Nephrology · General Medicine
Rapidly Progressive Glomerulonephritis
Also known as Rapidly progressive glomerulonephritis · RPGN · Crescentic glomerulonephritis · ANCA-associated vasculitis · Goodpasture syndrome · Pulmonary-renal syndrome
Rapidly progressive glomerulonephritis (RPGN) is the most aggressive form of glomerulonephritis and a true renal emergency: a syndrome of rapid loss of renal function (loss of over 50 per cent of GFR within under 3 months), unified by the histological hallmark of crescents — fibrin and proliferating parietal epithelial cells — in Bowman's space. Untreated it progresses to end-stage kidney disease within weeks. It is classified by immunofluorescence into three types: Type I anti-GBM (linear IgG; Goodpasture syndrome with pulmonary haemorrhage), Type II immune-complex (granular; lupus, post-infectious, IgA) and Type III pauci-immune (ANCA-associated vasculitis — GPA, microscopic polyangiitis; the commonest form in adults). Presentation is a rapidly rising creatinine with haematuria and dysmorphic red cells / red-cell casts, often with systemic vasculitic features or haemoptysis (the pulmonary-renal syndrome). Work-up is ANCA (anti-PR3, anti-MPO), anti-GBM antibody, complement, and urgent renal biopsy. The overriding principle is treat first, refine later — start high-dose IV methylprednisolone on suspicion before biopsy results, then add cyclophosphamide or rituximab, with plasma exchange for anti-GBM disease and selected severe ANCA (dialysis-dependent or life-threatening alveolar haemorrhage), followed by maintenance rituximab or azathioprine to prevent ANCA relapse. Speed of treatment determines whether the kidney survives — once crescents become fibrous the damage is irreversible.
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags
Overview & Definition
Rapidly progressive glomerulonephritis (RPGN) is not a single disease but a syndrome of rapid glomerular destruction, defined clinically by loss of more than 50 per cent of glomerular filtration rate (GFR) within under 3 months, and unified histologically by the finding of crescents — half-moon-shaped accumulations of fibrin, proliferating parietal epithelial cells and macrophages in Bowman's space — in over 50 per cent of glomeruli on renal biopsy. Untreated, the syndrome progresses to end-stage kidney disease (ESKD) within days to weeks.[1][4]
Because the window for salvage is narrow and the damage is largely irreversible once crescents become fibrous, the guiding principle is treat first, refine later: begin high-dose intravenous methylprednisolone on clinical suspicion of crescentic GN, arrange urgent renal biopsy and serology, then tailor therapy — adding cyclophosphamide or rituximab, and plasma exchange where the type dictates. The three immunofluorescence-based types span very different diseases that converge on the same histological endpoint:[8]
- Type I — anti-GBM disease (Goodpasture): linear IgG against the alpha-3 chain of type IV collagen; pulmonary haemorrhage; plasma exchange is essential.
- Type II — immune-complex: granular deposits; lupus nephritis, post-infectious GN with crescents, IgA nephropathy; treat the underlying disease.
- Type III — pauci-immune (ANCA-associated vasculitis): minimal deposits; granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA); the commonest cause of RPGN in adults; treat with steroids plus rituximab or cyclophosphamide.[4][11]
Classification
RPGN is classified by immunofluorescence pattern on renal biopsy, which reflects the underlying immunopathogenic mechanism and determines treatment. The three types differ in antibody, complement, IF pattern, and the role of plasma exchange.[8]

Type I — Anti-GBM
Linear IgG; Goodpasture
- **Anti-alpha3(IV)NC1 antibody** — non-collagenous domain of the alpha-3 chain of type IV collagen in the GBM and alveolar basement membrane
- **Immunofluorescence: LINEAR IgG** along the GBM (ribbon-like, continuous)
- **Goodpasture syndrome** — pulmonary haemorrhage (diffuse alveolar haemorrhage) plus crescentic GN
- **Complement normal**; ANCA usually negative (double-positive patients exist)
- **Plasma exchange is ESSENTIAL** (removes circulating tissue-fixed antibody) plus methylprednisolone + cyclophosphamide
- Rare (about 0.5 to 1 per million per year); bimodal (young men with pulmonary haemorrhage, older women renal-limited)
Type II — Immune complex
Granular deposits
- **GRANULAR immune-complex deposits** on immunofluorescence (full-house in lupus, IgA-dominant in IgA nephropathy, subepithelial humps in PSGN)
- Causes: **lupus nephritis class III/IV**, **post-infectious GN with crescents**, **IgA nephropathy with crescents**, cryoglobulinaemic GN, endocarditis-associated GN
- **Complement LOW** in lupus and post-infectious (classical pathway); may be normal in IgA
- Treat the **underlying disease** (lupus induction, supportive PSGN, steroids ± cyclophosphamide for crescentic IgA)
- Plasma exchange rarely indicated except in severe lupus or rapidly progressive cryoglobulinaemia
- Commonest cause in **children** (crescentic PSGN, IgA vasculitis) and in **young women** (lupus nephritis)
Type III — Pauci-immune
ANCA vasculitis; commonest
- **Minimal or no immunoglobulin/complement deposition** on immunofluorescence (pauci-immune) — the injury is against soluble neutrophil proteins, not fixed tissue
- **ANCA positive** — anti-PR3 (cytoplasmic c-ANCA, GPA) or anti-MPO (perinuclear p-ANCA, MPA)
- **Necrotising crescentic GN** on light microscopy; **necrotising small-vessel vasculitis** systemically
- Subtypes: **GPA** (ENT and lung granulomatous), **MPA** (renal-predominant, pulmonary haemorrhage), **EGPA** (asthma, eosinophilia)
- **Commonest cause of RPGN in adults** (about 60 to 70 per cent); peak age 60 to 70; drug causes — hydralazine, propylthiouracil, levamisole-cocaine, minocycline
- Treat with **steroids plus rituximab or cyclophosphamide**; plasma exchange only if dialysis-dependent or life-threatening alveolar haemorrhage (PEXIVAS)
Special phenotypes
Double-positive & drug-induced
- **Double-positive (ANCA + anti-GBM)** — manage as anti-GBM (add plasma exchange); ANCA positivity predicts a **relapsing course** once anti-GBM is suppressed
- **Drug-induced ANCA vasculitis** — hydralazine, propylthiouracil, levamisole-adulterated cocaine, minocycline; usually anti-MPO with anti-histone and anti-MPO co-positivity; stop the drug
- **Renal-limited vasculitis** — ANCA-positive crescentic GN with no extra-renal disease; treat as GPA/MPA
- **EGPA (Churg-Strauss)** — asthma, eosinophilia over 1.5 x10^9/L, sinusitis, mononeuritis; anti-MPO in about 40 per cent; mepolizumab (anti-IL-5) for eosinophilic/ANCA-negative phenotype
- **Idiopathic pauci-immune crescentic GN** — ANCA-negative but pauci-immune on biopsy; treated as ANCA disease
Defining the syndrome — clinical and histological
| Criterion | Detail |
|---|---|
| Clinical | Loss of over 50 per cent of GFR within under 3 months, with an active urinary sediment (haematuria, dysmorphic RBCs, RBC casts) |
| Histological | Crescents in over 50 per cent of glomeruli on renal biopsy |
| Crescent composition | Fibrin, parietal epithelial cells, macrophages, detached podocytes; cellular (early, treatable) → fibrocellular → fibrous (irreversible) |
| IF discriminator | Linear IgG (Type I); granular (Type II); pauci-immune (Type III) |
Epidemiology & Risk Factors
Incidence and relative frequency of the three types
The relative frequency of the RPGN types has shifted over the past two decades with better ANCA testing:[4]
- Type III (pauci-immune / ANCA-associated vasculitis) is now the commonest cause of RPGN in adults, accounting for about 60 to 70 per cent of cases in most series. The peak age is 60 to 70 years.[4]
- Type II (immune-complex) accounts for about 20 to 30 per cent. In children the dominant causes are crescentic post-infectious GN and IgA vasculitis (Henoch-Schonlein purpura) nephritis; in young women lupus nephritis class IV dominates.
- Type I (anti-GBM) is the rarest at about 10 to 20 per cent, with an incidence of about 0.5 to 1 case per million population per year. It has a characteristic bimodal age distribution: a first peak in young men (20 to 30 years) who present with pulmonary haemorrhage, and a second peak in older women (over 60 years) with more renal-limited disease.[1][3]
ANCA-associated vasculitis subtypes — demographics and antibodies
GPA
Granulomatosis with polyangiitis (Wegener)
- **Anti-PR3 (cytoplasmic c-ANCA)** in about 75 to 90 per cent
- **Granulomatous ENT and lung disease** — nasal crusting, epistaxis, saddle-nose deformity, sinusitis, subglottic stenosis, pulmonary nodules/cavities
- **Renal** crescentic GN; ocular disease (scleritis, orbital pseudotumour); mononeuritis multiplex
- Peak age 40 to 60; relapsing course (PR3 positivity predicts relapse)
- **Staphylococcus aureus nasal carriage** is associated with relapse — co-trimoxazole has a role in localised disease
MPA
Microscopic polyangiitis
- **Anti-MPO (perinuclear p-ANCA)** in about 50 to 70 per cent
- **Renal-predominant** disease with **pulmonary haemorrhage** (capillaritis) — NO granulomatous ENT disease
- Palpable purpura, mononeuritis multiplex, gastrointestinal involvement
- Peak age 60 to 70; slightly more common in Asian populations
- Less likely to relapse than PR3/GPA
EGPA
Eosinophilic granulomatosis with polyangiitis (Churg-Strauss)
- Adult-onset **asthma**, **eosinophilia** (over 1.5 x10^9/L or over 10 per cent), **sinusitis**, **mononeuritis multiplex**
- **Anti-MPO** in about 40 per cent
- Cardiac involvement (eosinophilic myocarditis) is a leading cause of death
- **Mepolizumab** (anti-IL-5 receptor) for eosinophilic/ANCA-negative phenotype; steroids plus rituximab/cyclophosphamide for ANCA-positive severe disease
Risk factors
- Genetic: HLA-DRB1*15 and HLA-DRB4 (anti-GBM); HLA-DP variants and SERPINA1 (alpha-1 antitrypsin deficiency, strongly associated with GPA); HLA-DQ (MPO-ANCA).[1]
- Environmental: Silica, hydrocarbon and farm-pesticide exposure increase the risk of ANCA vasculitis; smoking increases the risk of pulmonary haemorrhage in anti-GBM disease.[4]
- Drug-induced ANCA: hydralazine, propylthiouracil, levamisole-adulterated cocaine, minocycline, allopurinol — typically anti-MPO with anti-histone and sometimes anti-elastase co-positivity.[4][5]
- Chronic infection: hepatitis B and C, subacute bacterial endocarditis, deep abscess/shunt infection (immune-complex RPGN).
- Chronic inflammatory disease: systemic lupus erythematosus, rheumatoid arthritis.
- Malignancy: a modest association with ANCA and with membranoproliferative patterns.
- Recent infection: upper respiratory infection is a common precipitant of GPA relapse and of IgA nephropathy flares.
Pathophysiology
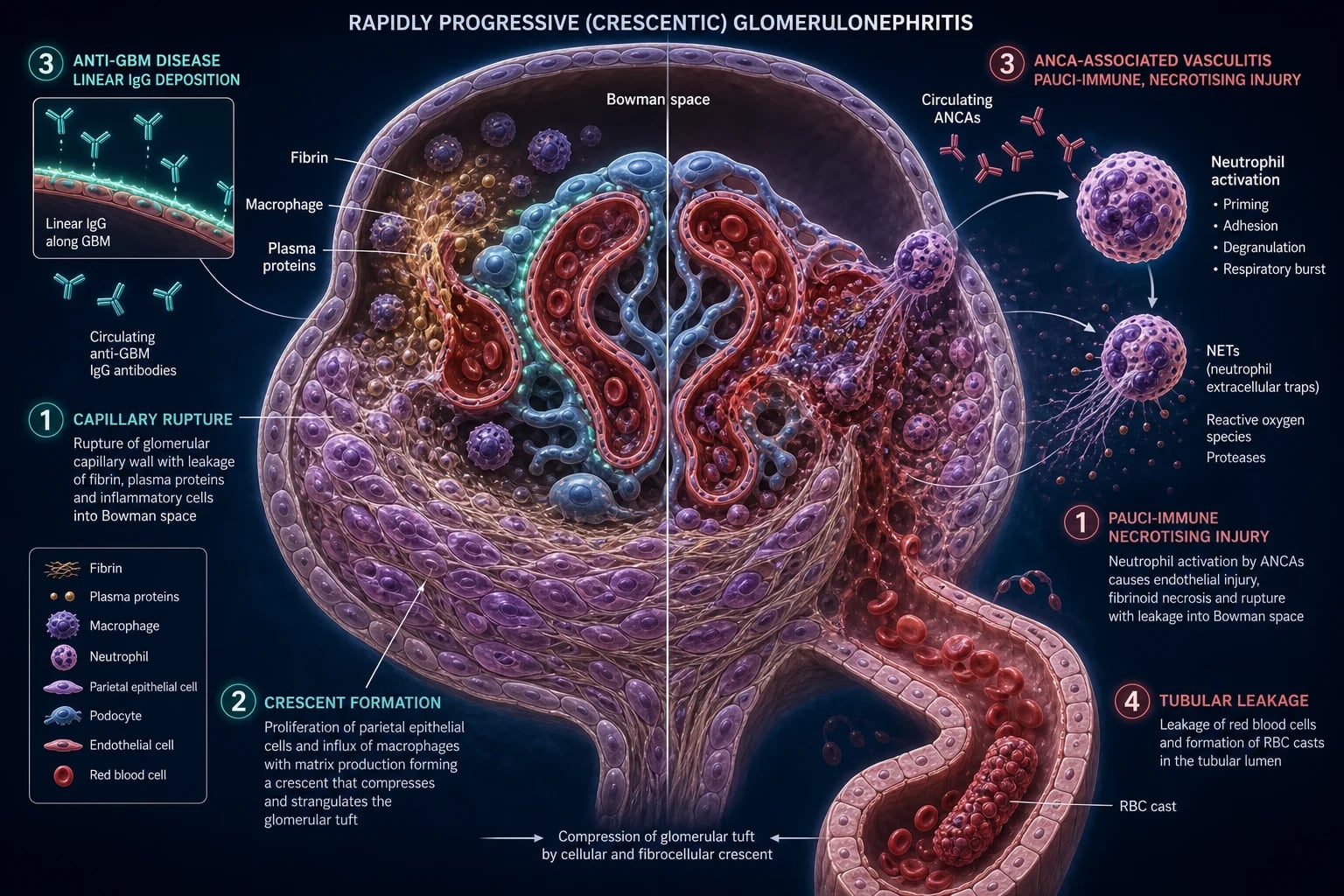
The glomerular filtration barrier and how it is breached
The normal filtration barrier has three layers: a fenestrated endothelium (charge and size selectivity via the glycocalyx), the glomerular basement membrane (GBM) rich in type IV collagen (alpha-3, -4, -5 chains — the target of anti-GBM disease), and podocyte foot processes joined by the slit diaphragm (nephrin, podocin). In RPGN the injury is to the endothelium, GBM and capillary wall: severe inflammation — whether antibody-mediated (anti-GBM), immune-complex-driven (Type II) or pauci-immune (ANCA) — physically ruptures the capillary wall, and the breach is what allows fibrin and cells into Bowman's space, igniting crescent formation.[1]
Crescent formation — the convergent endpoint
Whatever the trigger, severe capillary-wall injury tears the GBM. The breach allows:[8]
- Fibrin, macrophages and plasma proteins to enter Bowman's space.
- Parietal epithelial cells lining Bowman's capsule — and podocytes that detach and adopt a proliferative phenotype — to divide, forming a crescent-shaped cellular and fibrocellular mass that compresses and ultimately obliterates the glomerular tuft.
- Tubular atrophy and interstitial fibrosis from secondary ischaemia as the tuft collapses.[1]
Crescents evolve from cellular (early, potentially salvageable) to fibrocellular to fibrous (irreversible). This evolution underpins the cardinal rule of RPGN: treat fast, before the crescent scars.[1][11]
Type I — anti-GBM disease (Goodpasture): the linear antibody
The pathogenesis is a textbook example of a tissue-fixed antibody disease:[12]
- Autoantibodies (IgG, rarely IgA or IgM) are produced against the non-collagenous domain of the alpha-3 chain of type IV collagen — the alpha3(IV)NC1 antigen — which is normally sequestered within the mature GBM and the alveolar basement membrane.[1]
- These antibodies deposit in a continuous, ribbon-like LINEAR pattern along the GBM (and the alveolar BM), visible on direct immunofluorescence.
- Binding activates complement (C3) and triggers Fc-receptor-mediated neutrophil and macrophage injury, which ruptures the capillary wall, producing crescents in the kidney and alveolar haemorrhage in the lung.
- The lung is affected in about 40 to 70 per cent of cases (Goodpasture syndrome); the alveolar basement membrane shares the same alpha-3(IV)NC1 target. Smoking, respiratory infection and fluid overload increase the risk of pulmonary bleeding.
Because the pathogenic antibody is fixed in tissue and also circulates, removing the circulating antibody by plasma exchange is central to treatment — unlike pauci-immune ANCA disease, where the antibody is against soluble neutrophil granule proteins and plasma exchange has a narrower role.[2][3]
Type III — ANCA vasculitis: the NET/alternative-complement amplification loop
ANCA are IgG autoantibodies against neutrophil granule proteins — myeloperoxidase (MPO), giving a perinuclear (p-ANCA) pattern, and proteinase 3 (PR3), giving a cytoplasmic (c-ANCA) pattern. The currently accepted pathogenesis is the NET/alternative-complement-pathway amplification loop:[4]
- Priming — cytokines (IL-1, TNF, G-CSF) during infection or inflammation cause resting neutrophils to translocate MPO and PR3 from intracellular granules to the cell surface.
- Binding — circulating ANCA bind surface MPO/PR3 and cross-link them, activating the neutrophil via Fc-gamma receptors.
- Effector release — activated neutrophils degranulate, release reactive oxygen species, proteases and neutrophil extracellular traps (NETs) — webs of DNA, histones and granule proteins that damage the endothelium.
- Amplification — NETs activate the alternative complement pathway, generating C5a, which recruits more primed neutrophils — a self-sustaining loop that produces necrotising small-vessel vasculitis and crescentic GN.
- Pauci-immune appearance — because the antibody is directed against soluble neutrophil granule proteins, not fixed tissue antigen, little immunoglobulin or complement deposits in the glomerulus — hence the "pauci-immune" immunofluorescence.[4]
This mechanism explains why avacopan (a C5a receptor blocker) works as a steroid-sparing agent in ANCA vasculitis (ADVOCATE trial): it interrupts the amplification loop at the C5a-neutrophil recruitment step.[4][12]
Type II — immune-complex RPGN: granular deposition
Here the mechanism depends on the parent disease, but the unifying feature is circulating immune complexes depositing in the mesangium, subendothelium and subepithelium, activating the classical complement pathway, recruiting neutrophils and macrophages, and rupturing the capillary wall to form crescents:[4]
- Lupus nephritis class IV — defective clearance of apoptotic debris drives autoantibodies to nuclear antigens (dsDNA, nucleosomes); immune complexes deposit and produce full-house IF (IgG, IgA, IgM, C3, C1q, C4); complement is low (C3 and C4).
- Post-infectious GN with crescents — nephritogenic streptococcal antigens (SpeB, NAPlr) form subepithelial humps; complement C3 is low.
- IgA nephropathy with crescents — galactose-deficient IgA1 immune complexes deposit in the mesangium; IgA-dominant IF.[4]
The deposits appear granular on IF, in contrast to the linear pattern of anti-GBM and the absence of deposits in pauci-immune disease.[1]
Mechanism of the renal failure and the pulmonary-renal link
Crescents compress the tuft and collapse glomerular capillaries, reducing the filtering surface area — producing oliguria and a rapidly rising creatinine. Tubular atrophy and interstitial fibrosis from secondary ischaemia compound the damage. Diffuse alveolar haemorrhage occurs in anti-GBM and ANCA disease because the alveolar capillary bed shares the same antibody/inflammatory mechanism as the kidney (anti-alpha3(IV)NC1 in anti-GBM; capillaritis in ANCA). Immune-complex disease (lupus, post-infectious) generally spares the alveolus because the immune complexes deposit in the kidney, not the alveolar BM.[12]
Clinical Presentation
Classical RPGN presentation
- Rapidly progressive AKI — rising creatinine and falling urine output over days to weeks; this temporal signature distinguishes RPGN from acute GN (often self-limiting over weeks) and from chronic GN (slow CKD over years).
- Haematuria — macroscopic ("cola", "smoky", "tea-coloured") or microscopic; phase-contrast microscopy shows dysmorphic red cells (acanthocytes) and red-cell casts (the cardinal finding).
- Subnephrotic proteinuria — typically under 3.5 g/day; may be nephrotic-range in lupus nephritis.
- Oedema — periorbital (worse in the morning), sacral, pedal; from sodium and water retention (reduced GFR with intact tubular reabsorption — the overfill mechanism).
- Hypertension — from volume expansion and intrarenal renin-angiotensin system activation.
- Constitutional — malaise, fatigue, fever, weight loss, myalgia, arthralgia (especially prominent in ANCA vasculitis).[7]
The pulmonary-renal syndrome — recognise early
The combination of haemoptysis (or rapidly progressive dyspnoea from diffuse alveolar haemorrhage) with rapidly progressive GN is a medical emergency. The dominant causes are anti-GBM disease (Goodpasture) and ANCA-associated vasculitis (GPA, MPA); lupus and cryoglobulinaemia are rarer causes. Immediate management is plasma exchange plus high-dose steroids plus cyclophosphamide (or rituximab) — delay costs the kidney and the lung.[1][3][4]
Cause-specific systemic clues (the "look beyond the kidney")
Anti-GBM (Goodpasture)
Type I
- **Acute haemoptysis** and dyspnoea — diffuse alveolar haemorrhage
- **Iron-deficiency anaemia** from chronic pulmonary bleeding
- Rapidly progressive renal failure; bimodal age (young men, older women)
- **Smoking** and respiratory infection precipitate pulmonary haemorrhage
GPA (Wegener)
PR3/c-ANCA
- **ENT disease** — nasal crusting, epistaxis, **saddle-nose deformity**, sinusitis, **subglottic stenosis**, conductive hearing loss
- **Pulmonary** — nodules, cavities, infiltrates, haemorrhage
- **Ocular** — scleritis, episcleritis, orbital pseudotumour, proptosis
- **Mononeuritis multiplex**; palpable purpura
- Relapsing course; Staphylococcus aureus nasal carriage linked to relapse
MPA
MPO/p-ANCA
- **Renal-predominant** — crescentic GN is often the presenting feature
- **Pulmonary haemorrhage** (capillaritis) but NO granulomatous ENT disease
- **Palpable purpura** of lower limbs, **mononeuritis multiplex**, GI involvement
- Peak age 60 to 70; less likely to relapse than GPA
Immune-complex (Type II)
Lupus, post-infectious, IgA
- **Lupus** — malar rash, photosensitivity, arthritis, oral ulcers, alopecia, serositis (low C3 AND C4)
- **Post-infectious** — recent sore throat or skin infection; cola-coloured urine; low C3 normalising by 6 to 8 weeks
- **IgA nephropathy** — synpharyngitic (concurrent with URTI) gross haematuria; normal complement
Atypical presentations
- Elderly — may present with fatigue, anorexia, weight loss or unexplained AKI rather than visible haematuria; ANCA vasculitis is the dominant cause of crescentic GN in this group. Atypical presentation delays diagnosis; a low threshold for ANCA/anti-GBM testing in an elderly patient with unexplained AKI is essential.[4]
- Diabetic patients — may have diabetic nephropathy with superimposed crescentic GN (anti-GBM, ANCA or IgA). Biopsy is warranted if the AKI is rapid, RBC casts are present, complement is low, or there is no diabetic retinopathy (a discordant picture).
- Children — crescentic post-infectious GN and IgA vasculitis (Henoch-Schonlein purpura) nephritis dominate; anti-GBM is rare.
- Pregnancy — lupus nephritis may flare (especially postpartum); pre-eclampsia can mimic RPGN and must be distinguished by complement, serology and RBC casts (pre-eclampsia has no RBC casts, no dysmorphic RBCs, no ANCA/anti-GBM).
- Double-positive (ANCA + anti-GBM) — presents as a pulmonary-renal syndrome but should be managed as anti-GBM (add plasma exchange); ANCA positivity predicts a relapsing course once anti-GBM is suppressed.[1]
Differential Diagnosis
The differential is organised first by immunofluorescence type and serology, then by tempo and pattern to distinguish RPGN from non-glomerular causes of rapidly rising creatinine.[1]
The three-type serological split
The RPGN serology panel — what splits the differential
Distinguishing anti-GBM from ANCA vasculitis
| Feature | Anti-GBM (Goodpasture) | ANCA vasculitis (GPA/MPA) |
|---|---|---|
| IF pattern | LINEAR IgG along GBM | Pauci-immune (minimal deposition) |
| Antibody | Anti-alpha3(IV)NC1 (anti-GBM) | Anti-PR3 (c-ANCA) or anti-MPO (p-ANCA) |
| Complement | Normal | Normal |
| Pulmonary | Diffuse alveolar haemorrhage | Granulomas/nodules (GPA) or haemorrhage (MPA) |
| ENT | Usually spared | Saddle nose, sinusitis, subglottic stenosis (GPA) |
| Course | Usually monophasic | Relapsing (especially PR3/GPA) |
| Plasma exchange | Essential | Only if dialysis-dependent or life-threatening haemorrhage |
Distinguishing GPA from MPA from EGPA
| Feature | GPA (Wegener) | MPA | EGPA (Churg-Strauss) |
|---|---|---|---|
| ANCA | Anti-PR3 (c-ANCA) 75 to 90 per cent | Anti-MPO (p-ANCA) 50 to 70 per cent | Anti-MPO about 40 per cent |
| Granulomas | Yes (ENT, lung) | No | Yes (eosinophilic) |
| ENT | Saddle nose, sinusitis, subglottic stenosis | Absent | Sinusitis, nasal polyps |
| Lung | Nodules, cavities, infiltrates | Haemorrhage | Asthma, infiltrates |
| Bloods | Raised inflammatory markers | Raised inflammatory markers | Eosinophilia over 1.5 x10^9/L |
| Nerves | Mononeuritis multiplex | Mononeuritis multiplex | Mononeuritis multiplex |
Distinguishing RPGN from non-glomerular causes of rapidly rising creatinine
- Acute tubular necrosis (ATN) — ischaemic or toxic context; muddy brown granular casts, no dysmorphic RBCs, no RBC casts, bland urine in early phase. The commonest mimic; the presence of RBC casts redirects to a glomerular cause.
- Acute interstitial nephritis — drug history (beta-lactams, NSAIDs, PPIs); eosinophiluria, WBC casts, fever, rash; no RBC casts.
- Bilateral urinary tract obstruction — anuria or fluctuating output; renal ultrasound shows hydronephrosis; treat the obstruction.
- Renal vein thrombosis — flank pain, nephrotic-range proteinuria (nephrotic syndrome complication); Doppler or CT venography.
- Bilateral renal infarction — severe flank pain, LDH raised, angiography; embolic or hypercoagulable context.
- Thrombotic microangiopathy (HUS/TTP) — microangiopathic haemolytic anaemia (schistocytes), thrombocytopaenia, Coombs-negative; complement may be low (atypical HUS).[3]
The double-positive patient — a critical special case
About 20 to 40 per cent of patients with anti-GBM antibody are also ANCA-positive (usually MPO-ANCA). These double-positive patients should be managed as anti-GBM disease (add plasma exchange) because the tissue-fixed antibody drives early pulmonary and renal mortality. ANCA positivity predicts a relapsing course once the anti-GBM antibody is suppressed, so maintenance immunosuppression (as for ANCA disease) is warranted after the acute episode.[1][2]
Clinical & Bedside Assessment
Focused examination
- Volume status and BP — accurate blood pressure (often raised), JVP, weight, serial fluid balance; periorbital, sacral and pedal oedema; basal crackles and pulmonary oedema.
- Urinalysis — dipstick for blood and protein (blood 2+ to 4+ with proteinuria).
- Respiratory — oxygen saturation, respiratory rate; haemoptysis, basal crackles, effusion; suspect diffuse alveolar haemorrhage with falling saturations and rising respiratory rate (anti-GBM, MPA).[1]
The "look beyond the kidney" vasculitis screen
A systematic search for extra-renal disease often reveals the diagnosis before serology returns:[7]
- ENT — nasal crusting, epistaxis, saddle-nose deformity, sinus tenderness, oral ulcers, subglottic stenosis (stridor) → GPA.[4]
- Eyes — scleritis, episcleritis, conjunctivitis, orbital pseudotumour/proptosis, retinal changes → GPA, anti-GBM.
- Skin — palpable purpura of lower limbs (leucocytoclastic vasculitis), livedo reticularis, necrotic lesions, splinter haemorrhages, digital gangrene → ANCA, cryoglobulinaemia, endocarditis.
- Chest — crackles (pulmonary oedema), alveolar haemorrhage, pleural effusion (serositis in lupus).
- Heart — new murmur (endocarditis; the source of immune-complex RPGN), pericardial rub (lupus, uraemia).
- Abdomen — hepatosplenomegaly (chronic infection), ascites (serositis), abdominal tenderness (IgA vasculitis, mesenteric vasculitis).
- Neurological — mononeuritis multiplex (wrist drop, foot drop) → ANCA vasculitis; hypertensive encephalopathy (confusion, papilloedema).
- Fundoscopy — hypertensive retinopathy, choroiditis (lupus), retinal vasculitis.
Targeted history
- Time course — when did the creatinine start rising? (RPGN is days to weeks.)
- Recent infection — sore throat, skin infection (PSGN with crescents); sinusitis, otitis (GPA).
- Haemoptysis — anti-GBM, ANCA (diffuse alveolar haemorrhage).
- Drug exposure — hydralazine, propylthiouracil, levamisole-adulterated cocaine, minocycline, allopurinol (drug-induced ANCA).
- Chronic infection — hepatitis B/C, HIV, endocarditis.
- Family history — Alport carrier (anti-GBM risk after transplant), lupus.
- Smoking — increases risk of pulmonary haemorrhage in anti-GBM.
- Occupation — silica, farm and solvent exposure (ANCA).[4]
Life-threats to assess at the bedside
- Pulmonary haemorrhage — haemoptysis, falling saturations, rising respiratory rate (anti-GBM, MPA).
- Pulmonary oedema — crackles, raised JVP, hypoxia.
- Hyperkalaemia — peaked T waves, ECG changes.
- Hypertensive encephalopathy — confusion, papilloedema, visual disturbance, seizures.[1]
Investigations
Urine — the cardinal specimen
| Test | Finding in RPGN |
|---|---|
| Dipstick | Blood 2+ to 4+ with proteinuria (subnephrotic, under 3.5 g/day; occasionally nephrotic in lupus) |
| Phase-contrast microscopy | Dysmorphic red cells (acanthocytes) and red-cell casts — the cardinal finding of glomerular bleeding |
| Protein quantification | Urine PCR or ACR; 24-hour protein typically 1 to 3.5 g/day |
Red-cell casts are the single most discriminating microscopy finding: their presence confirms a glomerular source of bleeding and mandates a glomerular work-up. Their absence does not exclude RPGN — in focal ANCA disease the lesion may be patchy.[1]
Baseline bloods
- Urea, creatinine, eGFR — quantify the AKI; trend daily.
- CBC — anaemia of chronic disease or iron deficiency (pulmonary bleeding in anti-GBM); eosinophilia (EGPA); thrombocytopaenia (lupus, TMA).
- ESR and CRP — markedly raised in ANCA vasculitis (a clue).
- Albumin — often low.
- Electrolytes including bicarbonate — hyperkalaemia, metabolic acidosis.
- LFTs, glucose/HbA1c, bone profile.
- Blood cultures and echocardiography if endocarditis suspected (immune-complex RPGN).[2]
The serological workup — splits the three types
The serology panel is the bedside discriminator of the three RPGN types:[1]
RPGN serology — the key panels
Practical points:[1]
- Antigen-specific immunoassay (anti-PR3, anti-MPO) is now preferred over indirect immunofluorescence (IIF) alone, as it is more specific; IIF patterns (cytoplasmic c-ANCA, perinuclear p-ANCA) remain useful but can be confounded by non-specific ANCA (e.g. inflammatory bowel disease, infection).
- Complement C3 and C4 — low in immune-complex RPGN (lupus — both low; post-infectious — C3 low, C4 often normal); normal in anti-GBM and pauci-immune disease.
- Anti-GBM antibody titre correlates with disease activity and is used to monitor plasma exchange (continue until undetectable).[9]
Renal ultrasound
- Exclude obstruction.
- Assess kidney size — normal to slightly enlarged in acute disease; small, scarred kidneys (under 9 cm) indicate chronic, irreversible damage and contraindicate aggressive immunosuppression.
- Doppler to exclude renal vein thrombosis where flank pain and heavy proteinuria coexist.[7]
Renal biopsy — the gold standard
Indications: any suspected RPGN is an indication for urgent renal biopsy — the IF pattern is the discriminator of the three types and dictates treatment. Correct coagulopathy before biopsy (vitamin K, fresh frozen plasma); consider transjugular biopsy if anticoagulated.[8]
Histological findings by type:[7]
| Type | Light microscopy | Immunofluorescence | Electron microscopy |
|---|---|---|---|
| I — anti-GBM | Cellular crescents; segmental necrosis | LINEAR IgG along GBM (ribbon-like); C3 may be present | GBM disruption; no deposits |
| II — immune complex | Crescents + proliferative pattern | GRANULAR deposits — full-house (lupus), IgA-dominant (IgA), C3-dominant with humps (PSGN) | Deposits in mesangium, subendothelium (lupus), subepithelial humps (PSGN) |
| III — pauci-immune | Segmental necrotising crescentic GN; fibrinoid necrosis | Minimal or no deposition (pauci-immune) | No electron-dense deposits |
The percentage of glomeruli with crescents and the proportion of fibrous (chronic) crescents are the most powerful prognostic features on biopsy — a high proportion of fibrous crescents predicts poor renal recovery.[8]
Diffuse alveolar haemorrhage — confirm at the bedside
- Bronchoalveolar lavage (BAL) — progressively bloodier serial aspirates (pathognomonic).
- Chest X-ray / CT — bilateral alveolar infiltrates (typically perihilar, sparing apices).
- Diffusion capacity (DLCO) — raised from haemoglobin in the alveoli.[1]
Activity and prognostic scores
Two vasculitis scores you must know
B-F
Birmingham Vasculitis Activity Score — quantifies disease activity (0 to 63); tracks response to treatment and relapse
Five-Factor Score — age over 65, renal insufficiency (creatinine over 150 umol/L), cardiac, GI, and absence of ENT involvement; each adds 1 point; predicts 5-year mortality
Management — Resuscitation
The overriding principle: treat first, refine later
The single most important rule in RPGN is do not wait for biopsy or serology. If the clinical picture (rapidly rising creatinine, haematuria with RBC casts, rapidly progressive AKI) is consistent with crescentic GN, start high-dose IV methylprednisolone immediately while arranging urgent biopsy and serology. The window for salvage is narrow because crescents become fibrous and irreversible within days to weeks.[1][11]
ABC and life-threats
- Airway/Breathing — high-flow oxygen; secure the airway if massive pulmonary haemorrhage (anti-GBM/ANCA) with anaesthetic and intensive-care support; protective lung ventilation if intubated.
- Circulation — IV access; treat volume overload (pulmonary oedema) with oxygen, sit upright and IV furosemide 40 to 80 mg; renal replacement therapy if refractory.
- Hyperkalaemia — calcium gluconate 10 mL of 10 per cent IV (cardioprotection), insulin-dextrose (10 units soluble insulin in 25 g dextrose), nebulised salbutamol 10 to 20 mg, then removal (dialysis or gut potassium binders).[8]
Hypertensive emergency
Reduce BP gradually — no more than 25 per cent of mean arterial pressure in the first hour, then to 160/100 mmHg over 2 to 6 hours to avoid cerebral and coronary hypoperfusion.[1]
- IV labetalol 20 to 80 mg bolus every 10 minutes, or infusion 0.5 to 2 mg/min.
- IV nicardipine infusion 5 to 15 mg/hour (titrate).
- Avoid sublingual nifedipine (unpredictable, can cause precipitous falls).
- In pregnancy, IV hydralazine or labetalol are first-line.[8]
Indications for urgent renal replacement therapy — AEIOU
When to dialyse in AKI — AEIOU
AEIOU
Refractory metabolic acidosis (pH under 7.1) unresponsive to bicarbonate
Refractory hyperkalaemia (K+ over 6.5 mmol/L) unresponsive to medical therapy
Toxins or drug overdose amenable to dialysis (lithium, salicylate, metformin)
Refractory volume overload (pulmonary oedema unresponsive to diuretics)
Symptomatic uraemia — pericarditis, encephalopathy, uraemic bleeding
Dialysis-dependence at presentation is a poor prognostic sign and, in ANCA vasculitis, is one of the indications for adding plasma exchange to induction.[8][9]
Steroid cover — PJP, bone, glucose, gastric
High-dose steroids demand prophylaxis:[8]
- Pneumocystis jirovecii (PJP) prophylaxis — co-trimoxazole 480 mg once daily (or 960 mg three times weekly) for the duration of high-dose steroids plus cyclophosphamide/rituximab. Essential — PJP pneumonia is a leading cause of death in immunosuppressed vasculitis patients.
- Gastric protection — proton pump inhibitor (e.g. oral pantoprazole 40 mg daily).
- Bone protection — calcium and vitamin D; bisphosphonate if long-term steroids.
- Glucose monitoring — steroid-induced hyperglycaemia.
- Infection surveillance — hold cyclophosphamide/rituximab for active infection.[6]
Management — Definitive & Stepwise
Definitive treatment is type-specific but shares the principle of urgent induction immunosuppression (steroids plus a cytotoxic/B-cell agent), with plasma exchange layered in for anti-GBM and selected ANCA disease, followed by maintenance to prevent relapse.[9]

Step 1 — Glucocorticoid induction (all types)
- Methylprednisolone 500 to 1000 mg IV daily for 3 days (pulse), then
- Oral prednisolone 1 mg/kg/day (max 60 to 80 mg) tapering over weeks to months to a maintenance dose of about 5 to 10 mg by 3 to 6 months.[4][5]
- Reduce the pulse dose in the elderly, in diabetics and in active infection; consider avacopan to spare steroids in ANCA disease (ADVOCATE — see below).
- PJP prophylaxis (co-trimoxazole 480 mg daily) and gastric/bone/glucose cover throughout.
Step 2 — Add an immunosuppressant (cyclophosphamide or rituximab)
For ANCA vasculitis (Type III): add rituximab OR cyclophosphamide — both are guideline-endorsed first-line.[4][5]
- Rituximab — 375 mg/m2 IV weekly for 4 weeks (RAVE regimen) OR 1 g IV at day 0 and day 14 (RITUXVAS regimen). Preferred in relapsing disease, in women of childbearing potential (less gonadal toxicity), and where cyclophosphamide is contraindicated.
- Cyclophosphamide — 2 mg/kg/day oral (max 200 mg/day) adjusted for age and renal function, OR IV pulse 15 mg/kg every 2 weeks (CYCLOPS). Co-prescribe mesna and hydration to protect the bladder, and counsel on gonadal toxicity and infertility (offer sperm/ova cryopreservation).[7]
RAVE trial (Stone 2010): rituximab was non-inferior to cyclophosphamide for induction and superior in relapsing disease — establishing rituximab as first-line for ANCA vasculitis.[6]
RITUXVAS trial (Jones 2010, 2-year 2015): rituximab plus cyclophosphamide was non-inferior to cyclophosphamide alone in renal-limited ANCA vasculitis, with sustained remission at 2 years.[7]
For anti-GBM disease (Type I): add cyclophosphamide 2 mg/kg/day oral (dose-reduce for age and renal function) to suppress ongoing antibody production, alongside plasma exchange.[1][3]
For immune-complex RPGN (Type II): treat the underlying disease — lupus induction (mycophenolate or cyclophosphamide plus steroids plus voclosporin), supportive PSGN, steroids ± cyclophosphamide for crescentic IgA nephropathy.[7]
Step 3 — Plasma exchange (selected patients)
For anti-GBM disease — plasma exchange is ESSENTIAL.[1][3]
- Daily or alternate-day plasma exchange.
- 4-litre exchanges with 5 per cent human albumin; add fresh frozen plasma (150 to 300 mL per session) if there is active bleeding or within 72 hours of renal biopsy.
- Continue until anti-GBM antibody is undetectable — typically 2 to 3 weeks (9 to 12 sessions).
- Rationale: removes the circulating pathogenic antibody (a tissue-fixed antibody disease).[9]
For ANCA vasculitis — plasma exchange is now RESTRICTED.[9]
- MEPEX trial (Jayne 2007): plasma exchange was superior to methylprednisolone pulse for renal recovery in severe renal ANCA vasculitis — the historical basis for plasma exchange in dialysis-dependent disease.[8]
- PEXIVAS trial (Walsh 2020): plasma exchange did NOT reduce the composite of death or ESKD in severe ANCA vasculitis — narrowing its routine use.[9]
- Current indications (KDIGO 2024, ACR/VF 2021): plasma exchange is reserved for (i) anti-GBM or double-positive disease; (ii) ANCA vasculitis with dialysis-dependence and rapidly falling GFR (creatinine over 5.7 mg/dL or 500 umol/L) where there is a chance of renal recovery; and (iii) life-threatening diffuse alveolar haemorrhage with hypoxaemia.[4][5][12]
Risks of plasma exchange: central venous catheter infection and bleeding, hypocalcaemia (citrate anticoagulant — give calcium replacement), allergic reaction to fresh frozen plasma, hypotension, and mild coagulopathy. Monitor calcium, fibrinogen and platelets.[9]
Step 4 — Maintenance (ANCA vasculitis)
ANCA vasculitis relapses in 30 to 50 per cent of patients within 5 years without maintenance; relapse risks irreversible renal damage. MAINRITSAN trial (Guillevin 2014): rituximab was superior to azathioprine for maintenance, reducing relapse.[10]
- Rituximab — 1 g IV at day 0 and day 14, then 500 mg to 1 g every 4 to 6 months for 18 to 36 months (longer if PR3-positive and high relapse risk). Guideline-preferred.[4][10]
- Azathioprine — 2 mg/kg/day oral (alternative if rituximab unavailable).
- Methotrexate or mycophenolate — alternatives if azathioprine not tolerated.
- Co-trimoxazole — for localised upper-respiratory GPA and to reduce Staphylococcus aureus carriage.
Anti-GBM disease generally does not require long-term maintenance (it is usually monophasic), but double-positive patients (ANCA + anti-GBM) should receive ANCA-style maintenance because of the relapsing ANCA component.[1]
Step 5 — Avacopan (C5a receptor blocker) — steroid-sparing
ADVOCATE trial: the C5a receptor antagonist avacopan (10 mg twice daily) was glucocorticoid-sparing, non-inferior for remission at 26 weeks and superior for sustained remission at 52 weeks in ANCA vasculitis. It interrupts the alternative-complement-pathway amplification loop identified in pathogenesis. KDIGO 2024 endorses avacopan as a steroid-sparing option in ANCA vasculitis.[4][12]
Step 6 — Supportive care (all types)
- RAAS blockade — ACE inhibitor (e.g. ramipril 2.5 to 10 mg daily) or ARB (e.g. losartan 50 to 100 mg daily) to reduce proteinuria and intraglomerular pressure, once the creatinine has stabilised; monitor potassium.
- SGLT2 inhibitor — for proteinuric CKD.
- BP target under 130/80 mmHg.
- Salt restriction under 2 g/day; fluid restriction to urine output plus 500 mL insensible losses if oliguric.
- Smoking cessation (reduces pulmonary haemorrhage risk in anti-GBM).
- Vaccination (influenza, pneumococcal, COVID-19, hepatitis B) — ideally before immunosuppression; avoid live vaccines during treatment.
- Statins for cardiovascular risk.
- Bone, PJP, gastric prophylaxis as above.[1]
Specific Subtypes & Scenarios
Anti-GBM disease (Goodpasture syndrome) — Type I
- Pathogenesis: IgG against alpha3(IV)NC1 in the GBM and alveolar BM; linear IF.
- Presentation: acute haemoptysis, rapidly progressive renal failure, iron-deficiency anaemia from pulmonary bleeding; bimodal age (young men with pulmonary haemorrhage; older women renal-limited).
- Management — triple therapy: (i) plasma exchange — daily or alternate-day, 4 L exchanges with 5 per cent albumin and FFP if bleeding, until anti-GBM antibody undetectable (about 2 to 3 weeks, 9 to 12 sessions); (ii) methylprednisolone pulse 500 to 1000 mg IV daily for 3 days then oral prednisolone 1 mg/kg/day tapering; (iii) cyclophosphamide 2 mg/kg/day oral (dose-reduce for age and renal function).[1][3]
- Prognosis: high ESKD risk if oligoanuric at presentation (over 50 per cent); significant early mortality from pulmonary haemorrhage.
- Transplant: wait until anti-GBM antibody undetectable for at least 6 months to minimise recurrence (which is rare but devastating).
- Relapse: uncommon (monophasic) — but double-positive patients may relapse as ANCA disease.[1][2]
Granulomatosis with polyangiitis (GPA) — Type III
- Antibody: anti-PR3 (cytoplasmic c-ANCA) in 75 to 90 per cent.
- Features: ENT (nasal crusting, epistaxis, saddle-nose deformity, sinusitis, subglottic stenosis), pulmonary (nodules, cavities, infiltrates, haemorrhage), ocular (scleritis, orbital pseudotumour), renal (crescentic GN), mononeuritis multiplex.
- Induction: methylprednisolone pulse then oral prednisolone PLUS rituximab (preferred) or cyclophosphamide.
- Maintenance: rituximab 1 g every 4 to 6 months for 18 to 36 months; PR3 positivity predicts relapse.
- Localised upper respiratory disease: may respond to co-trimoxazole and reduced immunosuppression (Staphylococcus aureus carriage drives relapse).
- Relapse rate: 30 to 50 per cent over 5 years without maintenance.[4]
Microscopic polyangiitis (MPA) — Type III
- Antibody: anti-MPO (perinuclear p-ANCA) in 50 to 70 per cent.
- Features: renal-predominant disease (crescentic GN often the presenting feature), pulmonary haemorrhage (capillaritis) but NO granulomatous ENT disease, palpable purpura, mononeuritis multiplex, GI involvement.
- Induction and maintenance: as for GPA. Less likely to relapse than GPA.[10]
Eosinophilic granulomatosis with polyangiitis (EGPA, Churg-Strauss) — Type III
- Triad: adult-onset asthma, eosinophilia (over 1.5 x10^9/L or 10 per cent), sinusitis; plus mononeuritis multiplex and (often) cardiac involvement (eosinophilic myocarditis — a leading cause of death).
- Antibody: anti-MPO in about 40 per cent.
- Treatment: mepolizumab (anti-IL-5 receptor) for the eosinophilic/ANCA-negative phenotype; steroids plus rituximab or cyclophosphamide for ANCA-positive severe or organ-threatening disease.[6]
Crescentic lupus nephritis (class IV) — Type II
- Serology: low C3 AND C4, ANA and anti-dsDNA positive.
- Biopsy: full-house IF (IgG, IgA, IgM, C3, C1q, C4); wire-loop deposits; crescents.
- Induction (3 to 6 months): mycophenolate mofetil 2 to 3 g/day OR low-dose IV cyclophosphamide (Euro-Lupus 500 mg every 2 weeks for 6 doses) PLUS glucocorticoids (methylprednisolone pulse then prednisolone 0.5 to 0.8 mg/kg/day tapering), with voclosporin 23.7 mg twice daily or tacrolimus as triple therapy, and hydroxychloroquine 200 to 400 mg daily throughout.
- Maintenance: mycophenolate 1 to 2 g/day or azathioprine 2 mg/kg/day plus low-dose steroids.[8]
Crescentic IgA nephropathy — Type II
- Presentation: synpharyngitic gross haematuria; mesangial IgA-dominant deposits on IF.
- Management: supportive RAAS blockade; steroids (and cyclophosphamide for fulminant crescentic disease with rapidly falling GFR).[6]
The double-positive (ANCA + anti-GBM) patient
- Manage as anti-GBM disease — add plasma exchange, methylprednisolone and cyclophosphamide.
- ANCA positivity predicts a relapsing course once anti-GBM antibody is suppressed — give maintenance rituximab or azathioprine after the acute episode.[1][2]
Drug-induced ANCA vasculitis
- Causative drugs: hydralazine, propylthiouracil, levamisole-adulterated cocaine, minocycline, allopurinol.
- Pattern: typically anti-MPO with anti-histone and sometimes anti-elastase co-positivity.
- Management: stop the offending drug; immunosuppression as for idiopathic ANCA if severe.[9]
Complications & Pitfalls
Acute complications
- Irreversible ESKD requiring dialysis — especially anti-GBM with oligoanuria at presentation and dialysis-dependent ANCA.
- Diffuse alveolar haemorrhage and respiratory failure — anti-GBM and ANCA; a leading cause of early death.
- Hypertensive encephalopathy and intracerebral haemorrhage — from severe hypertension.
- Pulmonary oedema — from volume overload and oliguria.
- Hyperkalaemia and metabolic acidosis — from AKI.
- Opportunistic infection — Pneumocystis jirovecii pneumonia (prevent with co-trimoxazole), cytomegalovirus, fungal infection, from immunosuppression; a leading cause of death.
- Thromboembolism — renal vein, deep vein thrombosis, pulmonary embolism (vasculitic vasculopathy plus nephrotic-range proteinuria).
- Cyclophosphamide toxicity — haemorrhagic cystitis, bladder cancer (long-term risk), infertility, cytopaenias, infection.[7]
Chronic complications
- Progression to CKD/ESKD — particularly anti-GBM with oligoanuria at presentation, dialysis-dependent ANCA, crescentic lupus nephritis, IgA with persistent proteinuria.
- Cardiovascular disease — accelerated by CKD and chronic inflammation.
- Long-term steroid effects — osteoporosis, diabetes, cataracts, hypertension, infection, avascular necrosis.
- Cyclophosphamide effects — gonadal toxicity and infertility, bladder cancer, myelodysplasia, secondary malignancy.
- Relapse — especially PR3-ANCA/GPA (30 to 50 per cent over 5 years without maintenance).[7]
Classic pitfalls
- Delaying treatment while awaiting biopsy or serology — the cardinal error; start high-dose steroids on suspicion.
- Assuming all rapidly progressive AKI is ATN and missing RBC casts — the active sediment redirects to a glomerular cause.
- Treating ANCA with routine plasma exchange after PEXIVAS — restrict to dialysis-dependent crescentic ANCA with rapidly falling GFR and life-threatening alveolar haemorrhage.
- Failing to PJP-prophylax steroid- and cyclophosphamide-treated patients — PJP is a leading cause of death.
- Missing a double-positive (ANCA + anti-GBM) patient — these need plasma exchange (manage as anti-GBM).
- Biopsying small, scarred kidneys aggressively — small kidneys (under 9 cm) indicate chronic irreversible damage and contraindicate aggressive immunosuppression.
- Underestimating the mortality of diffuse alveolar haemorrhage — secure the airway, involve ITU early.
- Forgetting cryopreservation before cyclophosphamide in younger patients — offer sperm/ova banking.[8]
Cyclophosphamide-specific risks and mitigations
| Risk | Mitigation |
|---|---|
| Haemorrhagic cystitis | Mesna; vigorous hydration; avoid evening doses |
| Bladder cancer (long-term) | Surveillance urinalysis for haematuria; cystoscopy if persistent |
| Infertility / gonadal toxicity | Sperm/ova cryopreservation before treatment; consider gonadotropin-releasing hormone agonist in women |
| Cytopaenias, infection | Monitor CBC; dose-reduce for renal function and age; growth factor support |
| Malignancy (long-term) | Minimise cumulative dose; switch to rituximab where possible |
Plasma exchange — risks
- Central venous catheter infection and bleeding.
- Hypocalcaemia from citrate anticoagulant (paraesthesia, tetany) — give calcium replacement.
- Allergic reaction to fresh frozen plasma.
- Hypotension and mild coagulopathy — monitor fibrinogen and platelets.[8]
Prognosis & Disposition
Predictors of outcome
Prognosis depends on:[11]
- Speed of treatment — the single most important modifiable factor; once crescents are fibrous the damage is irreversible.
- Percentage of glomeruli with crescents and the proportion of fibrous (chronic) crescents on biopsy.
- GFR at presentation and dialysis-dependence at presentation.
- Antibody type — anti-GBM and dialysis-dependent ANCA do worst.
- Patient factors — age (elderly worse), comorbidity, infection.[5]
By cause
| Cause | Renal recovery / prognosis |
|---|---|
| ANCA vasculitis, dialysis-dependent at presentation | About 50 to 70 per cent renal recovery with prompt induction; dialysis-independence at 3 months predicts long-term renal survival(see MEPEX, PEXIVAS)[8][9] |
| ANCA vasculitis, GFR preserved at presentation | Over 80 per cent remission with induction; 30 to 50 per cent relapse over 5 years without maintenance[4][10] |
| Anti-GBM disease | Over 50 per cent ESKD risk if oligoanuric at presentation; significant early mortality from pulmonary haemorrhage; monophasic (relapse rare if antibody stays negative)[1][3] |
| Crescentic lupus nephritis | Variable; 5- and 10-year renal survival improved with modern induction (MMF/cyclophosphamide + voclosporin) |
| Crescentic IgA nephropathy | Moderate; worse with sustained proteinuria over 1 g/day and interstitial fibrosis |
| Post-infectious crescentic GN | Generally good in children; worse in adults with heavy crescent burden |
Patient survival
- ANCA vasculitis — 1-year mortality about 10 to 20 per cent; infection and active vasculitis are the leading causes of death, followed by cardiovascular disease and malignancy.[4]
- Anti-GBM disease — significant early mortality from pulmonary haemorrhage; survives into long-term remission if the acute episode is weathered.
Relapse prediction in ANCA disease
- PR3-ANCA positivity, GPA phenotype, persistent ANCA positivity after induction, and upper respiratory involvement predict relapse.
- Relapse rate is 30 to 50 per cent over 5 years without maintenance; maintenance rituximab (MAINRITSAN) markedly reduces this.[10]
Indications for nephrology referral and admission
- Emergency admission for any suspected RPGN or pulmonary-renal syndrome (haemoptysis, rapidly progressive GN); severe hypertension or hypertensive encephalopathy; pulmonary oedema; hyperkalaemia.
- Urgent nephrology referral for all suspected crescentic GN — for urgent biopsy and induction immunosuppression.[8]
Indications for renal replacement therapy and transplant
- ESKD requiring dialysis or transplant.
- Anti-GBM transplant: wait until anti-GBM antibody undetectable for at least 6 months to minimise recurrence.[1]
- ANCA transplant: possible after remission; ANCA recurrence in the graft is uncommon but possible.
Special Populations
Children
- Post-infectious crescentic GN and IgA vasculitis (Henoch-Schonlein purpura) nephritis dominate; anti-GBM is rare.
- Treatment principles are the same with weight-based dosing of steroids, cyclophosphamide and rituximab.
- Long-term outcome of crescentic IgA vasculitis is generally good; crescentic PSGN usually resolves with supportive care.[6]
Elderly
- ANCA vasculitis is the dominant cause of crescentic GN; atypical presentation with fatigue, weight loss or unexplained AKI.
- Reduced tolerance to cyclophosphamide — dose-reduce for age and renal function.
- Higher infection and mortality risk — consider reduced-dose regimens and early avacopan to spare steroids.
- Comorbidity (cardiac, vascular) complicates both induction and prognosis.[7]
Pregnancy
- Lupus nephritis may flare (especially postpartum); cyclophosphamide is teratogenic and contraindicated — use azathioprine (up to 2 mg/kg/day) and steroids, with calcineurin inhibitors (tacrolimus, ciclosporin) where needed.
- Plasma exchange is safe in pregnancy.
- Pre-eclampsia can mimic RPGN — distinguish by complement, serology and RBC casts (pre-eclampsia has no RBC casts, no dysmorphic RBCs, no ANCA/anti-GBM, and typically low platelets and raised liver enzymes after 20 weeks).[9]
Immunocompromised (HIV, transplant)
- HIV-associated immune-complex kidney disease (HIVICK) and collapsing FSGS.
- Post-transplant recurrent disease (IgA, MPGN/C3 glomerulopathy; lupus rarely recurs).
- Drug interactions with calcineurin inhibitors and rituximab.[7]
Anticoagulated patients
- Weigh bleeding risk against biopsy — correct INR with vitamin K or fresh frozen plasma, or use transjugular renal biopsy.
- Plasma exchange requires careful anticoagulation management (citrate, FFP) and catheter-site vigilance.[8]
Evidence, Guidelines & Regional Differences
Landmark trials
RAVE (2010)
Stone et al., NEJM
- **Rituximab 375 mg/m2 weekly x4 vs cyclophosphamide then azathioprine** for ANCA vasculitis induction
- **Rituximab non-inferior to cyclophosphamide and SUPERIOR in relapsing disease** — established rituximab as first-line induction
RITUXVAS (2010/2015)
Jones et al.
- Rituximab plus cyclophosphamide vs cyclophosphamide alone in renal-limited ANCA vasculitis
- **Non-inferior for remission; sustained at 2 years** — supports rituximab in renal ANCA
MEPEX (2007)
Jayne et al., JASN
- **Plasma exchange vs methylprednisolone pulse** as adjunct for severe renal ANCA vasculitis
- **Plasma exchange superior for renal recovery at 3 months** — historical basis for PLEX in dialysis-dependent disease
PEXIVAS (2020)
Walsh et al., NEJM
- Largest trial (704 patients) of plasma exchange + reduced-dose vs standard-dose steroids in severe ANCA
- **Plasma exchange did NOT reduce death or ESKD** — narrowed routine PLEX to anti-GBM, dialysis-dependent crescentic ANCA, life-threatening haemorrhage
MAINRITSAN (2014)
Guillevin et al., NEJM
- **Rituximab 500 mg day 0/14 then 6/12/18 months vs azathioprine** for ANCA maintenance
- **Rituximab superior to azathioprine — fewer relapses** — established rituximab as preferred maintenance
ADVOCATE (avacopan)
C5a receptor blocker
- Avacopan 10 mg twice daily as steroid-sparing in ANCA vasculitis
- **Non-inferior for remission at 26 weeks, superior for sustained remission at 52 weeks** with less steroid toxicity — KDIGO 2024 endorsed
Guidelines
- KDIGO 2024 Clinical Practice Guideline for the Management of ANCA Vasculitis — the current international standard; endorses rituximab-first induction, avacopan as steroid-sparing, and a restricted role for plasma exchange.[12]
- KDIGO 2021 Glomerular Diseases Guideline and 2024 IgA nephropathy update — the international standard for evaluation and treatment, with the supportive-care-first philosophy for IgA and the immunosuppressive induction regimens for RPGN and lupus.[11]
- ACR/VF 2021 Guideline for Management of ANCA-Associated Vasculitis — American recommendations for induction, maintenance and disease monitoring.[5]
- EULAR 2022 recommendations for ANCA-associated vasculitis management — European position, broadly concordant with ACR/VF.
Regional differences
- India and resource-limited settings: limited biopsy access mandates empirical treatment on clinical and serological grounds; endemic post-infectious crescentic GN and lupus are common; hydralazine- and propylthiouracil-induced ANCA is over-represented; cost and availability barriers to rituximab and plasma exchange push toward cyclophosphamide-based regimens and supportive care.
- United States/Europe: rituximab-first induction, avacopan steroid-sparing, and protocolised maintenance with rituximab are standard.
- Globally: the principle of empirical high-dose steroids before biopsy holds everywhere; the absolute speed of treatment determines outcome.[6]
Controversies
- Plasma exchange in ANCA after PEXIVAS — restricted, but many nephrologists still use it for dialysis-dependent crescentic ANCA in the first weeks, hoping for renal recovery.
- Duration of maintenance — rituximab for 18 vs 36 months in high-relapse-risk (PR3, GPA) disease.
- Avacopan access — cost and availability vary; its place alongside (rather than instead of) steroids is evolving.
- Treatment of ANCA-negative pauci-immune crescentic GN — managed as ANCA disease on biopsy phenotype.[8]
Exam Pearls
- RPGN = renal emergency; crescents on biopsy; AKI over days to weeks with RBC casts.
- Three immunofluorescence types: Type I anti-GBM (linear IgG, Goodpasture, pulmonary haemorrhage); Type II immune-complex (granular; lupus, post-infectious, IgA); Type III pauci-immune (ANCA — GPA, MPA; commonest in adults).[1][4]
- RBC casts = glomerular bleeding — the single most discriminating microscopy finding.
- ANCA: anti-PR3 (c-ANCA) = GPA (ENT and lung); anti-MPO (p-ANCA) = MPA (renal-predominant).
- Anti-GBM targets alpha3(IV)NC1 — the non-collagenous domain of the alpha-3 chain of type IV collagen in the GBM and alveolar basement membrane.
- Four drug causes of ANCA vasculitis: hydralazine, propylthiouracil, levamisole-adulterated cocaine, minocycline.
- Crescents in over 50 per cent of glomeruli defines RPGN histologically; fibrous crescents are irreversible — speed of treatment determines outcome.
- Treat empirically with high-dose IV methylprednisolone BEFORE biopsy results.
- Add cyclophosphamide or rituximab; plasma exchange for anti-GBM and selected ANCA (dialysis-dependent, life-threatening alveolar haemorrhage).[3][9]
- RAVE: rituximab non-inferior to cyclophosphamide and superior in relapse. PEXIVAS: plasma exchange does NOT reduce death/ESKD in severe ANCA. MAINRITSAN: rituximab superior to azathioprine for maintenance. MEPEX: plasma exchange superior to methylprednisolone for renal recovery in dialysis-dependent ANCA.[6][9][10][8]
- Double-positive (ANCA + anti-GBM) = manage as anti-GBM (add plasma exchange); ANCA predicts a relapsing course.
- Two prognostic scores: BVAS (disease activity) and FFS (Five-Factor Score — age over 65, renal insufficiency, cardiac, GI, absence of ENT involvement).
- Anti-GBM transplant rule: wait until antibody undetectable for at least 6 months.
Exam application bank (NEET-PG / INICET)
One-line answer
Rapidly progressive glomerulonephritis (RPGN) is the most aggressive form of glomerulonephritis and a true renal emergency: a syndrome of rapid loss of renal function (loss of over 50 per cent of GFR within under 3 months), unified by the histological hallmark of crescents — fibrin and proliferating parietal epithelial cells — in Bowman's space. Untreated it progresses to end-stage kidney disease within weeks. It is classified by immunofluorescence into three types: Type I anti-GBM (linear IgG; Goodpasture syndrome with pulmonary haemorrhage), Type II immune-complex (granular; lupus, post-infectious, IgA) and Type III pauci-immune (ANCA-associated vasculitis — GPA, microscopic polyangiitis; the commonest form in adults). Presentation is a rapidly rising creatinine with haematuria and dysmorphic red cells / red-cell casts, often with systemic vasculitic features or haemoptysis (the pulmon
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard.[8]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes.[1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change.[1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each.[1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory.[1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Rapidly Progressive Glomerulonephritis.
References
- [1]McAdoo SP, Pusey CD. Anti-Glomerular Basement Membrane Disease. Clinical Journal of the American Society of Nephrology, 2017.PMID 28515156
- [2]Ponticelli C, Cresseri D, Tarantino A, Moroni G. Anti-glomerular basement membrane vasculitis. Autoimmunity Reviews, 2023.PMID 36252931
- [3]Reggiani F, Esposito P, Togliatto L, et al. Goodpasture syndrome and anti-glomerular basement membrane disease. Clinical and Experimental Rheumatology, 2023.PMID 36995324
- [4]Kronbichler A, Subramanian AK, Smith RM, Jayne DRW. Diagnosis and management of ANCA-associated vasculitis. The Lancet, 2024.PMID 38368016
- [5]Chung SA, Langford CA, Maz M, et al. 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the Management of Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis and Rheumatology, 2021.PMID 34235894
- [6]Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis (RAVE trial). The New England Journal of Medicine, 2010.PMID 20647199
- [7]Jones RB, Furuta S, Terveaert JWC, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis: 2-year results of a randomised trial (RITUXVAS). Annals of the Rheumatic Diseases, 2015.PMID 25739829
- [8]Jayne DR, Gaskin G, Rasmussen N, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis (MEPEX). Journal of the American Society of Nephrology, 2007.PMID 17582159
- [9]Walsh M, Merkel PA, Peh CA, et al. Plasma Exchange and Glucocorticoids in Severe ANCA-Associated Vasculitis (PEXIVAS). The New England Journal of Medicine, 2020.PMID 32053298
- [10]Guillevin L, Pagnoux C, Karras A, et al. Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis (MAINRITSAN). The New England Journal of Medicine, 2014.PMID 25372085
- [11]Rovin BH, Caster DJ, Cattran DC, et al. Management and treatment of glomerular diseases (part 2): conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney International, 2019.PMID 30665569
- [12]Kidney Disease: Improving Global Outcomes (KDIGO) ANCA Vasculitis Work Group. KDIGO 2024 Clinical Practice Guideline for the Management of Antineutrophil Cytoplasmic Antibody-Associated Vasculitis (corrigendum). Kidney International, 2025.PMID 39848754