Haematology · General Medicine
Lymphoma (Hodgkin & Non-Hodgkin)
Also known as Hodgkin lymphoma · Non-Hodgkin lymphoma · Lymphosarcoma
Lymphoma is a clonal malignancy of lymphocytes arising in lymphoid tissue, divided by the Reed-Sternberg cell into Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL). HL spreads contiguously, is driven by the CD15+/CD30+ Reed-Sternberg cell, presents with cervical/supraclavicular nodes and a mediastinal mass, and is treated with ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine). NHL is heterogeneous — follicular (indolent, t(14;18) BCL2), DLBCL (commonest aggressive, R-CHOP), mantle cell (t(11;14) cyclin D1, CD5+), Burkitt (t(8;14) MYC, fastest human tumour, Ki67 ~100 percent), MALT (H. pylori, gastric). Diagnosis = EXCISIONAL biopsy (not FNA — need architecture). Ann Arbor staging (Cotswolds) with B symptoms (fever over 38C, drenching night sweats, weight loss over 10 percent in 6 months). PET-CT for staging and Deauville 5-point scale for response. IPS (HL) and IPI (NHL) for prognosis. Emergencies: SVC syndrome, mediastinal-mass airway, tumour lysis, cord compression.
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags
Overview & Definition
Lymphoma is a clonal malignancy of lymphocytes (B cells in the great majority, less often T or NK cells) that arises in lymphoid tissue — lymph nodes, spleen, bone marrow, Waldeyer ring, mucosa-associated lymphoid tissue (MALT) — and may also present at extranodal sites. It is distinguished from leukaemia by its primary origin in solid lymphoid tissue (rather than the bone marrow and peripheral blood), and from reactive (infective) lymphadenopathy by its clonal, destructive, monotomous proliferation that effaces normal node architecture and persists.[1]
The single histological criterion that splits the entire family is the Reed-Sternberg cell: a large, often binucleate cell with owl-eye nucleoli surrounded by a reactive inflammatory background. If Reed-Sternberg cells are present, the disease is Hodgkin lymphoma (HL); if absent, it is non-Hodgkin lymphoma (NHL). This distinction is not cosmetic — it governs pattern of spread (contiguous in HL, disseminated in NHL), staging emphasis, the diagnostic cell, the first-line regimen (ABVD vs R-CHOP) and prognosis.[1][12]
Lymphoma is the commonest haematological malignancy. NHL vastly outnumbers HL (roughly 90 percent versus 10 percent of all lymphoma), is the commonest blood cancer overall, and its incidence rises with age; HL has a characteristic bimodal age distribution and is one of the most curable human cancers.[1]
Classification
Lymphoma is classified by histology (Hodgkin vs NHL), by cell of origin (B, T, NK), and by clinical behaviour (indolent, aggressive, highly aggressive). The WHO classification integrates morphology, immunophenotype, cytogenetics and molecular lesions — so subtyping is impossible on morphology alone and demands excisional biopsy with immunohistochemistry (IHC), flow cytometry and cytogenetics/FISH.[1][2]
Hodgkin lymphoma
Classical HL (cHL) — ~95 percent
- Reed-Sternberg cells present; CD15+ CD30+, CD20 negative or weak
- Reactive background of lymphocytes, eosinophils, fibrosis
- Nodular sclerosis (commonest, ~60-70 percent): mediastinal mass in young women, lacunar cells, fibrous bands
- Mixed cellularity (~20-25 percent): older patients, EBV-associated, more B symptoms
- Lymphocyte-rich: good prognosis
- Lymphocyte-depleted (rarest): older/HIV patients, worst prognosis
Nodular lymphocyte-predominant HL (NLPHL) — ~5 percent
- Popcorn (LP) cells, NOT classic Reed-Sternberg
- CD20+ CD45+, CD15 negative CD30 negative
- Grows slowly, isolated node, male predominance
- Treated with rituximab and/or involved-site radiotherapy
- Can transform to DLBCL (risk ~10 percent)
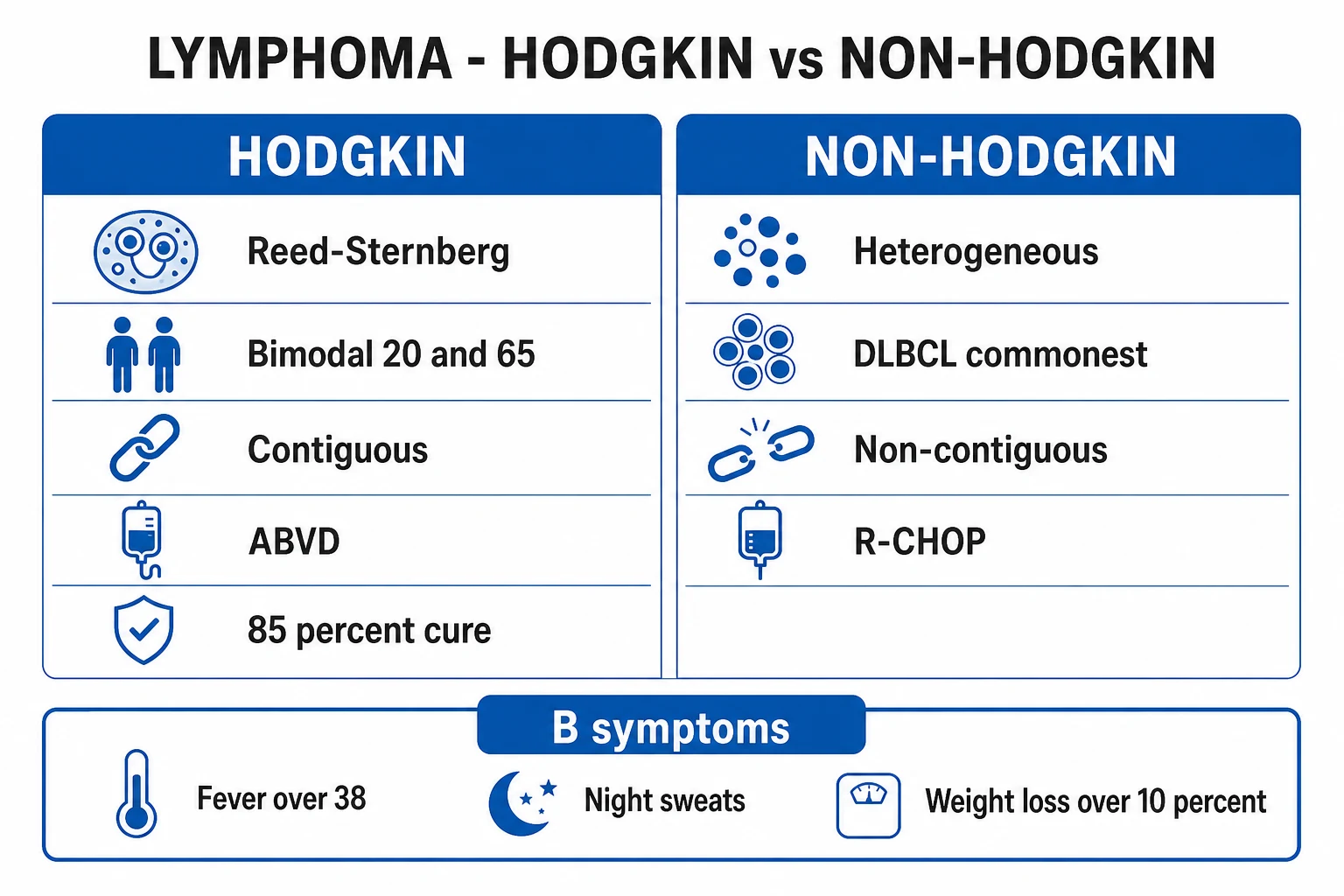
Non-Hodgkin lymphoma by clinical behaviour
Indolent (low-grade)
- Follicular lymphoma — commonest indolent NHL; t(14;18) BCL2; CD10+; slow, incurable, watch-and-wait or rituximab
- Marginal zone / MALT — H. pylori-driven gastric MALT (t(11;18) API2-MALT1); can be cured by eradication
- Lymphoplasmacytic (Waldenstrom) — MYD88 L265P, IgM paraprotein, hyperviscosity
- Small lymphocytic (SLL/CLL) — CD5+ CD23+
- Often widely disseminated at diagnosis; long natural history
Aggressive
- Diffuse large B-cell lymphoma (DLBCL) — commonest aggressive NHL and commonest NHL overall; R-CHOP; GCB subtype better than ABC
- Mantle cell lymphoma — t(11;14) cyclin D1 overexpression, CD5+ CD20+; aggressive course despite indolent appearance
- Often present as a rapidly enlarging mass; treated with curative intent
Highly aggressive
- Burkitt lymphoma — t(8;14) c-MYC; fastest-growing human tumour, Ki67 ~100 percent; endemic African jaw mass vs sporadic abdominal mass; intensive chemo + CNS prophylaxis
- Lymphoblastic lymphoma (B or T) — arises from lymphoblasts, often a mediastinal mass (T-cell), overlaps with acute leukaemia
- Very high tumour-lysis risk; treat as a medical emergency within hours
Cell of origin in DLBCL matters prognostically: the germinal-centre B-cell (GCB) subtype has a better outcome with R-CHOP than the activated B-cell (ABC) subtype, which relies on chronic active NF-κB signalling and relapses more often. T-cell NHLs (peripheral T-cell lymphoma NOS, anaplastic large cell, angioimmunoblastic) are rarer, biologically heterogeneous and generally carry a worse prognosis than B-cell NHL.[2]
Epidemiology & Risk Factors
Hodgkin lymphoma shows a characteristic bimodal age distribution: a first peak in young adults (about 20 years, nodular sclerosis) and a second peak in adults over 55 (mixed cellularity). Non-Hodgkin lymphoma incidence rises steadily with age (median about 65) and is the commonest haematological malignancy overall; DLBCL is the commonest aggressive subtype and follicular lymphoma the commonest indolent subtype.[1][7]
Infection-associated lymphomas are a high-yield theme — the examiner wants the bug-to-subtype pairings: [1]
Pathogen
- EBV (Epstein-Barr virus)
- Helicobacter pylori
- HTLV-1
- HHV-8 (human herpesvirus 8)
- Hepatitis C virus
- Borrelia burgdorferi / Chlamydia psittaci
Lymphoma linked
- Classical HL (especially mixed cellularity, HIV); Burkitt (endemic); DLBCL in HIV; post-transplant
- Gastric MALT lymphoma (and some DLBCL)
- Adult T-cell leukaemia/lymphoma (ATLL)
- Primary effusion lymphoma (and multicentric Castleman, in HIV)
- Marginal zone / lymphoplasmacytic lymphoma, mixed cryoglobulinaemia DLBCL
- Marginal zone / MALT (skin, ocular adnexa)
Other risk factors: immunosuppression — HIV (drives aggressive B-cell NHL and Burkitt), post-solid-organ or stem-cell transplant (post-transplant lymphoproliferative disorder, PTLD, often EBV-related), congenital immunodeficiency; autoimmune / chronic inflammatory disease — Sjogren syndrome and Hashimoto thyroiditis (MALT), rheumatoid arthritis and coeliac disease (enteropathy-associated T-cell lymphoma); prior chemotherapy or radiotherapy (secondary NHL/HL and myelodysplasia); chemical exposures (pesticides, herbicides such as Agent Orange, organic solvents, hair dyes); and family history. Male sex and Caucasian ancestry modestly increase HL risk.[1]
Pathophysiology
The malignant cell of classical Hodgkin lymphoma is the Reed-Sternberg cell — a large (20 to 50 micrometre), often binucleate or multinucleate giant cell with prominent owl-eye (eosinophilic inclusion-like) nucleoli and abundant amphophilic cytoplasm. It derives from a germinal-centre B cell that has undergone a crippled immunoglobulin gene rearrangement and would normally be deleted by apoptosis, but is rescued by constitutive NF-κB, JAK/STAT3 and PI3K/AKT signalling, often driven by EBV-encoded LMP1 (which mimics CD40 signalling) and amplified by amplification of 9p24.1 (the PD-L1/PD-L2 locus, the basis for PD-1 inhibitor activity). The Reed-Sternberg cell is rare within the tumour — it secretes cytokines (IL-5, IL-13, eotaxin, TGF-beta) that recruit the characteristic reactive background of T lymphocytes (rosetting), eosinophils, plasma cells, histiocytes and fibrosis. This background is what you actually see under the microscope; the neoplastic cells are a minority.[1][12]
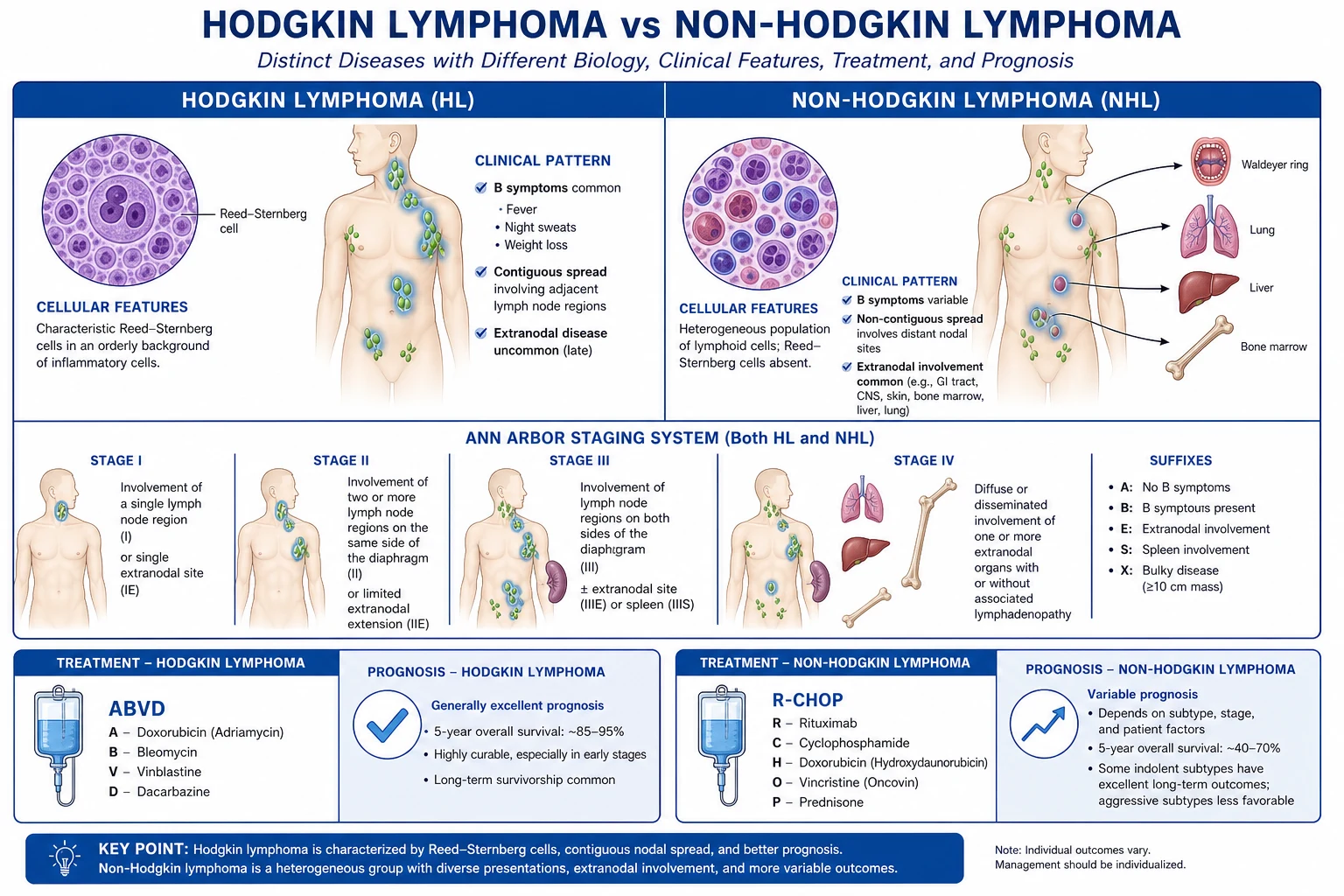
Pattern of spread distinguishes HL from NHL. Hodgkin lymphoma spreads centiguously and orderly from one node group to the next (cervical → supraclavicular → mediastinal), making staging the key determinant of therapy and radiotherapy relevant. NHL spreads non-contiguously and haematogenously, often skipping node groups and seeding extranodal sites (GI tract, marrow, CNS, skin, testis, Waldeyer ring), so it is systemic from early on and chemotherapy-centred.[1]
The defining molecular lesions of NHL — each translocation fuses a proto-oncogene to the immunoglobulin heavy-chain locus on chromosome 14, driving its constitutive expression: [1]
Subtype
- Follicular
- Mantle cell
- Burkitt
- Gastric MALT
- Lymphoplasmacytic (Waldenstrom)
- DLBCL (subset)
Molecular lesion
- t(14;18) — BCL2 overexpression (anti-apoptotic)
- t(11;14) — cyclin D1 overexpression (CCND1)
- t(8;14) — c-MYC overexpression (proliferation)
- t(11;18)(q21;q21) — API2-MALT1 fusion
- MYD88 L265P (and CXCR4) mutations
- BCL6 rearrangement, MYC/BCL2 'double-hit' or 'double-expressor'
Mechanism of rituximab. Rituximab is a chimeric monoclonal antibody against CD20 (a transmembrane protein on pre-B to mature B cells, absent on stem cells and plasma cells). Binding triggers complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC) and direct apoptosis of CD20+ B cells. Adding it to chemotherapy (R-CHOP) transformed DLBCL from a 40 percent cure to a roughly 60 to 70 percent cure disease because virtually all B-cell NHL express CD20; plasma cells survive (so immunoglobulin is partly preserved) and haematopoietic stem cells are spared (so count recovery is intact).[6][11]
Tumour lysis syndrome (TLS). Highly proliferative tumours — especially Burkitt (doubling time about 24 to 48 hours, Ki67 ~100 percent) and high-burden aggressive NHL — release massive amounts of intracellular contents on therapy: potassium (hyperkalaemia), phosphate (hyperphosphataemia → secondary hypocalcaemia), and nucleic acids (metabolised to uric acid, causing acute urate nephropathy and AKI). The risk is highest in the first 24 to 72 hours of treatment, which is why Burkitt is treated as a haemato-oncology emergency within hours of diagnosis with prophylaxis already running.[8]
Clinical Presentation
The classic presentation of lymphoma is painless, rubbery lymphadenopathy, most often cervical (followed by supraclavicular, axillary, inguinal), that persists beyond 4 to 6 weeks and progressively enlarges. Nodes are typically non-tender, firm and rubbery, and may become matted. Up to a third of patients have B symptoms, which must be elicited and recorded verbatim because they alter stage and prognosis.[1]
Hodgkin lymphoma patterns: cervical/supraclavicular nodes with a mediastinal mass (nodular sclerosis in young women) producing cough, dyspnoea or SVC syndrome; alcohol-induced pain in involved nodes (rare but classical); and generalised pruritus. Spread is centiguous.[1]
Non-Hodgkin lymphoma patterns are extranodal-focussed:[2]
- Gastrointestinal tract (commonest extranodal site) — abdominal mass, pain, bleeding, obstruction or intussusception (ileocaecal Burkitt).
- Bone marrow — cytopenias, fatigue, infection.
- Central nervous system — primary CNS lymphoma (periventricular, EBV-related in HIV), cranial neuropathy, leptomeningeal disease.
- Skin — mycosis fungoides and its leukaemic variant Sezary syndrome (cutaneous T-cell lymphoma with Pautrier microabscesses).
- Testis — primary testicular NHL (older men, relapses in CNS and contralateral testis).
- Waldeyer ring — tonsillar/nasopharyngeal mass, often associated with GI involvement (rule out both).
- Bone — lytic lesion with pain. [1]
Burkitt lymphoma has two faces: endemic (African) — a jaw or facial (orbit) mass in a child, strongly EBV-associated; and sporadic — an abdominal/ileocaecal mass in a child or young adult, presenting with obstruction, intussusception and florid tumour lysis.[8]
Atypical presentations: in the elderly — fatigue, weight loss, B symptoms may be muted, and a lower threshold to biopsy is warranted; in the immunocompromised (HIV) — aggressive DLBCL, Burkitt or primary effusion lymphoma, often extranodal and advanced; in pregnancy — cervical/supraclavicular node or mediastinal mass discovered incidentally, requiring staging adapted to the fetus.[1]
Differential Diagnosis
A painless node has a wide differential. The task is to distinguish lymphoma from its benign and malignant mimics using tempo, character, distribution and (definitively) histology.[1]
Reactive / infective
- Tender, mobile, often cervical with viral/bacterial URTI; resolves in 2-3 weeks
- EBV (infectious mononucleosis): widespread nodes, atypical lymphocytes, heterophile antibody
- Toxoplasmosis, CMV, HIV seroconversion: generalised nodes
- Cellulitis-related regional nodes are hot, tender, erythematous
Tuberculous lymphadenitis (scrofula)
- Painless matted cervical nodes, often with overlying skin discolouration/sinus
- Constitutional symptoms (fever, night sweats) overlap with B symptoms
- Distinguish by AFB/TB-PCR/culture of aspirate or biopsy; caseating granulomas
- May coexist — exclude TB before immunosuppressive chemo
Metastatic carcinoma (Virchow node)
- Hard (stony), fixed, left supraclavicular node drains the abdomen (gastric, pancreatic, testicular)
- Primary usually evident (breast, lung, thyroid, GI, head-and-neck squamous)
- Cytology of FNA often sufficient; architecture less critical than for lymphoma
Chronic lymphocytic leukaemia / SLL
- Older adult, painless generalised nodes with absolute lymphocytosis on blood film
- CD5+ CD23+ clonal B cells; marrow involvement
- Overlaps with lymphoma — distinguished by leukaemic phase and immunophenotype
Sarcoidosis / autoimmune / drug
- Bilateral hilar lymphadenopathy, erythema nodosum, uveitis (sarcoid)
- Phenytoin can cause a pseudolymphoma; HIV must be excluded in generalised nodes
- Diagnosis still ultimately rests on EXCISIONAL biopsy with IHC
Generalised lymphadenopathy (cervical, axillary, inguinal simultaneously) should trigger thought of HIV, EBV, sarcoidosis, autoimmune disease (SLE, rheumatoid), and drug reaction — and these must be excluded before committing to a lymphoma diagnosis.[1]
Clinical & Bedside Assessment
A systematic node examination covers all peripheral groups in a fixed order — submental, submandibular, cervical (anterior and posterior chain), supraclavicular, occipital, pre/post-auricular, axillary, epitrochlear, inguinal, and popliteal. Describe each node by site, size, consistency (rubbery in lymphoma, hard in carcinoma, soft in reactive), tenderness, mobility and matting. A lymphomatous node is classically rubbery, non-tender, mobile early then matted. Palpate the supraclavicular fossa carefully — a node here is always abnormal (drains the thoracic duct and abdomen).[1]
Beyond the nodes, assess for hepatosplenomegaly (involvement), tonsillar and Waldeyer ring enlargement, skin lesions (mycosis fungoides), and signs of bone marrow failure — pallor, purpura, infection. Examine for superior vena cava syndrome (facial plethora, jugular distension, arm swelling), mediastinal-mass airway compromise (stridor, orthopnoea) and spinal cord compression (back pain, sensory level, sphincter dysfunction).[1]
Document two things at the bedside that drive staging and prognosis: performance status (ECOG) and the presence of B symptoms.[2]
Lymphoma — high-yield numbers
Investigations
The diagnostic standard is an EXCISIONAL lymph node biopsy — removing an entire, representative node intact so that architecture, immunohistochemistry, flow cytometry, cytogenetics and molecular studies can all be performed. Fine-needle aspiration (FNA) is inadequate for a primary lymphoma diagnosis: it destroys architecture, cannot subtype reliably, and yields too little material for the full workup. Core biopsy is acceptable when a node is surgically inaccessible, but excisional biopsy remains gold-standard.[1][2]
Histological workup of the excised node: [1]
- Morphology (H&E) — architecture (nodular vs diffuse), cell size, Reed-Sternberg cells, mitotic/Ki67 rate.
- Immunohistochemistry (IHC) — the decisive discriminator: classical HL is CD15+ CD30+, CD20 negative or weak; NLPHL is CD20+ CD45+, CD15− CD30− (popcorn cells); B-cell NHL is CD20+, CD19+, CD79a+, with further markers (CD10, BCL6, BCL2, CD5, cyclin D1); T-cell NHL is CD3+, CD4/CD8; Burkitt is CD20+, CD10+, BCL6+, BCL2 negative, Ki67 ~100 percent.
- Flow cytometry on a fresh fragment — defines clonality (light-chain restriction) and immunophenotype.
- Cytogenetics / FISH — the translocations: t(8;14) Burkitt, t(14;18) follicular, t(11;14) mantle cell, t(11;18) MALT.
- Molecular — clonal immunoglobulin / T-cell receptor gene rearrangement, MYD88 mutation (lymphoplasmacytic). [1]
Staging — Ann Arbor staging with Cotswolds modifications (reproduced verbatim):[2]
PET-CT (FDG) is the modern staging and response-assessment standard for FDG-avid lymphomas (virtually all HL, DLBCL, follicular, Burkitt, mantle cell). Lugano response criteria integrate PET, with the Deauville 5-point scale:[2]
Deauville
- 1 — no uptake
- 2 — uptake at or below mediastinum
- 3 — uptake above mediastinum but at or below liver
- 4 — uptake moderately above liver
- 5 — uptake markedly above liver and/or new lesions
Response meaning
- Scores 1, 2, 3 = complete metabolic response (CMR) — PET-negative
- Score 4 or 5 = residual metabolic disease (partial/no response)
- Score X — new areas of uptake unlikely to be lymphoma (e.g. infection)
- Drives PET-adapted therapy: stop or de-escalate if CMR, escalate if not
Baseline bloods and supportive tests: full blood count and film (cytopenias from marrow involvement), ESR/CRP, U&E and creatinine, LFTs, LDH (tumour burden and a component of IPI), beta-2 microglobulin (prognosis), hepatitis B (surface antigen and core antibody — rituximab can reactivate HBV), hepatitis C and HIV serology, bone marrow aspirate and trephine (when blood counts abnormal or PET/bone lesions suggest involvement — increasingly omitted for early-stage PET-negative HL/DLBCL), ECG and echocardiogram before anthracyclines (doxorubicin is cardiotoxic), and pulmonary function tests before bleomycin (pulmonary fibrosis). Pregnancy test in women of childbearing age. Sperm/egg cryopreservation is offered before treatment because ABVD/R-CHOP cause infertility.[1]
International Prognostic Index (IPI) — NHL (reproduced verbatim)
One point each for:[4]
- Age 60 years or older
- Performance status (ECOG) 2 or higher
- Stage III or IV (Ann Arbor)
- Extranodal involvement in 2 or more sites
- Serum LDH elevated (above normal) [1]
Risk groups: low (0 to 1, score 0-1) — 5-year survival about 73 percent; low-intermediate (2) — about 51 percent; high-intermediate (3) — about 43 percent; high (4 to 5) — about 26 percent. The revised IPI (R-IPI) for the R-CHOP era collapses these into very good (0, 4-year PFS 94 percent), good (1 to 2, 80 percent) and poor (3 to 5, 53 percent).[4][5]
Hasenclever International Prognostic Score (IPS) — Hodgkin lymphoma (reproduced verbatim)
One point each for:[3]
- Serum albumin below 40 g/L
- Haemoglobin below 105 g/L
- Male sex
- Age 45 years or older
- Stage IV disease
- Leucocytosis (white cell count at least 15 times 10^9/L)
- Lymphocytopenia (lymphocyte count below 0.6 times 10^9/L, or below 8 percent of the WCC, or both) [1]
A score of 0 carries the best prognosis (rate of freedom from progression at 5 years about 84 percent); the rate falls steadily to about 42 percent for a score of 5 or more.[3]
Management — Resuscitation
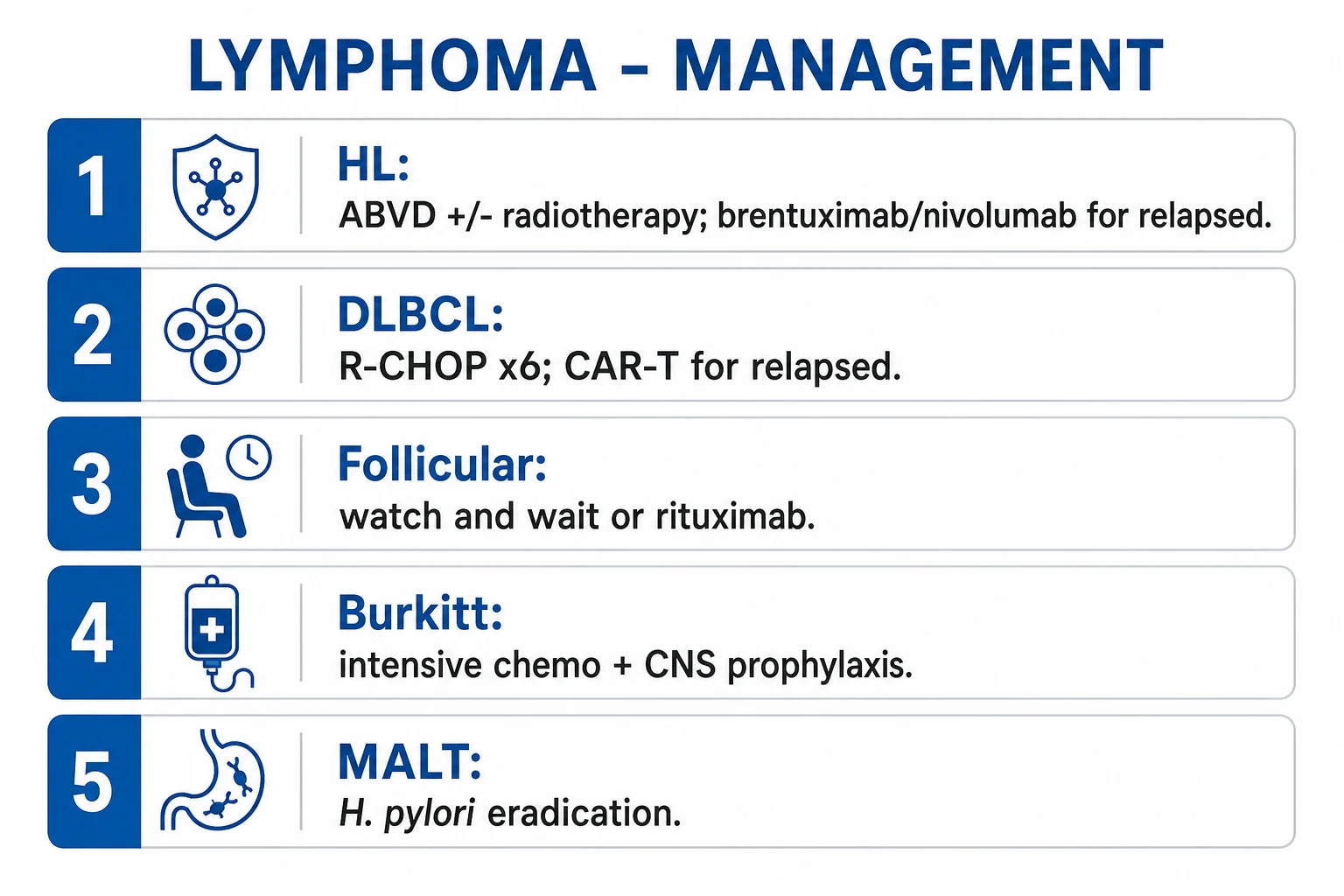
Treat the presenting oncological emergencies first, then start systemic therapy. Four emergencies recur:[1]
Lymphoma emergencies at presentation
MASS
stridor, orthopnoea — sit upright, avoid sedation, secure airway with anaesthetics, give steroids, defer general anaesthetic biopsy to a controlled setting
facial/arm swelling, venous distension — elevate head of bed, oxygen, IV dexamethasone, urgent haemato-oncology; often resolves with chemo
back pain + neuro deficit — IV dexamethasone 10 mg then 16 mg/day, urgent MRI whole spine, radiotherapy/neurosurgery
Burkitt/high-burden NHL — IV hydration, rasburicase/allopurinol, monitor K+/phosphate/Ca2+/urate/renal
Tumour lysis syndrome prophylaxis and treatment. Start before the first chemotherapy dose in any high-risk patient (Burkitt, high-burden aggressive NHL, LDH markedly raised, bulky disease):[8]
- Aggressive intravenous hydration — isotonic saline 3 L/m2 per 24 hours (or 2 to 3 L/m2/day), aiming for urine output at least 100 mL/m2/h; alkalinisation is no longer routine.
- Rasburicase (recombinant urate oxidase) 0.15 to 0.2 mg/kg IV once daily — for high-risk or established hyperuricosaemia; contraindicated in G6PD deficiency (haemolysis).
- Allopurinol 300 mg orally daily (lower dose in renal impairment) — for moderate-risk patients.
- Monitor potassium, phosphate, calcium, urate, creatinine every 4 to 6 hours for the first 24 to 72 hours; treat hyperkalaemia (calcium gluconate, insulin-dextrose, potassium binders) and hyperphosphataemia (phosphate binders) as they arise. [1]
Febrile neutropenia from chemotherapy is treated with empirical broad-spectrum IV antibiotic within one hour of presentation (per local policy — typically an anti-pseudomonal beta-lactam such as piperacillin-tazobactam 4.5 g IV every 6 to 8 hours or ceftazidime), blood cultures first, and reassessment for source.[1]
Management — Definitive & Stepwise
Therapy is histology-, stage- and risk-adapted. The regimens below are the MBBS high-yield core — know the acronym expansion, the agents, and at least the key doses.[1][2]
Hodgkin lymphoma — ABVD
Drug
- A — Doxorubicin (Adriamycin)
- B — Bleomycin
- V — Vinblastine
- D — Dacarbazine (DTIC)
Dose / route / timing
- 25 mg/m2 IV days 1 and 15
- 10 units/m2 IV days 1 and 15
- 6 mg/m2 IV days 1 and 15
- 375 to 650 mg/m2 IV days 1 and 15
ABVD is given every 28 days for 2 to 6 cycles depending on stage and PET response. PET-adapted therapy (the modern standard): a positive interim PET drives escalation to escalated BEACOPP; a negative interim PET allows de-escalation (omit bleomycin to avoid lung toxicity). Limited (early) stage disease typically receives 2 cycles of ABVD plus involved-site radiotherapy (ISRT); advanced disease receives 6 cycles of ABVD, or escalated BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, prednisone) for unfavourable advanced disease in some centres. Brentuximab vedotin (anti-CD30 antibody-drug conjugate, 1.8 mg/kg IV every 3 weeks) with doxorubicin/vinblastine/dacarbazine is a bleomycin-sparing frontline alternative. Nivolumab (anti-PD-1) exploits the 9p24.1 PD-L1 amplification.[1][11][12]
Diffuse large B-cell lymphoma — R-CHOP
Drug
- R — Rituximab
- C — Cyclophosphamide
- H — Doxorubicin (Hydroxydaunorubicin)
- O — Vincristine (Oncovin)
- P — Prednisone
Dose / route / timing
- 375 mg/m2 IV day 1 (anti-CD20)
- 750 mg/m2 IV day 1
- 50 mg/m2 IV day 1
- 1.4 mg/m2 IV day 1 (capped at 2 mg total)
- 100 mg orally daily days 1 to 5
R-CHOP is given every 21 days for 6 cycles (standard); limited-stage non-bulky DLBCL may receive 3 cycles of R-CHOP plus involved-site radiotherapy. Adding rituximab to CHOP (established by the GELA LNH-98.5 and MInT trials) improved complete response and overall survival in all age groups and is now non-negotiable first-line. Six cycles of intrathecal methotrexate CNS prophylaxis is added for high-CNS-risk DLBCL (testicular, renal/adrenal, paranasal sinus, bone marrow, high-IPI, double-hit). DA-EPOCH-R (dose-adjusted etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, rituximab) is an alternative, especially for double-hit / MYC-rearranged DLBCL.[5][6]
Follicular lymphoma — watch-and-wait or rituximab
Follicular lymphoma is indolent and, in advanced stage, incurable but with a long natural history (median survival now well over 10 to 15 years). Asymptomatic advanced-stage disease can be managed with watch-and-wait (active surveillance) without survival disadvantage — therapy starts at symptoms, cytopenias, organ compromise, bulky disease or transformation. When treatment is needed: rituximab alone (375 mg/m2 weekly for 4 doses) for low-burden, or rituximab plus chemotherapy — R-CHOP, R-CVP (cyclophosphamide, vincristine, prednisone) or BR (bendamustine, rituximab) — followed by rituximab maintenance every 2 months for 2 years. Watch for histological transformation to DLBCL (rising LDH, rapid nodal growth) — treated as aggressive NHL.[7]
Burkitt lymphoma — intensive short-course chemotherapy
Burkitt is the fastest-growing human tumour (doubling time about 24 to 48 hours) and is treated as an emergency with intensive, short-course chemotherapy that includes CNS prophylaxis (intrathecal methotrexate/cytarabine) and tumour-lysis prophylaxis from the first cycle. Regimens include CODOX-M/IVAC (cyclophosphamide, vincristine, doxorubicin, high-dose methotrexate / ifosfamide, etoposide, high-dose cytarabine) and DA-EPOCH-R. With modern intensive therapy, cure rates exceed 80 to 90 percent in children and adults. Low-risk localised disease may need fewer cycles.[8]
Mantle cell lymphoma
Mantle cell lymphoma carries t(11;14) with cyclin D1 overexpression, is CD5+ CD20+ and has an aggressive (though sometimes clinically indolent-looking) course. Fit younger patients receive intensive induction (e.g. R-CHOP or NORDIC/alternating regimens) followed by consolidation with autologous stem-cell transplant and rituximab maintenance. Older/frail patients receive BR with rituximab maintenance. Targeted agents — bortezomib (proteasome inhibitor), ibrutinib/acalabrutinib (BTK inhibitors) and lenalidomide — are effective in relapse.[11]
Gastric MALT lymphoma — H. pylori eradication
For early-stage, H. pylori-positive gastric MALT lymphoma, H. pylori eradication therapy can achieve complete durable remission and cure — the clearest example of an infection-driven malignancy reversed by treating the infection. Eradication uses a triple/quadruple regimen (e.g. proton-pump inhibitor + amoxicillin + clarithromycin, with metronidazole replacing amoxicillin in penicillin-allergic patients) for 14 days, followed by endoscopic surveillance. The t(11;18) API2-MALT1 translocation predicts poor response to eradication and the need for radiotherapy/chemotherapy. H. pylori-negative or resistant disease receives involved-site radiotherapy or rituximab.[9]
Relapsed and refractory disease
Hodgkin lymphoma
- Salvage chemo then autologous stem-cell transplant (ASCT) — gold standard for chemo-sensitive relapse
- Brentuximab vedotin (anti-CD30 ADC) before and after ASCT
- Nivolumab / pembrolizumab (anti-PD-1) — exploits 9p24.1 PD-L1 amplification
- Allogeneic SCT for second relapse
DLBCL / aggressive NHL
- Salvage chemo then autologous SCT for chemo-sensitive relapse
- CAR-T cell therapy: axicabtagene ciloleucel, tisagenlecleucel, lisocabtagene maraleucel (anti-CD19)
- Polatuzumab vedotin (anti-CD79b ADC) + bendamustine + rituximab
- Tafasitamab (anti-CD19), loncastuximab (anti-CD19), bispecifics (glofitamab, epcoritamab)
Follicular / indolent
- Rituximab ± chemo re-treatment, lenalidomide, obinutuzumab
- Autologous SCT or allogeneic SCT for multiply-relapsed disease
- CAR-T (axi-cel, tisa-cel) approved for relapsed FL
Specific Subtypes & Scenarios
- Nodular sclerosis classical HL — the commonest HL subtype, young women, mediastinal mass, lacunar cells and fibrous bands; ABVD with excellent cure rates.[1]
- NLPHL — popcorn cells, CD20+ CD15− CD30−; slow-growing, isolated node; treated with rituximab and/or involved-site radiotherapy; watch for transformation to DLBCL.[1]
- DLBCL — commonest aggressive NHL and commonest NHL overall; R-CHOP; GCB subtype better than ABC; double-hit (MYC + BCL2/BCL6 rearrangements) and double-expressor (high MYC + BCL2 protein) fare worse and need DA-EPOCH-R.[5]
- Follicular lymphoma — t(14;18) BCL2, indolent, incurable but long survival; watch-and-wait or rituximab ± chemotherapy.[7]
- Mantle cell lymphoma — t(11;14) cyclin D1, CD5+; aggressive course; R-CHOP + maintenance rituximab, intensive regimens + ASCT, or targeted (BTK inhibitors, bortezomib).[11]
- Burkitt lymphoma — t(8;14) MYC, Ki67 ~100 percent, fastest-growing human tumour; endemic African jaw mass (EBV) vs sporadic abdominal mass; intensive chemo + CNS prophylaxis.[8]
- Gastric MALT lymphoma — H. pylori association; eradication therapy for early-stage H. pylori-positive disease.[9]
- Primary CNS lymphoma — typically diffuse large B-cell, periventricular, strongly EBV-associated in HIV; treated with high-dose methotrexate-based chemotherapy (avoid radiotherapy first-line in older patients because of neurotoxicity); iritinib (MYD88) in some.[10]
- Primary mediastinal large B-cell lymphoma — young women, mediastinal mass, mimics HL; treated with DA-EPOCH-R or R-CHOP.
- Mycosis fungoides / Sezary syndrome — cutaneous T-cell lymphoma; Pautrier microabscesses, epidermotropism; skin-directed therapy then systemic.[2]
Complications & Pitfalls
Treatment complications are themselves heavily examined:[1][11]
- Bleomycin lung toxicity — pneumonitis and pulmonary fibrosis; a reason for PET-adapted de-escalation and pre-treatment lung function tests.
- Anthracycline (doxorubicin) cardiotoxicity — dose-dependent congestive heart failure; baseline echo and ECG.
- Febrile neutropenia — empirical IV antibiotic within one hour.
- Infertility — offer sperm/egg cryopreservation before ABVD/R-CHOP.
- Secondary malignancies in HL survivors — breast cancer (from chest radiotherapy), lung cancer, acute leukaemia/MDS (from alkylators/radiotherapy), non-melanoma skin cancer.
- Cardiovascular disease — accelerated atherosclerosis and valve disease from mediastinal radiotherapy, a leading cause of late death in HL survivors.
- Thyroid dysfunction — hypothyroidism after neck radiotherapy.
- Rituximab — hepatitis B reactivation (screen HBsAg and anti-HBc; antiviral prophylaxis such as entecavir if positive), infusion reactions, late neutropenia.
- Brentuximab vedotin — peripheral neuropathy; nivolumab — immune-related adverse effects.
- CAR-T — cytokine release syndrome and immune-effector-cell neurotoxicity syndrome (ICANS). [1]
Classic diagnostic pitfalls: relying on FNA instead of excisional biopsy; missing HIV (a fundamental co-pathology); omitting PET-CT for staging/response; under-treating Burkitt or failing to prophylax tumour lysis; not screening for hepatitis B before rituximab; forgetting CNS prophylaxis in high-risk DLBCL; and failing to plan fertility preservation before curative chemo.[1]
Disease complications: superior vena cava syndrome, airway compromise from a mediastinal mass, spinal cord compression from epidural lymphoma, bone marrow failure, and bowel obstruction from an abdominal mass.[1]
Prognosis & Disposition
Hodgkin lymphoma is one of the most curable human cancers — overall survival exceeds 80 to 85 percent, and early-stage disease approaches 95 percent. Stage, B symptoms, bulk, age and the IPS modify prognosis; the challenge in HL is now minimising late toxicity (second cancers, cardiovascular disease, infertility) in long-term survivors through PET-adapted de-escalation.[1][3]
DLBCL is cured in roughly 60 to 70 percent of patients with R-CHOP, with the IPI/R-IPI predicting outcome (R-IPI very-good group 4-year PFS 94 percent, poor group 53 percent).[5] Follicular lymphoma is incurable but indolent (median survival over 10 to 15 years), with the FLIPI (follicular lymphoma IPI) stratifying risk.[7] Burkitt is curable in over 80 to 90 percent with modern intensive therapy but rapidly fatal untreated.[8]
[1]Disposition is to a haemato-oncology unit; the safety-net for the discharging clinician is patient education on febrile neutropenia (temperature over 38C = emergency) and tumour-lysis symptoms.[1]
Special Populations
- Pregnancy — diagnostic staging is adapted (avoid CT abdomen/pelvis, use MRI and shielded X-ray; PET-CT deferred if possible in first trimester). Treatment: ABVD is acceptable in pregnancy (doxorubicin, bleomycin, vinblastine, dacarbazine — none clearly teratogenic in the 2nd/3rd trimester); avoid radiotherapy to the fetus; vinblastine and rituximab have been used in the 2nd/3rd trimester; multidisciplinary timing (defer until 2nd trimester where possible; deliver then treat).[1]
- HIV — aggressive B-cell NHL (DLBCL, Burkitt, primary effusion lymphoma, primary CNS lymphoma) is AIDS-defining. Treat the lymphoma with appropriate regimen plus antiretroviral therapy and watch for immune reconstitution inflammatory syndrome (IRIS) and drug interactions.[1]
- Elderly — comorbidity-adapted, lower-intensity regimens (R-mini-CHOP, BR); R-CHOP remains tolerable in fit older adults; CAR-T increasingly used for relapse.
- Children and adolescents — Burkitt and lymphoblastic lymphoma predominate; paediatric-inspired protocols (less radiotherapy, emphasising chemotherapy) have excellent survival; fertility preservation is essential before gonadotoxic therapy.[8]
- Post-transplant (PTLD) — often EBV-driven B-cell proliferation; first step is reduction of immunosuppression ± rituximab.[11]
Evidence, Guidelines & Regional Differences
Guidelines: NCCN (US) and ESMO (Europe) provide the contemporary framework for staging and treatment, with the Lugano classification (Cheson 2014) standardising staging and PET response assessment, and the Deauville scale standardising PET scoring.[2] The British Society for Haematology (BSH)/BHOPA guidance applies in the UK.[1]
- Hasenclever & Diehl (NEJM 1998) — the IPS for advanced HL, seven factors predicting freedom from progression.[3]
- International Non-Hodgkin's Lymphoma Prognostic Factors Project (NEJM 1993) — the original IPI.[4]
- Sehn et al. (Blood 2007) — the R-IPI, recalibrated for the rituximab era.[5]
- GELA LNH-98.5 and MInT trials — R-CHOP superior to CHOP in elderly and young good-prognosis DLBCL respectively.[6]
- PET-adapted therapy trials (e.g. RATHL, CALGB 50604) — interim PET guiding escalation/de-escalation.
- ECHELON-1 (brentuximab + AVD frontline HL) and CHECKMATE 205 (nivolumab in relapsed HL).[11]
- ZUMA-1, JULIET, TRANSCEND — CAR-T (axicabtagene, tisagenlecleucel, lisocabtagene) in relapsed DLBCL.
Regional deltas: PET-CT, CAR-T, brentuximab and PD-1 inhibitors are routine in high-income settings but access-limited in India and other low/middle-income countries, where cost drives treatment toward generic CHOP, ABVD, thalidomide-based and palliative regimens and transplant capacity is constrained; H. pylori-driven gastric MALT is over-represented in regions of high H. pylori prevalence.[1]
Controversies: the extent of PET-adapted de-escalation (how little therapy is safe); the role of radiotherapy versus its late effects; watch-and-wait versus early rituximab in asymptomatic follicular lymphoma; the optimal frontline regimen for double-hit DLBCL; and timing of CAR-T (earlier in relapse vs after salvage).[2]
Exam Pearls
[1]IPS — seven adverse factors in Hodgkin lymphoma
ALMOSTS
below 40 g/L
below 0.6 times 10^9/L (or below 8 percent)
one of seven IPS factors
45 years or older
advanced stage
leucocytosis at least 15 times 10^9/L
haemoglobin below 105 g/L
- R-CHOP expansion = Rituximab, Cyclophosphamide, hydroxyDaunorubicin (doxorubicin), vincristine (Oncovin), Prednisone.
- ABVD expansion = Adriamycin (doxorubicin), Bleomycin, Vinblastine, Dacarbazine.
- Double-hit DLBCL = MYC + BCL2 (or BCL6) rearrangement; worse prognosis; DA-EPOCH-R.
- Brentuximab vedotin = anti-CD30 antibody-drug conjugate (HL); rituximab = anti-CD20 (B-cell NHL); daratumumab = anti-CD38 (myeloma — don't confuse).
- Hepatitis B screening before rituximab (HBsAg + anti-HBc) — reactivation can be fatal.
- Myeloma versus lymphoma marker pitfall: CD20 = lymphoma; CD138/CD38 = myeloma (plasma cells); CD5+ CD23+ = CLL/SLL; CD5+ cyclin D1+ = mantle cell.
- Deauville 1-3 = complete metabolic response (PET-negative); 4-5 = residual disease. [1]
Exam application bank (NEET-PG / INICET)
One-line answer
Lymphoma is a clonal malignancy of lymphocytes arising in lymphoid tissue, divided by the Reed-Sternberg cell into Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL). HL spreads contiguously, is driven by the CD15+/CD30+ Reed-Sternberg cell, presents with cervical/supraclavicular nodes and a mediastinal mass, and is treated with ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine). NHL is heterogeneous — follicular (indolent, t(14;18) BCL2), DLBCL (commonest aggressive, R-CHOP), mantle cell (t(11;14) cyclin D1, CD5+), Burkitt (t(8;14) MYC, fastest human tumour, Ki67 ~100 percent), MALT (H. pylori, gastric). Diagnosis = EXCISIONAL biopsy (not FNA — need architecture). Ann Arbor staging (Cotswolds) with B symptoms (fever over 38C, drenching night sweats, weight loss over 10 percent in 6 months). PET-CT for staging and Deauville 5-point scale for response. IPS (HL) and IPI (NHL) for pr
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard. [1]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes. [1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change. [1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each. [1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory. [1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Lymphoma (Hodgkin & Non-Hodgkin).
References
- [1]Connors JM, Cozen W, Steidl C, et al. Hodgkin lymphoma Nat Rev Dis Primers, 2020.PMID 32703953
- [2]Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification J Clin Oncol, 2014.PMID 25113753
- [3]Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin's disease. International Prognostic Factors Project on Advanced Hodgkin's Disease N Engl J Med, 1998.PMID 9819449
- [4]The International Non-Hodgkin's Lymphoma Prognostic Factors Project. A predictive model for aggressive non-Hodgkin's lymphoma N Engl J Med, 1993.PMID 8141877
- [5]Sehn LH, Berry B, Chhanabhai M, et al. The revised International Prognostic Index (R-IPI) is a better predictor of outcome than the standard IPI for patients with diffuse large B-cell lymphoma treated with R-CHOP Blood, 2007.PMID 17105812
- [6]Pfreundschuh M, Trumper L, Osterborg A, et al. CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: a randomised controlled trial by the MabThera International Trial (MInT) Group Lancet Oncol, 2006.PMID 16648042
- [7]Carbone A, Roulland S, Gloghini A, et al. Follicular lymphoma Nat Rev Dis Primers, 2019.PMID 31831752
- [8]Lopez C, Klein B, Mestre C, et al. Burkitt lymphoma Nat Rev Dis Primers, 2022.PMID 36522349
- [9]Zucca E, Bertoni F. Marginal zone lymphomas Hematol Oncol, 2023.PMID 37294969
- [10]Ferreri AJM, Bruno-Ventre M, Donadoni G, et al. Primary central nervous system lymphoma Nat Rev Dis Primers, 2023.PMID 37322012
- [11]Batlevi CL, Matsuki E, Brentjens RJ, Younes A. Novel immunotherapies in lymphoid malignancies Nat Rev Clin Oncol, 2016.PMID 26525683
- [12]Ansell SM, Rathmell WK, Gorog A, et al. Hodgkin lymphoma: 2025 update on diagnosis, risk-stratification, and management Am J Hematol, 2024.PMID 39239794