 or T4 | N2 (≥4 nodes) or any N | M0 | ~5% |
| III | T4 (SMA or CA encasement) | N0 | M0 | ~5% |
| IV | any T | any N | M1 | ~1% (median 6–11 mo) |
Resectability criteria (NCCN/AHPBA/SSO consensus)[1]
- Resectable:
- No distant metastases.
- No radiographic evidence of SMA or coeliac axis abutment or encasement.
- No radiographic evidence of SMV/PV distortion (clear fat plane around these vessels).
- ≤180° contact with SMV/PV without vein deformity.
- Borderline resectable (BR-PC):
- SMV/PV: >180° contact, or ≤180° contact with vein deformity, or short-segment occlusion with reconstructible vessel above and below.
- SMA / coeliac axis: ≤180° contact of the circumference.
- No distant metastases.
- Locally advanced (unresectable):
- SMA / coeliac axis: >180° encasement.
- SMV/PV: non-reconstructible occlusion.
- No distant metastases.[1]
Landmark trials at a glance[1]
- PRODIGE 4/ACCORD 11 (Conroy 2011, NEJM) — FOLFIRINOX vs gemcitabine in metastatic PDAC, 342 patients, ECOG 0 to 1. Median OS 11.1 vs 6.8 months (HR 0.57), median PFS 6.4 vs 3.3 months, ORR 32% vs 9%. Established FOLFIRINOX as first-line for fit patients.[7]
- MPACT (Von Hoff 2013, NEJM) — gemcitabine + nab-paclitaxel vs gemcitabine in metastatic PDAC, 861 patients. Median OS 8.5 vs 6.7 months (HR 0.72), median PFS 5.5 vs 3.7 months, ORR 23% vs 7%. Established the doublet as first-line for patients not suitable for FOLFIRINOX.[8]
- ESPAC-4 (Neoptolemos 2017, Lancet) — adjuvant gemcitabine + capecitabine vs gemcitabine alone after R0/R1 resection, 732 patients. Median OS 28.0 vs 25.5 months (HR 0.82, p=0.05). Established the doublet as adjuvant option.[10]
- PRODIGE 24/ACCORD 24 (Conroy 2018, NEJM) — adjuvant mFOLFIRINOX vs gemcitabine after R0/R1 resection, 493 patients, ECOG 0 to 1. Median OS 54.4 vs 35.0 months (HR 0.64), median DFS 21.6 vs 12.8 months, 3-year DFS 39.7% vs 21.4%. The largest survival gain in adjuvant PDAC ever, and the new standard of care in fit patients.[9]
- POLO (Golan 2019, NEJM) — maintenance olaparib vs placebo in germline BRCA-mutated metastatic PDAC with no progression on ≥16 weeks of platinum-based chemotherapy, 154 patients. Median PFS 7.4 vs 3.8 months (HR 0.53). First precision-medicine indication in PDAC.[11]
- Murphy total neoadjuvant (2019, JAMA Oncol) — total neoadjuvant FOLFIRINOX + losartan + chemoradiotherapy in locally advanced PDAC. R0 resection rate 61%, median OS in resected patients 33 months. Strongest evidence for total neoadjuvant therapy.[12]
- PREOPANC (2018, J Clin Oncol) — neoadjuvant gemcitabine-based chemoradiotherapy vs upfront surgery in (borderline) resectable PDAC. R0 rate 71% vs 40%, median OS 17.1 vs 13.5 months (HR 0.74). The Dutch randomised trial that underpins the neoadjuvant paradigm in BR-PC.[5]
- ESPAC-1 (Neoptolemos 2004, JAMA) — adjuvant chemotherapy (5-FU/leucovorin) vs chemoradiotherapy vs surgery alone. Chemotherapy improved 5-year OS vs no chemo; chemoradiotherapy added no benefit and may have harmed. Established the modern adjuvant chemotherapy paradigm.[5]
- Waddell 2015 (Nature) — ICGC whole-genome sequencing of 100 PDACs; four genomic subtypes: stable, locally rearranged (BRCA/PALB2 enriched), scattered, and unstable. Foundation for precision-medicine approach.[5]
Specific Subtypes & Scenarios[1]
Periampullary carcinoma[1]
As noted, the umbrella term for tumours of the ampulla of Vater, distal CBD, or second part of the duodenum. Present identically to pancreatic head cancer (painless jaundice) but with a much better prognosis — 5-year survival 40 to 50% after Whipple (intestinal-type ampullary adenocarcinoma 50 to 70%; distal cholangiocarcinoma 30 to 40%; duodenal 40 to 50%). Treatment: pancreatoduodenectomy with regional lymphadenectomy, and adjuvant chemotherapy is debated (CAPOX or gemcitabine-based regimens). The differential is settled on histopathology and immunohistochemistry (CK7/CK20/CDX2 patterns).[2]
IPMN (intraductal papillary mucinous neoplasm)[1]
A mucin-producing cystic tumour of the pancreatic duct with potential for malignant transformation. Subtypes:[2]
- Main-duct IPMN — segmental or diffuse dilation of the main pancreatic duct >5 mm without another cause. Malignancy rate 40 to 70%; resect.
- Branch-duct IPMN — cystic dilation of side branches. Malignancy rate 5 to 15%. Resect if high-risk stigmata (obstructive jaundice, enhancing solid component, main pancreatic duct ≥10 mm) or worrisome features (pancreatitis, cyst ≥3 cm, thickened enhancing cyst walls, non-enhancing mural nodule <5 mm, main pancreatic duct 5 to 9 mm, abrupt change in MPD calibre with distal pancreatic atrophy) — the Fukuoka/Kyoto criteria.[2][5]
Surveillance of unresected IPMN: MRI/MRCP or CT every 1 to 2 years.[2]
Pancreatic neuroendocrine tumour (pNET)[1]
pNETs are 3 to 5% of pancreatic neoplasms. They are hypervascular on CT (a useful discriminator from hypoenhancing PDAC), are graded by Ki-67 index and mitotic count (G1, G2, G3), and may be functional (hormone-excess syndrome) or non-functional.[2]
- Insulinoma — most common functional pNET; Whipple triad of fasting hypoglycaemia (<2.5 mmol/L), neuroglycopenic symptoms, and relief with glucose. Enucleate for small benign lesions; resect for larger or suspicious ones.
- Gastrinoma (Zollinger-Ellison syndrome) — refractory peptic ulceration, gastric acid hypersecretion, secretin-stimulated gastrin rise. Associated with MEN1 in 25%. Localise with SRS / Ga-68 DOTATATE PET; resect; somatostatin analogues for symptom control.
- Glucagonoma — necrolytic migratory erythema, diabetes, weight loss, DVT. Resect if localised; somatostatin analogues.
- VIPoma — WDHA syndrome (watery diarrhoea, hypokalaemia, achlorhydria). Resect; somatostatin analogues.
- Somatostatinoma — diabetes, gallstones, steatorrhoea. Resect.[2]
Surgical resection is curative for localised pNET; somatostatin analogues (octreotide, lanreotide) control hormone syndromes and slow progression (PROMID, CLARINET trials). For metastatic G1/G2 pNET, everolimus and sunitinib are approved; for G3, platinum-based chemotherapy (cisplatin/etoposide) or PRRT (177Lu-DOTATATE) for somatostatin-receptor-positive disease.[2][5]
Acinar cell carcinoma[1]
A rare, aggressive variant presenting with the Schmid triad of subcutaneous fat necrosis, polyarthralgia, and eosinophilia from lipase hypersecretion. Resection when localised; gemcitabine-based chemotherapy for metastatic disease.[2]
Autoimmune pancreatitis (IgG4-related)[1]
Type 1 AIP is part of the IgG4-related disease spectrum. The pancreas appears "sausage-shaped" with a peripancreatic halo, and patients have raised serum IgG4 and may have other organ involvement (sialadenitis, retroperitoneal fibrosis, Riedel thyroiditis). The most important exam trap: AIP can mimic PDAC radiologically and clinically, including obstructive jaundice, but it is steroid-responsive. A short trial of prednisolone (30 to 40 mg/day for 2 to 4 weeks) reverses the mass and jaundice in 80% of cases. Distinguishing features: elevated IgG4 (>2× upper limit), multi-organ involvement, older male patient. The Whipple specimen is the diagnosis made too late.[2]
Hereditary pancreatic cancer[1]
The high-risk surveillance clinic is now a standard offering. Candidates for surveillance include: (a) carriers of germline mutations with high PDAC penetrance (BRCA2, PALB2, ATM, STK11, CDKN2A, PRSS1, MLH1/MSH2/MSH6), (b) individuals with ≥2 first-degree relatives with PDAC ("familial PDAC"), and (c) those with hereditary pancreatitis.[3][14]
- Surveillance modality: annual MRI/MRCP (and/or EUS) starting at age 50 (or 40 for STK11 carriers, 35 for PRSS1), or 10 years before the youngest affected relative's age at diagnosis. The CAPS (Cancer of the Pancreas Screening) consortium studies show surveillance detects PDAC at stage I in over 80% of cases, vs <20% in symptomatic presentation. CT adds radiation burden and is reserved for EUS/MRI abnormalities.[14]
- Germline testing is now recommended for all patients with pancreatic cancer (NCCN 2020), not just those with a family history, because of the implications for relatives and for PARP-inhibitor therapy.
- Screening in chronic pancreatitis (non-hereditary) is debated; CA 19-9 and annual CT or MRI are used in some centres.
Pancreatic cancer in pregnancy[1]
Exceedingly rare (1 in 1,000,000 pregnancies). The challenge is that abdominal pain, nausea, and weight loss are common in pregnancy and may delay diagnosis. MRI without gadolinium is the imaging of choice. Surgery is ideally deferred to the second trimester (or postpartum) if curative, and chemotherapy (FOLFIRINOX or gemcitabine) is contraindicated in the first trimester. Most cases are diagnosed at an advanced stage.[2]
Complications & Pitfalls[1]
Of the disease[1]
- Obstructive jaundice — pruritus, malabsorption (steatorrhoea from lack of bile), coagulopathy (vitamin K deficiency), cholangitis (Charcot triad).
- Duodenal obstruction — vomiting, dehydration, malnutrition.
- Intractable pain — from coeliac/superior mesenteric plexus invasion; the dominant symptom of body/tail tumours and advanced disease.
- Venous thromboembolism / Trousseau syndrome — paraneoplastic hypercoagulability; high VTE risk (8× general population).
- Malnutrition and cachexia — exocrine insufficiency (steatorrhoea, weight loss), anorexia, catabolic state; PERT and dietitian support are under-utilised.
- Diabetes — from tumour destruction of islets and from paraneoplastic insulin resistance.
- Hepatic and peritoneal metastases — the commonest end-state.[1]
Of the Whipple procedure[1]
- Pancreatic fistula (POPF, 10 to 20%) — leak from the pancreaticojejunostomy; the most feared complication. Drains are left at surgery; drain amylase >3× serum on POD 3 confirms the diagnosis. Management: percutaneous drainage of any collection, octreotide (50 to 100 μg SC 8-hourly), antibiotics if infected, enteral or TPN nutrition, completion pancreatectomy only as a last resort. Mortality of grade C POPF up to 30%.[1][2]
- Delayed gastric emptying (DGE, 10 to 30%) — prolonged NG drainage, intolerance of oral intake beyond POD 7 to 14. Treatment: prokinetics (erythromycin 250 mg IV 8-hourly, metoclopramide), NG decompression, patience. Resolves in nearly all cases within 4 to 6 weeks.
- Post-pancreatectomy haemorrhage (PPH) — early (within 24 hours, surgical anastomosis) or late (after POD 5 to 14, pseudoaneurysm from POPF eroding the gastroduodenal artery stump). A sentinel bleed (small haematemesis or drain bleed) before massive haemorrhage is the classic warning. Urgent mesenteric angiography with coil embolisation; surgery only if unstable. Mortality of untreated rupture >50%.[1]
- Anastomotic leak (hepaticojejunostomy, gastrojejunostomy) — uncommon; reoperation or percutaneous drainage.
- Intra-abdominal abscess — secondary to POPF; percutaneous drainage.
- Wound infection, pneumonia, DVT/PE — generic post-op complications.
- Pancreatic exocrine insufficiency — common (>50% after Whipple); lifelong PERT.
- New-onset diabetes — after Whipple (10 to 25%) or total pancreatectomy (100%); insulin-dependent and brittle (loss of glucagon response).
Classic pitfalls[1]
- Missing Courvoisier's sign — a palpable non-tender gallbladder in a jaundiced patient is cancer until proven otherwise.
- Mistaking painless jaundice for gallstones without imaging — gallstones are by far the commonest cause of jaundice in a 70-year-old, but painless progressive jaundice is malignant until proven otherwise.
- Not doing staging laparoscopy before Whipple — occult peritoneal disease is missed in 10 to 20%.
- Not offering FOLFIRINOX to fit patients with metastatic disease — depriving them of 4 to 6 months additional survival.
- Not relieving jaundice before surgery — stent first to normalise bilirubin and coagulation.
- Not considering pancreatic cancer in new-onset diabetes in an older, thin patient with weight loss.
- Treating autoimmune pancreatitis (AIP) as inoperable cancer — a steroid trial is diagnostic and may avoid Whipple.
- Confusing ampullary carcinoma with pancreatic head cancer — they present identically but have a markedly different prognosis; histology is the only way to distinguish.
- Missing sentinel bleed after Whipple — dismiss at your peril; urgent angiography.
- Not testing BRCA in patients with PDAC — denies them and their relatives the benefits of germline testing and PARP inhibitors.
- Forgetting to vaccinate before or after splenectomy (pneumococcus, meningococcus, Hib).
- Ignoring Lewis-negative CA 19-9 — normal CA 19-9 in 5 to 10% of the population is uninformative, not reassuring.[1]
Prognosis & Disposition[1]
Pancreatic cancer has the worst overall prognosis of any major solid malignancy, with an overall 5-year survival of approximately 10% in most series (12% in the most recent SEER data, 2023). The principal reason is that over 80% of patients present at an unresectable stage.[1][2][5]
- Overall 5-year survival: under 10% (the main reason: over 80% present at an unresectable stage).
- After curative R0 resection + adjuvant mFOLFIRINOX: median OS 54 months, 5-year survival 30 to 40% (improving with PRODIGE-24 era).[9]
- After R0 + gemcitabine/capecitabine: 5-year 25 to 30%.[10]
- After R0 + gemcitabine alone: 5-year 20 to 25%.[1]
- Locally advanced, unresectable: median OS 15 to 20 months with chemotherapy ± chemoradiotherapy.[5]
- Metastatic, treated with FOLFIRINOX: median OS 11 months, 5-year 3%.[7]
- Metastatic, treated with gemcitabine/nab-paclitaxel: median OS 8.5 months.[8]
- Metastatic, best supportive care: median OS 3 to 6 months.[1]
Adverse prognostic factors (multivariate analyses):[1]
- Advanced stage (T3/T4, N+, M+).
- Positive resection margin (R1/R2) — arguably the single most powerful factor after stage.
- Lymph node involvement (number of positive nodes; N2 worse than N1).
- Elevated CA 19-9 (pre-treatment >500 U/mL is a strong adverse factor; post-treatment CA 19-9 normalisation is a favourable sign).
- Large tumour size.
- Perineural, lymphovascular, or venous invasion.
- Poor performance status (ECOG ≥2).
- Weight loss >10% of body weight.
- High modified Glasgow Prognostic Score (high CRP, low albumin).
- SMAD4 loss — associated with metastatic phenotype.
- Basal-like / squamous molecular subtype — worse prognosis.
- New-onset diabetes (paraneoplastic, often signals more aggressive disease).[1]
Follow-up after resection is variable across guidelines but generally includes clinical review, CA 19-9, and CT every 3 to 6 months for the first 2 years, then every 6 months. Recurrence is common (over 70% within 2 years), most often in the liver, peritoneum, or locally at the surgical bed.[1]
Disposition: patients with resected disease are followed in surgical oncology; unresectable or metastatic patients need parallel oncology and palliative care input from the first encounter (early palliative care improves outcomes, per Temel 2010 and replicated in PDAC cohorts).[1]
Special Populations[1]
Hereditary risk (BRCA2, Peutz-Jeghers, Lynch, hereditary pancreatitis)[1]
- Germline testing is now recommended for all patients with pancreatic cancer (NCCN 2020 onward), regardless of family history, because (a) BRCA1/2/PALB2/ATM are found in 4 to 10% of unselected PDAC, (b) results inform PARP-inhibitor use, and (c) results affect first-degree relatives.[3][14]
- Surveillance for high-risk carriers: annual MRI/MRCP ± EUS from age 50 (40 for STK11, 35 for PRSS1), or 10 years before the youngest affected relative. Biannual surveillance is increasingly considered; CAPS data show 12-monthly detects 80% of stage I disease.[14]
- Therapeutic implications: olaparib for germline BRCA1/2-mutated metastatic PDAC after platinum response; platinum-based chemotherapy is preferred first-line in BRCA-mutated disease (the SWOG S1505 and POLO subset data both support); immune checkpoint inhibitors for MSI-H/Lynch.
New-onset diabetes in an older, thin patient with weight loss[1]
A clinical pearl that has moved from anecdote to evidence: new-onset diabetes in a patient over 50 with weight loss has approximately a 1% probability of underlying PDAC and warrants cross-sectional imaging (pancreatic-protocol CT or MRI). Studies using the ratio of CA 19-9 to age, or a combined risk model (the ENDPAC score: weight loss, smoking, age at diabetes onset), identify a sub-group with 4 to 5% PDAC prevalence — well above the threshold for further investigation. Resection in such patients often yields early-stage disease.[1][3]
Elderly and frail[1]
- Surgery is feasible in selected elderly patients (octogenarians) without comorbidity, but outcomes are worse. The decision to proceed to Whipple should be based on physiological age, performance status, and patient preference, not chronological age alone.
- Chemotherapy: dose-reduced gemcitabine (800 mg/m² weekly) or gemcitabine/nab-paclitaxel (dose-reduced). FOLFIRINOX is generally avoided in ECOG ≥2, age >75, or bilirubin >1.5 × ULN.
- Best supportive care is appropriate for ECOG 3 to 4.[1]
Anticoagulated[1]
- Warfarin is often switched to LMWH during chemotherapy because of drug interactions (capecitabine, 5-FU) and INR instability.
- Direct oral anticoagulants (DOACs) are used cautiously; rivaroxaban and apixaban are reasonable, but interactions with enzyme inhibitors (e.g. azoles) and vomiting/diarrhoea limit use.
- Therapeutic anticoagulation is required for Trousseau syndrome; LMWH is the standard.[1]
Pregnant[1]
PDAC in pregnancy is exceptionally rare. MRI without gadolinium is the imaging of choice. Surgery is ideally deferred to the second trimester (or postpartum) if curative; chemotherapy is contraindicated in the first trimester. Most cases are diagnosed at an advanced stage, and prognosis is poor.[1]
Renal or hepatic impairment[1]
- Irinotecan is contraindicated in bilirubin >2 × ULN and requires dose reduction in significant renal impairment.
- Gemcitabine is renally cleared; dose reduction in eGFR <30 mL/min/1.73 m².
- FOLFIRINOX is generally avoided in significant organ failure.[1]
Evidence, Guidelines & Regional Differences[1]
NCCN (US, 2024)[1]
NCCN is the most widely cited guideline for PDAC. The 2024 update positions:[1]
- Resectable: surgery first, then adjuvant mFOLFIRINOX for 12 cycles (fit) or gemcitabine/capecitabine (less fit).
- Borderline resectable: neoadjuvant FOLFIRINOX or gem/nab for 4 to 6 months, restage, then surgery if response.
- Locally advanced: induction FOLFIRINOX or gem/nab, then chemoradiotherapy if response; surgery in highly selected.
- Metastatic, fit: FOLFIRINOX first-line; alternative gem/nab; olaparib maintenance in germline BRCA after platinum; pembrolizumab in MSI-H.
- Palliative: biliary stenting, coeliac plexus block, PERT, early palliative care referral.
- Germline testing for all patients; somatic NGS on the tumour to identify actionable alterations.[1]
ESMO (Europe, 2023 update)[1]
ESMO broadly follows NCCN with minor differences: emphasises total neoadjuvant therapy in BR-PC, recommends FOLFIRINOX as first-line in fit metastatic patients, and is more conservative on SBRT for locally advanced disease.[1]
NICE (UK, 2023)[1]
NICE NG85 (pancreatic cancer in adults) recommends:[1]
- Adjuvant mFOLFIRINOX as first option after resection in fit patients; gemcitabine + capecitabine for those who cannot tolerate FOLFIRINOX; single-agent gemcitabine for less fit.
- Neoadjuvant chemotherapy for borderline resectable disease (the All-Party Parliamentary Group on Pancreatic Cancer report of 2023 calls for universal access).
- FOLFIRINOX for fit metastatic patients.
- Routine germline testing is now offered through the NHS Genomic Medicine Service.
- EUS-FNA for tissue diagnosis before therapy.[1]
ASCO guidelines[1]
- Adjuvant mFOLFIRINOX for fit patients (strong recommendation).
- Germline testing for all PDAC patients.
- Routine assessment of sarcopenia and frailty in older patients before surgery.
- Early integration of palliative care.[1]
ICMR (India)[1]
The Indian Council of Medical Research guidelines (2023) recognise that:[1]
- Tropical (nutritional) pancreatitis is a distinct, geographically-confined precursor in southern India (Kerala, Karnataka, Tamil Nadu, Andhra Pradesh); patients present in their 30s to 50s with diabetes, steatorrhoea, and large ductal calculi; lifetime PDAC risk is high.
- Cost and access limit the use of FOLFIRINOX; gemcitabine-based regimens remain the practical standard in much of public-sector practice, with capecitabine added when feasible.
- Late presentation and limited access to high-volume surgical centres are the dominant barriers; centralisation to high-volume centres is recommended.
- Pancreatic enzyme replacement therapy is under-utilised; PERT is recommended in all PDAC patients with weight loss or steatorrhoea.
- Pain control is a major quality-of-life issue; coeliac plexus block availability is patchy; oral morphine is the mainstay.[1]
Japan / JSHP[1]
Japanese surgeons take a more aggressive surgical approach: extended lymphadenectomy, arterial resection and reconstruction in selected patients, and aggressive neoadjuvant chemoradiotherapy. The JASPAC-01 trial (Uesaka 2016, JAMA) showed adjuvant S-1 (an oral fluoropyrimidine) superior to gemcitabine after resection in Japanese patients (2-year OS 70% vs 53%), but S-1 is not registered for use in Europe or the US, and the results have not been replicated in Western populations.[1]
High-volume centres[1]
Whipple procedure mortality is significantly lower in high-volume centres (under 3%) than in low-volume centres (over 10%). Centralisation of pancreatic surgery to centres performing at least 20 to 30 Whipples per year is recommended by all major guidelines. The patient should be referred to a hepatobiliary multidisciplinary team (MDT) at a high-volume centre for all resectable and borderline-resectable cases.[1][5]
Key trials and what they changed[1]
- Conroy 2011 (PRODIGE 4/ACCORD 11) — FOLFIRINOX in metastatic PDAC: 11.1 vs 6.8 months. Changed practice to FOLFIRINOX first-line in fit patients.[7]
- Von Hoff 2013 (MPACT) — gemcitabine + nab-paclitaxel in metastatic PDAC: 8.5 vs 6.7 months. Established the doublet as an alternative first-line.[8]
- Conroy 2018 (PRODIGE 24/ACCORD 24) — adjuvant mFOLFIRINOX: 54.4 vs 35.0 months. Changed practice to mFOLFIRINOX adjuvant for fit patients.[9]
- Neoptolemos 2017 (ESPAC-4) — adjuvant gem-cap: 28.0 vs 25.5 months. Established the doublet adjuvant for less fit patients.[10]
- Golan 2019 (POLO) — olaparib maintenance in germline BRCA: PFS 7.4 vs 3.8 months. First precision-medicine indication in PDAC.[11]
- Murphy 2019 (JAMA Oncol) — total neoadjuvant FOLFIRINOX + losartan + CRT: 61% resection rate, 33-month median OS in resected patients. Best evidence for total neoadjuvant therapy.[12]
- Uesaka 2016 (JASPAC-01) — adjuvant S-1 vs gemcitabine in Japanese: 2-year OS 70% vs 53%. S-1 standard in Japan; not generalisable to Western populations.
Pancreatic cancer driver mutations — the cascade
KCpTS
earliest, G12D, RAS/MAPK, undruggable historically
cell-cycle brake removed
DNA-damage response lost
TGF-beta pathway lost; metastatic phenotype
Resectability categories — the four R's
RBLM
no vascular contact; upfront surgery + adjuvant mFOLFIRINOX
≤180° SMA / reconstructible vein; neoadjuvant then re-stage
>180° SMA encasement; chemo ± CRT; median OS 15-20 mo
FOLFIRINOX or gem/nab; median OS 8-11 mo treated
Exam Pearls[1]
- Painless obstructive jaundice + palpable non-tender gallbladder (Courvoisier's sign) = pancreatic head cancer until proven otherwise.[2]
- Courvoisier's law: jaundice + palpable gallbladder is unlikely to be gallstones (stones cause a contracted, fibrotic GB; cancer causes distension).[2]
- Smoking is the biggest modifiable risk factor. Others: chronic pancreatitis, diabetes, BRCA2, Peutz-Jeghers.[1]
- KRAS mutation in over 90% of PDAC. Cascade: KRAS → CDKN2A → TP53 → SMAD4. SMAD4 loss = metastatic.[5]
- Only 15 to 20% are resectable. Whipple for head tumours. Distal pancreatectomy + splenectomy for body/tail.[1]
- Whipple = pancreatoduodenectomy: remove head of pancreas, duodenum, distal CBD, gallbladder, sometimes distal stomach. Three anastomoses: pancreaticojejunostomy, hepaticojejunostomy, gastrojejunostomy.[1]
- mFOLFIRINOX adjuvant after resection (PRODIGE 24, 54 vs 35 mo OS). FOLFIRINOX or gem/nab for metastatic.[9][7][8]
- Whipple complications: pancreatic fistula (most feared, sentinel bleed), delayed gastric emptying, post-pancreatectomy haemorrhage.[1]
- 5-year survival under 10%. After R0 + mFOLFIRINOX: 30-40%. Fourth leading cause of cancer death.[1][5]
- Trousseau syndrome (migratory thrombophlebitis) is a paraneoplastic sign.[1]
- New-onset diabetes in older thin patient with weight loss → image the pancreas.[1]
- Olaparib maintenance for germline BRCA1/2-mutated metastatic PDAC (POLO trial, 7.4 vs 3.8 mo PFS).[11]
- CA 19-9: 5-10% are Lewis-negative and cannot produce it; it is for monitoring, not diagnosis.[2]
- Hypoenhancing mass on arterial phase = PDAC; hypervascular = pNET.[5]
- Sentinel bleed after Whipple = pseudoaneurysm; urgent angiography and embolisation.[1]
- Ampullary carcinoma has a much better prognosis than PDAC — do not lump them.[2]
- Germline testing is now recommended for ALL patients with PDAC.[3]
- Total neoadjuvant FOLFIRINOX in BR-PC and LA-PC; aim for R0 resection in 60-85%.[12]
- Autoimmune pancreatitis is a steroid-responsive mimic — a missed diagnosis costs a Whipple.[2]
- Centralise Whipple to high-volume centres (mortality <3% vs >10% at low-volume).[1]
- Vaccinate splenectomy patients against encapsulated organisms.[5]
- Capecitabine dosing: 1,000 mg/m² PO twice daily days 1-14, every 21 days, 6 cycles.[10]
- Gemcitabine + nab-paclitaxel: gem 1,000 mg/m² + nab 125 mg/m² days 1, 8, 15, q28d × 6 cycles.[8]
- FOLFIRINOX: oxaliplatin 85 + irinotecan 180 + leucovorin 400 + 5-FU bolus 400 + 5-FU 2,400 mg/m² over 46 h, every 14 days.[7]
- Olaparib: 300 mg PO BD (capsule) or 150 mg BD (tablet) until progression.[11]
References
- [1]Park W, Chawla A, O'Reilly EM Pancreatic Cancer: A Review. JAMA, 2021.PMID 34547082
- [2]McGuigan A, Kelly P, Turkington RC, et al. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World Journal of Gastroenterology, 2018.PMID 30487695
- [3]Zhao Z, Liu W Pancreatic Cancer: A Review of Risk Factors, Diagnosis, and Treatment. Technology in Cancer Research and Treatment, 2020.PMID 33357065
- [4]Kwon W, Heo JS, Han IW, et al. Features of T1 pancreatic cancer and validation of the eighth edition AJCC staging system definition using a Korean-Japanese joint cohort and the SEER database. J Hepatobiliary Pancreat Sci, 2023.PMID 36734142
- [5]Kamisawa T, Wood LD, Itoi T, Takaori K Pancreatic cancer. Lancet, 2016.PMID 26830752
- [6]Ryan DP, Hong TS, Bardeesy N Pancreatic adenocarcinoma. N Engl J Med, 2014.PMID 25207767
- [7]Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med, 2011.PMID 21561347
- [8]Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med, 2013.PMID 24131140
- [9]Conroy T, Hammel P, Hebbar M, et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N Engl J Med, 2018.PMID 30575490
- [10]Neoptolemos JP, Palmer DH, Ghaneh P, et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4). Lancet, 2017.PMID 28129987
- [11]Golan T, Hammel P, Reni M, et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N Engl J Med, 2019.PMID 31157963
- [12]Murphy JE, Wo JY, Ryan DP, et al. Total Neoadjuvant Therapy With FOLFIRINOX in Combination With Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer. JAMA Oncol, 2019.PMID 31145418
- [13]Rawla P, Sunkara T, Gaduputi V Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J Oncol, 2019.PMID 30834048
- [14]Wang Y, Cuggia A, Sapisochin G, et al. Is Biannual Surveillance for Pancreatic Cancer Sufficient in Individuals With Genetic Syndromes or Familial Pancreatic Cancer? J Natl Compr Canc Netw, 2022.PMID 35714671