Infectious Diseases · General Medicine
Leprosy (Hansen Disease)
Also known as Leprosy · Hansen disease · Hanseniasis · Mycobacterium leprae · Tuberculoid leprosy · Lepromatous leprosy
Leprosy (Hansen disease) is a chronic granulomatous mycobacterial infection caused by Mycobacterium leprae (and the related M. lepromatosis), an obligate intracellular, acid-fast bacillus with tropism for Schwann cells of peripheral nerves and dermal macrophages in cooler tissues (skin, peripheral nerves, nasal mucosa, eyes, testes). Transmission is via prolonged close contact through nasal droplets from untreated multibacillary cases, with a long incubation (2 to 12 years). The clinical spectrum is determined by host cell-mediated immunity — strong Th1 immunity gives the paucibacillary/tuberculoid pole (single anaesthetic hypopigmented patches with thickened nerves), whereas absent CMI gives the multibacillary/lepromatous pole (numerous symmetric lesions, nodules, leonine facies, madarosis, glove-and-stocking neuropathy, saddle nose). Lepra reactions — type 1 (reversal) and type 2 (erythema nodosum leprosum, ENL) — cause acute, irreversible nerve damage and disability. Diagnosis is clinical (anaesthetic patch + thickened nerve) plus slit-skin smear, skin biopsy (Fite-Faraco acid-fast bacilli) and PCR. Treatment is WHO multidrug therapy (MDT) — paucibacillary: rifampicin + dapsone for 6 months; multibacillary: rifampicin + dapsone + clofazimine for 12 months. Reactions are treated with corticosteroids (type 1) and thalidomide/clofazimine/steroids (type 2).
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags
Overview & Definition
Leprosy (Hansen disease, Hanseniasis) is a curable, chronic granulomatous infection of the skin and peripheral nerves caused by Mycobacterium leprae (and the related species M. lepromatosis, which produces a similar diffuse lepromatous phenotype and Lucio phenomenon). It is one of the oldest documented human diseases and remains a leading infectious cause of preventable disability and stigma.[1][2]
M. leprae is obligate intracellular, weakly acid-fast, slow-growing (doubling time about 14 days), and does not grow on artificial media (the historic research model is the mouse foot-pad and the nine-banded armadillo). It has a predilection for cooler tissues (30 to 33 °C), which explains its distribution to skin, peripheral nerves (superficial), nasal mucosa, anterior chamber of the eye, and testes.[3]
The clinical skill in leprosy is recognising the cardinal triad — (1) hypopigmented/erythematous skin lesion with definite loss of sensation, (2) thickened peripheral nerve with sensory or motor deficit, (3) positive skin smear — and then classifying the patient on the immunological spectrum (which decides treatment duration) and managing lepra reactions (which decide whether the patient is left with disability). WHO multidrug therapy (MDT) is curative; the residual challenge is late presentation, reaction-related nerve damage, and stigma.[1][8]
Leprosy at a glance
Classification
Two complementary classifications coexist. The Ridlëy–Jopling spectrum (1966) classifies disease along an immunological spectrum from the tuberculoid pole (strong cell-mediated immunity, paucibacillary) to the lepromatous pole (no CMI, multibacillary), with three unstable borderline groups in between. The WHO operational classification is used at the bedside to choose MDT duration: paucibacillary (PB, 1 to 5 lesions) versus multibacillary (MB, more than 5 lesions or smear-positive).[1][2]
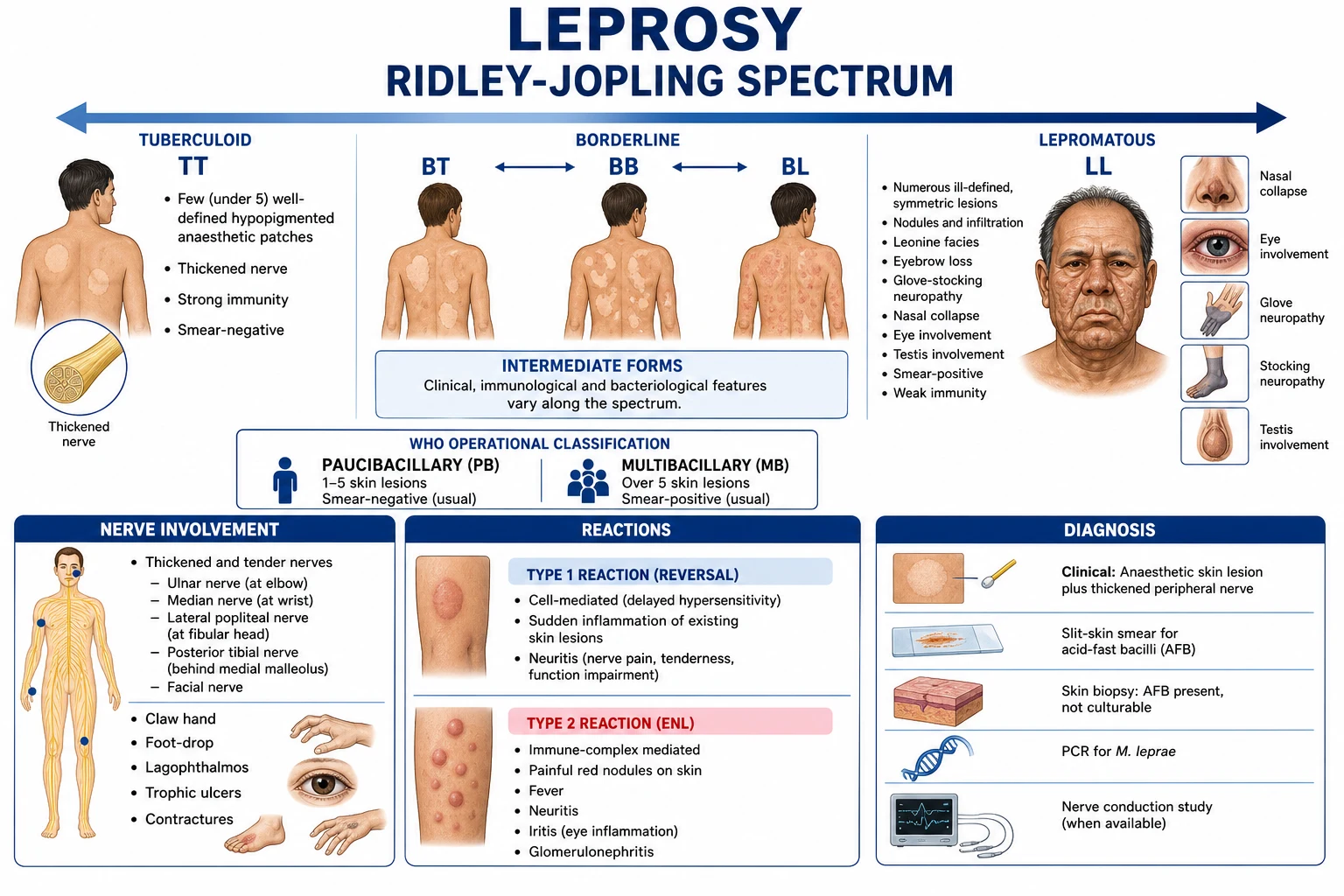
Two further clinical forms deserve recognition. Indeterminate leprosy is the earliest presentation — a faint hypopigmented macule with vague sensory impairment, often in a child — which then polarises to TT or LL or self-resolves. Pure neuritic leprosy (no skin lesion but a thickened tender sensory nerve with mononeuropathy or mononeuritis multiplex) is particularly common in India and frequently missed.[2]
Epidemiology & Risk Factors
Leprosy is reported from over 100 countries but is heavily concentrated in a handful of high-burden nations. India, Brazil and Indonesia together account for the majority of new cases worldwide; India alone contributes about 60% of the global total of new cases detected annually.[1][4] The global burden has fallen since the introduction of MDT in 1981–82, but new case detection has plateaued — driven by ongoing transmission, active case-finding revealing hidden cases, and the long incubation period.[4]
Transmission is via prolonged close contact with an untreated multibacillary case, principally through nasal droplets (M. leprae multiplies in nasal mucosa and is shed in secretions). The human is the major reservoir; in the Americas the nine-banded armadillo is a zoonotic source (genotype 3I). Skin-to-skin transmission is much less efficient.[1]
The incubation period is long — typically 2 to 12 years (mean about 5 years), occasionally longer — which explains why disease can appear years after exposure and why contact-tracing spans many years. Only about 5 to 10% of exposed individuals develop clinical leprosy, reflecting the dominant role of host immunity and genetic susceptibility (associations with HLA-DR2/DR3, PARK2/PACRG, NOD2/CARD15, TLR1 and TIRAP polymorphisms).[1]
Risk factors: household contact with an untreated MB case, poverty and overcrowding, malnutrition, geographic origin (tropical belt), genetic susceptibility, and immunosuppression. A bimodal age distribution is observed, with peaks in young adolescence (10–15 years) and adulthood (30–60 years); childhood disease reflects ongoing local transmission.[4]
The WHO Global Leprosy Strategy 2021–2030 ('Towards zero leprosy') targets: (1) interruption of transmission in 120 countries, (2) zero new grade-2 disability (visible deformity) in children, (3) repeal of all discriminatory laws. India's National Leprosy Eradication Programme (NLEP) implements active case-finding campaigns in endemic districts, SDR-PEP for contacts, Accompanied MDT, Disability Prevention and Medical Rehabilitation (DPMR), and education to combat stigma.[8]
Pathophysiology
Tropism: why skin and nerves
M. leprae enters via the respiratory route, multiplies in macrophages of the nasal mucosa, and is then disseminated haematogenously. Its predilection for cooler tissues (skin, peripheral nerves, nasal mucosa, anterior eye, testes — all 30–33 °C) reflects an intracellular optimum below core body temperature.[3]
The molecular basis for Schwann-cell tropism is now well described: the bacillus's surface phenolic glycolipid-1 (PGL-1) binds laminin-2 (merosin) and α-dystroglycan in the basal lamina of Schwann cells, allowing M. leprae to enter via α-dystroglycan-mediated endocytosis. Once inside a Schwann cell, M. leprae is sheltered from immune surveillance and from antibiotics, and replicates slowly, eventually lysing the cell and spreading to adjacent nerves and dermal macrophages.[1][5]
The immune spectrum
Position on the Ridley–Jopling spectrum is the host's immune decision, not the organism's:[1]
- Tuberculoid pole (TT) — a strong Th1 / cell-mediated response dominated by CD4+ T-helper-1 cells, IL-2, IFN-γ and TNF-α activates macrophages to kill intracellular bacilli and forms well-organised epithelioid granulomas with Langhans giant cells. Few bacilli survive (paucibacillary); the cost is bystander granulomatous destruction of nerves.
- Lepromatous pole (LL) — the Th1 response is switched off in favour of a Th2 / Treg response (IL-4, IL-5, IL-10, TGF-β). Macrophages fail to kill the organism, become foamy "Virchow" cells stuffed with bacilli (globi), and the patient mounts high-titre antibodies (which do not protect). Tissue destruction is paradoxically less, but bacillary load is enormous.[1][3]
The high humoral antibody titres (anti-PGL-1) in LL paradoxically coexist with anergy to M. leprae antigens (a "split tolerance").[3]
Nerve injury
Leprosy is the only human bacterial disease that regularly damages peripheral nerves. Mechanisms include:[5][7]
- Direct Schwann-cell infection and demyelination — the bacillus multiplies in Schwann cells causing segmental demyelination; in LL, bacilli disseminate along the nerve via the perineurium and endoneurial vessels.
- Granulomatous inflammation and caseation — in TT/BT, epithelioid granulomas form inside the nerve and caseate, destroying fascicles.
- Oedema and compression — the perineurium is a rigid sheath; intraneural oedema (during reactions) raises endoneurial pressure and produces ischaemic fascicular injury. This is why reversal reactions cause acute, compressive, salvageable nerve damage — high-dose steroids within 24 hours can reverse it.
- Vasa nervorum vasculitis — in BL/LL and ENL, immune-complex vasculitis of epineurial vessels produces infarction.
- Fibrosis — end-stage, the nerve is replaced by fibrous tissue (irreversible).[5]
The result is loss of autonomic, sensory and motor fibres in the affected distribution — the typical presentation of anaesthesia, anhidrosis, and muscle wasting and weakness in the territory of the involved peripheral nerve (not a dermatomal or stocking-glove pattern of metabolic neuropathy).[5]
Lepra reactions
Reactions are acute immunological flares superimposed on chronic disease. They are the most important cause of new nerve damage and must be recognised and treated within hours.[1][6]
- Type 1 (reversal) reaction — a delayed (type IV) hypersensitivity reaction to M. leprae antigens, occurring in borderline (BT/BB/BL) patients, often within months of starting MDT, in pregnancy, or with intercurrent infection. Existing skin lesions become erythematous, oedematous and tender, and there is acute neuritis (painful, tender, thickened nerve with new motor loss). There is no systemic inflammation (no fever). Treatment: corticosteroids.
- Type 2 reaction (erythema nodosum leprosum, ENL) — an immune-complex (type III) reaction in BL/LL patients, characterised by deposition of immune complexes in vessels with neutrophilic vasculitis and a TNF-α-driven systemic inflammatory response. Patients develop crops of tender erythematous nodules on the face and extensor limbs, with fever, malaise, arthralgia, iritis, dactylitis, orchitis, glomerulonephritis and neuritis. Episodes can be recurrent over years. Treatment: thalidomide, clofazimine, corticosteroids.[6]
Clinical Presentation
The three cardinal signs of leprosy (WHO): (1) hypopigmented or erythematous skin lesion(s) with definite sensory loss, (2) thickened peripheral nerves with sensory and/or motor deficit in the distribution of the affected nerve, (3) positive skin smear. Diagnosis can be made on the first two cardinal signs alone (most PB cases are smear-negative).[1]
Tuberculoid (TT) leprosy
The patient (often an adult from an endemic area) presents with a single (or up to three) sharply demarcated hypopigmented or erythematous patch anywhere on the body. The patch has loss of sensation (cotton wool, pinprick, temperature), loss of hair (alopecia) and loss of sweating (anhidrosis). A nearby peripheral nerve is often palpably thickened and tender (e.g. the ulnar, radial cutaneous, or posterior auricular nerve). Skin smear is negative.[2]
Borderline (BT, BB, BL) leprosy
Lesions are more numerous, asymmetric, polymorphic (macules, plaques, annular lesions with a punched-out centre). BT lesions resemble TT but are more numerous; BL lesions approach the lepromatous pattern but retain some asymmetry. Borderline patients are the most likely to develop type 1 reactions, because their CMI is unstable.[1]
Lepromatous (LL) leprosy
LL presents with numerous, symmetric, ill-defined lesions — macules, plaques, papules and nodules (erythema nodosum-like, but chronic). There is diffuse cutaneous infiltration; the face becomes thickened and corrugated — leonine facies — with eyebrow alopecia (madarosis), saddle-nose deformity (from nasal cartilage destruction), ear-lobe infiltration, and gynaecomastia (from testicular atrophy). The skin smear is strongly positive.[2]
Neurologically, LL produces symmetric, distal "glove-and-stocking" sensory loss (because of dermal and intraneural infiltration of multiple small cutaneous nerves) with preservation of tendon reflexes early on; motor loss is late. The nasal mucosa is heavily involved (chronic nasal congestion, epistaxis, septal perforation). The eye is involved via corneal anaesthesia (trigeminal) and lagophthalmos (facial nerve), leading to exposure keratitis; iridocyclitis is direct ocular infection. The testes are infiltrated, producing orchitis, atrophy, infertility and gynaecomastia.[1]
Nerve-palpation findings and the deformities they cause
The eight nerves to palpate
Other clinical forms
- Pure neuritic leprosy — no skin lesion, but a thickened tender peripheral nerve with sensory/motor deficit; common in India; diagnosed on nerve biopsy or by exclusion.[2]
- Indeterminate leprosy — an early faint hypopigmented macule, usually in a child, with vague sensory impairment; usually self-resolves or polarises.
- Lucio–Latapí leprosy — a rare diffuse form of LL seen in Mexico and Central America; Lucio phenomenon is a severe necrotising vasculitis with painful irregular skin ulcers and infarcts — a medical emergency.[1]
Differential Diagnosis
A hypopigmented or nodular skin lesion is not always leprosy. The decisive discriminator is sensation in the lesion and the presence of a thickened peripheral nerve.[2]
For peripheral neuropathy with thickened nerves, the leprosy-distinctive feature is the combination of cutaneous anaesthesia and a thickened, tender nerve — which diabetic mononeuropathy, vasculitic mononeuritis multiplex, hereditary neuropathy with pressure palsies (HNPP), and B12 neuropathy do not produce. Hereditary motor sensory neuropathy (Charcot-Marie-Tooth) can produce palpable nerves but is symmetric, slowly progressive, and lifelong with a family history.[7]
Clinical & Bedside Assessment
A focused bedside examination is the diagnostic core of leprosy.[1]
Skin examination — inspect the whole skin under good light; identify hypopigmented, erythematous, infiltrative or nodular lesions. For each lesion, test sensation:
- Light touch with a wisp of cotton wool or Semmes-Weinstein nylon monofilament (10 g) — ask the patient to close their eyes and point to where they feel the touch (compare lesion with normal contralateral skin).
- Temperature with two test-tubes (warm and cold).
- Pain with a sterile pin (compare with normal skin).
- A lesion with definite sensory loss plus a thickened nerve is leprosy.[5]
Nerve palpation — systematically palpate and assess the eight peripheral nerves (see mnemonic): ulnar at the olecranon groove, radial cutaneous at the wrist, median at the wrist, common peroneal at the fibular neck, posterior tibial behind the medial malleolus, facial branches, posterior auricular over the mastoid, and greater auricular across the sternocleidomastoid. Assess each for thickening, tenderness, consistency (soft, firm, hard), and the presence of an associated motor or sensory deficit.[5]
Voluntary muscle testing (VMT) — test the muscles supplied by each at-risk nerve (e.g. first dorsal interosseous for ulnar; abductor pollicis brevis for median; ankle dorsiflexion for common peroneal; orbicularis oculi closure for facial).[5]
WHO disability grading (reproduced verbatim):[1]
- Grade 0 — no anaesthesia, no visible deformity.
- Grade 1 — anaesthesia present, but no visible deformity or damage.
- Grade 2 — visible deformity or damage of the hands, feet or eyes (claw hand, foot drop, plantar ulcer, lagophthalmos, corneal opacity, visual impairment).
The eye–hand–foot (EHF) score (sum of WHO grades for the two eyes, two hands, two feet, range 0–12) is used to monitor disability over time and to flag patients at high risk of new nerve damage.[7]
Investigations
The diagnosis is clinical, supported by laboratory tests when the cardinal signs are equivocal or when classification is unclear.[1][3]
Slit-skin smear — the cornerstone laboratory test for multibacillary leprosy. A small (5 mm) incision is made in pinched skin (typically ear lobes and 3 to 4 active lesions), the dermis is scraped with the blade edge, and the scrapings are smeared, fixed, and stained with Ziehl–Neelsen or Fite-Faraco for acid-fast bacilli. The Bacterial Index (BI) is the mean log-10 bacillary load across sites, graded 0 to 6+ on the Ridley logarithmic scale:
- 0 = no bacilli in 100 oil-immersion fields
- 1+ = 1–10 bacilli per 100 fields
- 2+ = 1–10 per 10 fields
- 3+ = 1–10 per field (average)
- 4+ = 10–100 per field
- 5+ = 100–1000 per field
- 6+ = >1000 per field (globi/clumps)[1]
The Morphological Index (MI) is the percentage of uniformly stained, viable-appearing bacilli (vs granular, dead forms) and falls rapidly after starting rifampicin — a useful early marker of treatment response. PB leprosy is smear-negative; MB leprosy is smear-positive.[1]
Skin biopsy — a wedge biopsy (including subcutaneous fat) is taken from the active border of a lesion. Histopathology distinguishes the spectrum:[2][3]
- TT — well-formed epithelioid cell granulomas with Langhans giant cells in the dermis, often around nerves and skin appendages; caseation can occur; bacilli are scarce or absent; a subepidermal free zone is preserved.
- BT — epithelioid granulomas less well-formed, some bacilli may be found in nerves.
- BB — diffuse epithelioid infiltrate with scattered lymphocytes and macrophages; few bacilli.
- BL — macrophage predominance with some epithelioid foci; moderate bacillary load.
- LL — sheets of foamy "Virchow" macrophages filled with bacilli (often in clumps called globi); a characteristic subepidermal free (Grenz) zone separates the infiltrate from the epidermis; numerous plasma cells and lymphocytes.
Acid-fast staining of tissue requires the Fite-Faraco method (not plain Ziehl–Neelsen) because M. leprae's cell wall is more readily decolourised by acid than M. tuberculosis.[3]
Nerve biopsy — a sensory cutaneous nerve (e.g. sural, superficial radial, or posterior auricular) is biopsied when pure neuritic leprosy is suspected or the skin biopsy is non-diagnostic. Findings range from epithelioid granuloma (TT) to heavy bacillary infiltration (LL). Nerve biopsy can cause a small permanent sensory deficit at the biopsy site and is reserved for diagnosis-specific indications.[1]
Molecular diagnosis — PCR — M. leprae DNA (target RLEP, 16S rRNA or groEL) can be detected by PCR in skin smears, biopsies and nerve tissue. PCR is highly sensitive in MB but low sensitivity in PB (because few organisms are present); useful for confirmation and for species identification (distinguishing M. leprae from M. lepromatosis).[3]
Serology — anti-PGL-1 antibodies — high sensitivity in LL/BL but low sensitivity in TT/BT; useful for epidemiological surveys and as an adjunct for classification, not for individual diagnosis.[1]
Why culture is impossible — M. leprae does not grow on Lowenstein-Jensen or other artificial media; the historic research model is the mouse foot-pad (Shepard, 1960) and the nine-banded armadillo (Kirchheimer, 1971). The Mantoux test with tuberculin does not diagnose leprosy and reflects TB exposure/BGC, not leprosy.[3]
Baseline investigations at diagnosis: full blood count (dapsone can cause haemolysis/agranulocytosis), G6PD assay (especially in regions of high prevalence — dapsone is relatively contraindicated in severe deficiency), renal and liver function (rifampicin, dapsone hepatotoxicity), HIV and hepatitis B/C screen (co-infection affects management), urinalysis, BMI (malnutrition is a risk factor), and a chest X-ray if TB co-infection is suspected.[3]
Management — Resuscitation
Leprosy itself is rarely a resuscitation emergency, but three situations are time-critical:[1][6]
1. Acute type 1 (reversal) reaction with nerve function impairment — the single most important preventable cause of disability. A patient (typically borderline) presents with inflamed, erythematous, oedematous skin lesions and acute painful neuritis with new sensory or motor loss. Start oral prednisolone 40 to 60 mg once daily (or equivalent) within 24 hours of onset, continue MDT, refer to a specialised leprosy centre, immobilise the affected limb, and monitor with VMT and sensory testing daily. Steroids can be lifesaving for nerve function.[1]
2. Severe type 2 (ENL) reaction — the patient (BL/LL) has crops of painful erythematous nodules, fever, malaise, and may have iritis, orchitis, dactylitis, nephritis, hepatosplenomegaly or neuritis. Management: thalidomide (where licensed and NOT in a woman of childbearing potential without strict contraception) 100 to 300 mg/day, high-dose clofazimine (300 mg/day, reducing over months), NSAIDs/analgesia, antipyretics; short-course corticosteroids for severe systemic involvement; supportive care. Admit if systemic involvement is severe.[6]
3. Lucio phenomenon — a medical emergency with high mortality: painful, irregular, angulated skin ulcers and infarcts on a background of untreated diffuse LL. Management: urgent specialist referral, systemic corticosteroids, MDT, wound care, and treatment of secondary infection.[1]
Acute complications needing immediate intervention: lagophthalmos (eye lubrication + night-time eye shield; tarsorrhaphy if persistent), foot drop (ankle-foot orthosis to prevent falls and equinus contracture), claw hand (intrinsic-plus splint, daily passive extension), and plantar trophic ulcer (off-loading, debridement, protective footwear, treatment of secondary infection).[7]
Management — Definitive & Stepwise
WHO multidrug therapy (MDT) — curative
MDT is the single most important intervention. It is curative, free (donated by Novartis via WHO), blister-packed, and well tolerated. MDT uses three drugs with different mechanisms to prevent resistance.[1][2]
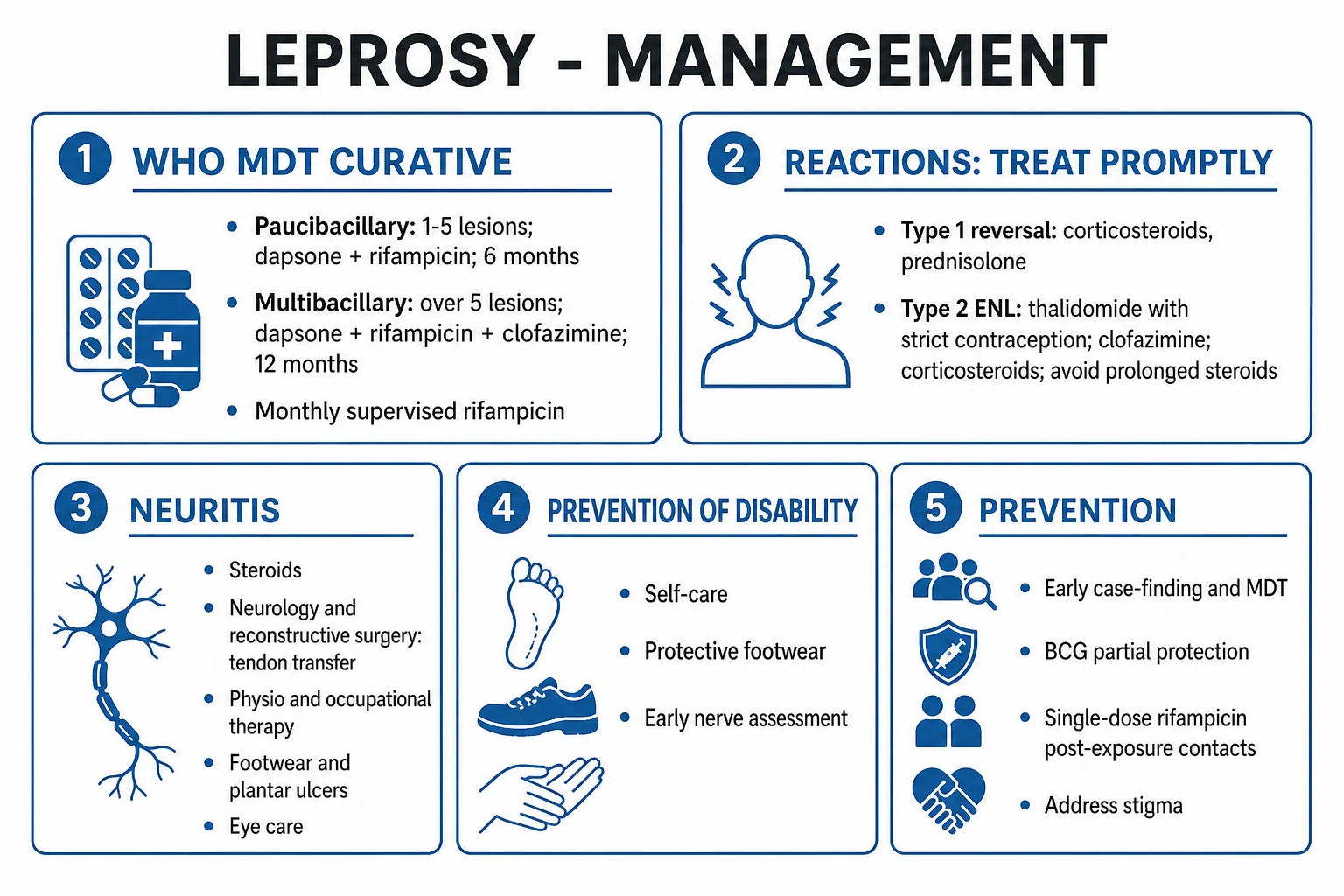
Paucibacillary (PB) MDT — 6 months[1]
- Rifampicin 600 mg orally once monthly (supervised/intake observed) — powerful bactericidal: a single 600 mg dose kills 99.9% of viable bacilli within days; the patient becomes essentially non-infectious after the first dose.
- Dapsone 100 mg orally once daily (self-administered) — bacteriostatic; cheap; oral.
- Duration: 6 months (blister pack of 4 weekly doses).
Multibacillary (MB) MDT — 12 months[1]
- Rifampicin 600 mg once monthly supervised (bactericidal).
- Clofazimine 300 mg once monthly supervised PLUS 50 mg once daily (self-administered) — weakly bactericidal plus anti-inflammatory (useful in ENL); reservoir effect.
- Dapsone 100 mg once daily (self-administered).
- Duration: 12 months (historical regimen was 24 months; shortened to 12 months in 1998).
MDT mnemonic
Rationale and pharmacology of each drug[1]
- Rifampicin (600 mg monthly) — inhibits DNA-dependent RNA polymerase; bactericidal; a single dose renders the patient non-infectious; given once monthly because of its long post-antibiotic effect on M. leprae. Side-effects: orange-red discoloration of urine/tears/sweat (counsel), hepatotoxicity (monitor LFTs), thrombocytopenia, 'flu' syndrome, gastrointestinal upset; potent inducer of hepatic enzymes — reduces the efficacy of oral contraceptives (counsel to use barrier too), warfarin, protease inhibitors.[1]
- Dapsone (100 mg daily) — folate antagonist (inhibits dihydropteroate synthase), bacteriostatic; cheap; oral. Side-effects: dose-related haemolysis (especially in G6PD deficiency — check before starting), methaemoglobinaemia, agranulocytosis (rare but serious), dapsone hypersensitivity syndrome (fever, rash, lymphadenopathy, hepatitis, eosinophilia — resembles DRESS, occurs in first 6 weeks), hepatitis, peripheral neuropathy (rare).[3]
- Clofazimine (300 mg monthly + 50 mg daily) — binds mycobacterial DNA; weakly bactericidal; anti-inflammatory; long tissue half-life (reservoir). Side-effects: reddish-brown skin pigmentation (counsel; resolves months after stopping), ichthyosis and dry skin, GI upset, and crystal deposition in mesenteric nodes. The pigmentation can cause stigma; counsel patients at the start.[1]
Special regimens[1]
- Single-dose rifampicin post-exposure prophylaxis (SDR-PEP) — a single dose of rifampicin given to contacts of an index case reduces the risk of leprosy by about 50–60% in the first 2 years; now endorsed by the WHO 2018 guideline. Contraindicated in pregnancy, in confirmed leprosy, and in contacts of PB single-lesion cases (low yield).[4]
- Uniform MDT (U-MDT) — a 6-month 3-drug regimen for both PB and MB — is being studied; not yet standard of care.[1]
- Monotherapy is forbidden — it selects for resistance. Historically, dapsone monotherapy (1960s–80s) produced dapsone-resistant relapse; rifampicin monotherapy rapidly selects rifampicin-resistant M. leprae. Always use combination MDT.[2]
Management of lepra reactions
Type 1 (reversal) reaction[1][5]
- Corticosteroids — oral prednisolone 40 to 60 mg/day (max 1 mg/kg/day), maintained for 2–4 weeks, then tapered over 12 to 24 weeks (avoid rapid taper — recurrence risk). Severe neuritis may need IV methylprednisolone.
- Continue MDT throughout the reaction.
- Rest the affected limb; immobilise if acute neuritis; treat precipitating causes (infection, pregnancy, anaemia).
- Surgical decompression of acutely swollen nerves is reserved for refractory cases and is controversial.
Type 2 reaction (ENL)[6]
- Thalidomide — 100 to 300 mg orally at night, reducing to maintenance; the most effective agent for ENL. Strict contraindications: pregnancy (severe teratogen — phocomelia) and women of childbearing potential not on rigorous contraception (use the Thalidomide Pharmion Risk Evaluation and Mitigation Strategy / pregnancy prevention programme). Monitor for peripheral neuropathy, somnolence, thrombosis.
- Clofazimine — 300 mg/day for several weeks, reducing to 100 mg/day; anti-inflammatory; useful for recurrent ENL.
- Corticosteroids — prednisolone for severe systemic involvement or neuritis; avoid prolonged courses (steroid toxicity, secondary immunosuppression).
- Alternatives / adjuncts: pentoxifylline, colchicine, ciclosporin, methotrexatum, tumour-necrosis-factor inhibitors for refractory cases.
- Continue MDT.
Management of established nerve damage and deformity
The goal of disability prevention and medical rehabilitation (DPMR) is to preserve nerve function and limit secondary damage:[7]
- Acute neuritis — corticosteroids (as above).
- Anaesthetic hands/feet — patient self-care education: daily inspection for cuts/blisters, soaking and oiling skin, protective footwear (microcellular rubber, MCR) for plantar anaesthesia, avoid burns and trauma (use long-handled utensils, test water temperature).
- Claw hand — intrinsic-plus splint, daily passive interphalangeal extension, hand therapy; reconstructive surgery (tendon transfer, e.g. extensor carpi radialis longus to lateral band) once disease is stable for ≥6 months.
- Foot drop — ankle-foot orthosis (AFO) to prevent falls and equinus contracture; tendon transfer (tibialis posterior to lateral cuneiform) once stable.
- Lagophthalmos — night-time eye shield and lubricants during the day; tarsorrhaphy or gold-weight upper-lid implant for permanent lagophthalmos.
- Plantar ulcer — off-loading (rest, crutches, total-contact cast), debridement, treatment of secondary infection, and after healing, MCR footwear.
Specific Subtypes & Scenarios
Borderline disease and type 1 reactions
Borderline patients (BT, BB, BL) carry unstable CMI: a sudden upswing in Th1 immunity (triggered by MDT killing bacilli and releasing antigen, or by pregnancy, anaemia, intercurrent infection) upgrades the patient toward the tuberculoid pole — clinically, a type 1 (reversal) reaction. New lesions may appear; existing lesions become inflamed and oedematous; acute neuritis is the feared complication. Treatment: corticosteroids as above. Reactions may recur and require a prolonged steroid taper.[1][5]
Lepromatous disease and type 2 reactions (ENL)
In LL/BL, the high bacillary load and antigenaemia precipitate immune-complex vasculitis — ENL. Patients are systemically unwell, with crops of painful nodules, fever, iritis, orchitis, nephritis and neuritis. ENL is recurrent and a major cause of morbidity; thalidomide is the most effective agent. Long-term amyloidosis (AA type) can complicate recurrent ENL, presenting as nephrotic syndrome or renal failure.[6]
Lucio phenomenon
A severe necrotising vasculitis of small dermal vessels seen in diffuse LL (Lucio–Latapí), endemic to Mexico and Central America. Painful, angulated, irregular skin ulcers and infarcts appear abruptly; systemic toxicity is marked; mortality is high. Management: systemic corticosteroids, MDT, wound care, infection control, supportive care.[1]
Pure neuritic leprosy
No skin lesion but a thickened, tender peripheral nerve with sensory or motor loss in its distribution — a common presentation in India. Diagnosis: nerve biopsy (or PCR), exclusion of other mononeuropathies. Treated as PB or MB based on smear and clinical judgement.[2]
Indeterminate leprosy
An early faint hypopigmented macule with vague sensory impairment in a child (often a household contact). Most resolve spontaneously; those that progress require MDT.[1]
Pregnancy and leprosy
Pregnancy is a recognised trigger of type 1 reactions (especially in the postpartum) and may unmask or flare disease due to immune modulation. MDT is safe in pregnancy (PB and MB). Thalidomide is absolutely contraindicated; rifampicin reduces OCP efficacy — counsel additional barrier contraception.[4]
Complications & Pitfalls
Nerve damage (the central complication)
The deformities of untreated leprosy — claw hand, foot drop, lagophthalmos, plantar ulcers, autoamputation, blindness — result from peripheral nerve damage. They are largely irreversible once established, which is why early diagnosis and prompt treatment of reactions is the single most important prevention.[7]
Classic pitfalls
- Missing leprosy because sensation was not tested in a hypopigmented patch — every patch is anaesthetic until proven otherwise.
- Mislabelling TT as vitiligo or pityriasis versicolor because the patch is hypopigmented — vitiligo and pityriasis are not anaesthetic.
- Failing to recognise a type 1 reaction as the cause of acute nerve damage in a borderline patient — every new weakness in known leprosy is a reversal reaction until proven otherwise; start steroids within 24 hours.
- Stopping MDT prematurely — relapse risk; MDT must be completed (PB 6 months, MB 12 months) even if lesions look "cured".
- Prescribing thalidomide to a pregnant woman (or one of child-bearing potential without rigorous contraception).
- Forgetting that dapsone causes haemolysis in G6PD deficiency — screen where feasible.
- Forgetting that rifampicin reduces OCP and warfarin efficacy — counsel and adjust.[5]
Prognosis & Disposition
Leprosy is curable with WHO MDT, with a bacteriological cure rate near 100% and a relapse rate of less than 1% per year after completion.[1] Patients become non-infectious after the first dose of rifampicin; isolation is never required. The prognosis for nerve function depends on how early treatment is started and how promptly reactions are treated — established nerve damage before treatment is largely irreversible. Patients with grade-2 disability at diagnosis have a worse functional outcome and require long-term DPMR.[7]
Surveillance after MDT: clinical and sensory examination (and serial slit-skin smears for MB) for 2 to 5 years to detect relapse (most relapses occur in the first 3 years) and new reactions. Relapse is treated with the same WHO MDT regimen (resistance is rare if MDT was used).[1]
Discharge criteria: completed MDT course, no active reaction, all reversible nerve function impairment treated, grade-2 disability managed with appropriate footwear/splint/eye care, and the patient educated in self-care. Patients should be advised to return promptly if they develop new skin lesions, nerve pain, weakness or new disability.[8]
Special Populations
Pregnancy
Leprosy may worsen during pregnancy and the postpartum (immune shift toward Th2 in pregnancy → disease activity; Th1 rebound postpartum → type 1 reactions). MDT (PB and MB) is safe in pregnancy — continue throughout. Thalidomide is absolutely contraindicated (teratogen). Rifampicin reduces OCP efficacy — counsel. Clofazimine crosses the placenta and is excreted in breast milk; it is compatible with breastfeeding but infants should be monitored for skin pigmentation and GI upset.[4]
Lactation
MDT is compatible with breastfeeding. Clofazimine is excreted in milk and may lightly pigment the infant's skin (cosmetic, reversible).[1]
Children
Children account for a significant fraction of new cases in endemic areas; grade-2 disability in a child is a public-health failure indicating ongoing transmission. Weight-based dosing is provided in WHO blister packs:
- 10–14 years — adult-dose MDT (PB and MB).
- Under 10 years — half the adult dose.
- Under 5 years / under 15 kg — quarter the adult dose (approximately). Children tolerate MDT well; counselling about rifampicin discoloration of urine is important for adherence.[1]
HIV co-infection
HIV does not appear to alter the natural history of leprosy in patients on antiretroviral therapy (ART), but immune reconstitution inflammatory syndrome (IRIS) after starting ART can unmask leprosy or trigger type 1 reactions. Co-management with HIV services is essential.[4]
G6PD deficiency
Dapsone causes dose-related haemolysis and methaemoglobinaemia, particularly in G6PD-deficient patients. Screen where feasible; if deficient, use MB regimen without dapsone (rifampicin + clofazimine + a fluoroquinolone such as ofloxacin, or minocycline) under specialist guidance.[3]
Elderly and immunocompromised
The elderly may present with more advanced disease (longer incubation, comorbidity, immune senescence) and tolerate steroids poorly. Immunosuppressive therapy (anti-TNF, transplant immunosuppression) can unmask or reactivate latent leprosy.[1]
Evidence, Guidelines & Regional Differences
WHO Guidelines for the Diagnosis, Treatment and Prevention of Leprosy (2018)[4]
The 2018 WHO guideline formalised the current standard of care:
- 3-drug MDT for all MB (rifampicin + clofazimine + dapsone, 12 months) and 2-drug MDT for all PB (rifampicin + dapsone, 6 months) — replacing the previous recommendation of 3-drug MB only for smear-positive cases. This simplified operational delivery.
- Single-dose rifampicin post-exposure prophylaxis (SDR-PEP) for contacts of an index case.
- Accompanied MDT — the patient may take home the full course to encourage completion; supervised monthly rifampicin where possible.
- No isolation; no routine separation of patients from family or community.[4]
WHO Global Leprosy Strategy 2021–2030 — "Towards zero leprosy"[8]
Three targets:
- 120 countries achieve interruption of transmission (no new autochthonous cases in children under 5).
- Zero new grade-2 disability in children.
- Repeal of all discriminatory legislation (e.g. India's repeal of the colonial-era Leprosy Act).[8]
India — National Leprosy Eradication Programme (NLEP)[8]
India's NLEP (run by the Ministry of Health and Family Welfare) delivers WHO MDT free through primary health centres, runs Leprosy Case Detection Campaigns (LCDC) in endemic districts, provides SDR-PEP to contacts, supports Disability Prevention and Medical Rehabilitation (DPMR) with reconstructive surgery camps, and operates a stigma-reduction public-awareness campaign. Accompanied MDT and ASHA-based referral are operational innovations.[8]
Controversies and research gaps
- MB MDT duration: 12 months is now standard; an older 24-month regimen and a proposed 6-month uniform MDT (U-MDT) for both PB and MB remain under study.
- Chemoprophylaxis vs vaccination: BCG gives partial protection (about 50%) and is more effective against PB than LL; SDR-PEP gives additive benefit. A specific leprosy vaccine is not yet available.
- Early case-finding tools: anti-PGL-1 serology, contact tracing, and PCR on nasal swabs are being evaluated for population screening.
- Treatment of leprosy reactions: thalidomide is teratogenic and neuropathic; alternative anti-TNF agents (thalidomide analogues, lenalidomide) are being explored for ENL.
- M. lepromatosis — a second species causing diffuse leprosy and Lucio phenomenon — is increasingly recognised and may require the same management.[4]
Exam Pearls
The seven pearls that decide a leprosy answer
One-liners that decide MCQs:
- Rifampicin renders the patient non-infectious after a single dose.
- Dapsone is contraindicated (or used with caution) in G6PD deficiency — causes haemolysis and methaemoglobinaemia.
- Thalidomide is absolutely contraindicated in pregnancy (phocomelia); women of childbearing potential need a rigorous contraception programme.
- Type 1 reaction: existing lesions inflame, no fever, treat with steroids. Type 2 reaction: new crops of nodules with fever, treat with thalidomide/clofazimine.
- Most common cause of blindness in leprosy: lagophthalmos + corneal anaesthesia → exposure keratitis.
- Pure neuritic leprosy is most common in India.
- Lucio phenomenon is associated with M. lepromatosis/diffuse LL in Mexico and is a medical emergency.
- Rifampicin induces hepatic enzymes → reduces OCP, warfarin, PI efficacy.
- Relapse after MDT is treated with the same WHO MDT regimen.
- India contributes about 60% of global leprosy cases.[4]
Exam application bank (NEET-PG / INICET)
One-line answer
Leprosy (Hansen disease) is a chronic granulomatous mycobacterial infection caused by Mycobacterium leprae (and the related M. lepromatosis), an obligate intracellular, acid-fast bacillus with tropism for Schwann cells of peripheral nerves and dermal macrophages in cooler tissues (skin, peripheral nerves, nasal mucosa, eyes, testes). Transmission is via prolonged close contact through nasal droplets from untreated multibacillary cases, with a long incubation (2 to 12 years). The clinical spectrum is determined by host cell-mediated immunity — strong Th1 immunity gives the paucibacillary/tuberculoid pole (single anaesthetic hypopigmented patches with thickened nerves), whereas absent CMI gives the multibacillary/lepromatous pole (numerous symmetric lesions, nodules, leonine facies, madarosis, glove-and-stocking neuropathy, saddle nose). Lepra reactions — type 1 (reversal) and type 2 (
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard.[5]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes.[1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change.[1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each.[1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory.[1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Leprosy (Hansen Disease).
References
- [1]Grijsen ML, Nguyen TH, Pinheiro RO, et al. Leprosy. Nature Reviews Disease Primers, 2024.PMID 39609422
- [2]White C, Franco-Paredes C. Leprosy in the 21st century. Clinical Microbiology Reviews, 2015.PMID 25567223
- [3]Mungroo MR, Khan NA, Siddiqui R. Mycobacterium leprae: Pathogenesis, diagnosis, and treatment options. Microbial Pathogenesis, 2020.PMID 32931893
- [4]Rodrigues LC, Lockwood DNj. Leprosy now: epidemiology, progress, challenges, and research gaps. Lancet Infectious Diseases, 2011.PMID 21616456
- [5]Scollard DM. Mechanisms of nerve injury in leprosy. Clinics in Dermatology, 2015.PMID 25432810
- [6]Kahawita IP, Lockwood DNj. Towards understanding the pathology of erythema nodosum leprosum. Transactions of the Royal Society of Tropical Medicine and Hygiene, 2008.PMID 18313706
- [7]Ebenezer GJ, et al. Treatment and Evaluation Advances in Leprosy Neuropathy. Neurotherapeutics, 2021.PMID 34799845
- [8]Lockwood DNj. Leprosy: too complex a disease for a simple elimination paradigm. Bulletin of the World Health Organization, 2005.PMID 15798849