Paediatrics · Paediatrics
Childhood Leukaemia
Also known as Childhood Leukaemia
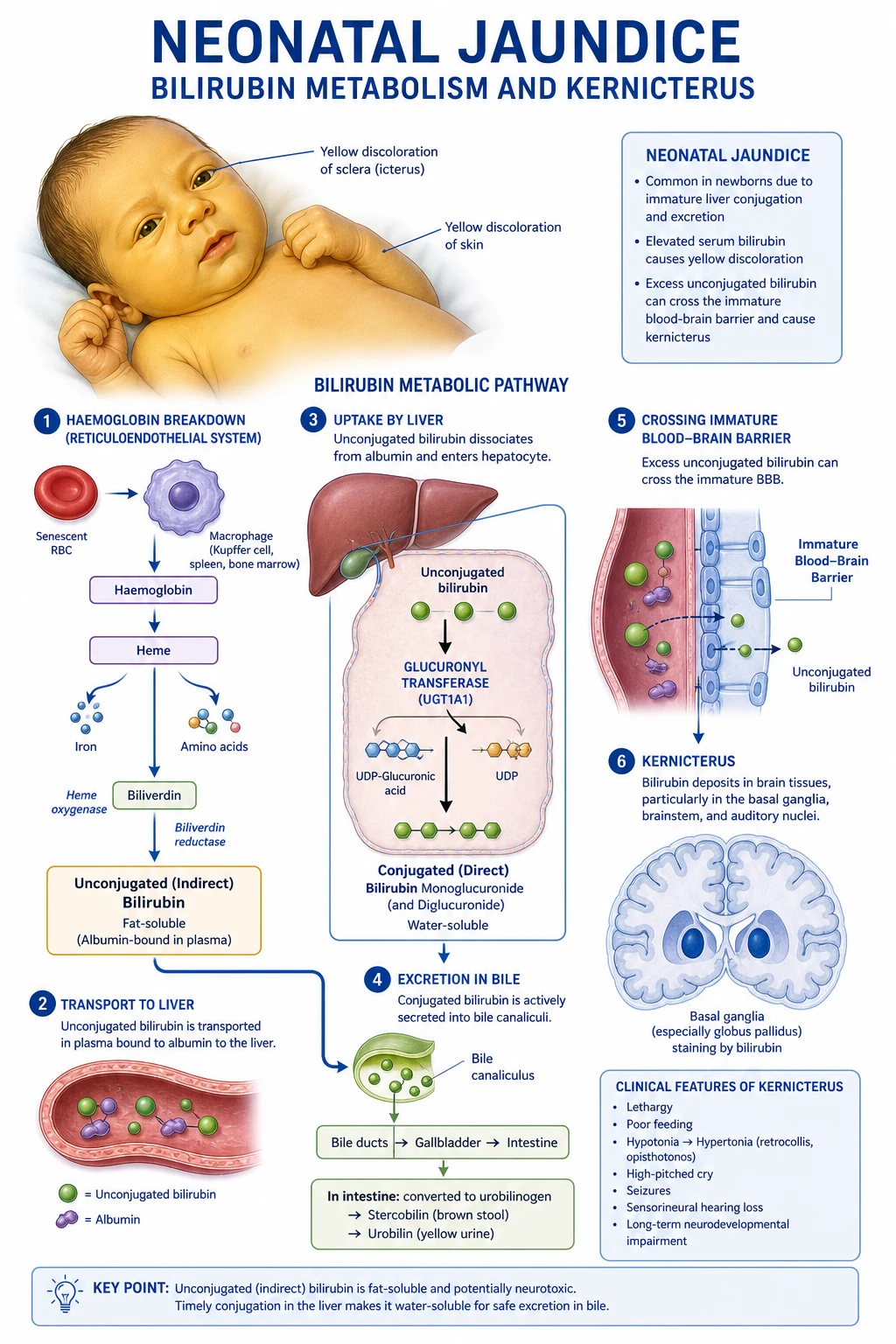
Childhood leukaemia is the most common childhood malignancy (30% of all childhood cancers). Acute lymphoblastic leukaemia (ALL) accounts for 75-80%; acute myeloid leukaemia (AML) 15-20%. Peak age: 2-5 years. Presentation: bone marrow failure (anaemia, infection, bleeding), organ infiltration (hepatosplenomegaly, lymphadenopathy, CNS). Diagnosis: FBC + blood film + bone marrow aspirate (morphology, flow cytometry, cytogenetics). Treatment: risk-stratified chemotherapy. 5-year survival ALL: 90%, AML: 65-70%.
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags
Overview
Leukaemia is the most common childhood cancer, accounting for 30% of all paediatric malignancies. [1]
Presentation
Bone Marrow Failure
- Anaemia: pallor, fatigue, lethargy, dyspnoea
- Infection/fever: neutropenia → bacterial/viral/fungal infections
- Bleeding/bruising: thrombocytopenia → petechiae, purpura, mucosal bleeding
Organ Infiltration
- Hepatosplenomegaly: common in ALL
- Lymphadenopathy: cervical, axillary, inguinal
- CNS signs: headache, vomiting, cranial nerve palsies (meningeal involvement)
- Bone pain: (especially legs), limping, refusal to walk
- Testicular swelling: leukaemic infiltration (T-ALL)
- Mediastinal mass: T-ALL (superior vena cava syndrome)
- Gum hypertrophy: AML (monocytic differentiation — M4/M5)
Diagnosis
| Investigation | Role |
|---|---|
| FBC + blood film | Blasts, pancytopenia, leukocytosis, leukopenia |
| Bone marrow aspirate + trephine | Gold standard: morphology, immunophenotyping (flow cytometry), cytogenetics, molecular testing |
| Lumbar puncture | CNS involvement (send CSF for cytology) |
| Coagulation | DIC screen (especially APL/M3) |
| Imaging (CXR) | Mediastinal mass (T-ALL), infections |
| LDH, urate | Tumour lysis syndrome risk |
| Infection screen | Blood, urine, viral (EBV, CMV) |
Diagnostic threshold: ≥20% blasts in the bone marrow (or peripheral blood) confirms acute leukaemia. The bone marrow aspirate and trephine provide morphology, and the minimum sample required includes immunophenotyping (flow cytometry), cytogenetics (karyotype, FISH) and molecular testing (PCR for fusion transcripts) — all essential for risk stratification and treatment assignment.[1]
Classification (WHO/FAB)

ALL (by immunophenotype)
- B-cell ALL (85%): better prognosis
- T-cell ALL (15%): mediastinal mass, higher WCC, poorer prognosis
AML (FAB M0-M7)
- M0: minimally differentiated; M1/M2: myeloid without/with maturation; M4: myelomonocytic (gum hypertrophy); M5: monoblastic (common in Down syndrome infants)
- M3 = Acute Promyelocytic Leukaemia (APL): t(15;17), DIC risk, treat with ATRA (all-trans retinoic acid) + arsenic trioxide + chemotherapy
- M6: erythroid; M7: megakaryoblastic (common in Down syndrome, transient abnormal myelopoiesis)
- Diagnosis: ≥20% blasts in marrow or peripheral blood; immunophenotyping via flow cytometry confirms lineage (myeloid markers CD13/CD33/CD117; lymphoid markers CD19/CD79a/CD10)[3]
Cytogenetics and Risk Stratification
| Cytogenetic Finding | Risk |
|---|---|
| Hyperdiploidy (over 50 chromosomes) | GOOD (ALL) |
| ETV6-RUNX1 (TEL-AML1) t(12;21) | GOOD (ALL) |
| Philadelphia chromosome t(9;22) BCR-ABL1 | POOR (ALL) — TKI (imatinib/dasatinib) added |
| Hypodiploidy (under 44 chromosomes) | POOR (ALL) |
| MLL rearrangement t(4;11) | POOR (infant ALL) |
| t(15;17) PML-RARA | APL — ATRA-responsive |
Minimal residual disease (MRD) at end of induction (day 29) is the single most powerful prognostic marker: MRD-negative (below 0.01%) → standard/low risk; MRD-positive → intensification. Combined with age (1-10 years = favourable), initial WCC (under 50 × 10⁹/L = standard risk), and cytogenetics, MRD drives risk-adapted therapy in modern protocols.[1]
Treatment
ALL Treatment (UKALL Protocol)
- Induction (4-6 weeks): dexamethasone/prednisolone, vincristine, asparaginase, daunorubicin → remission in 95%
- Consolidation: methotrexate, mercaptopurine, cytarabine
- CNS-directed therapy: intrathecal methotrexate (CNS sanctuary site)
- Maintenance: daily mercaptopurine, weekly methotrexate for 2-3 years
- Total duration: 2-3 years (boys longer than girls)
AML Treatment
- Intensive induction (2 cycles): cytarabine + daunorubicin/etoposide
- Consolidation: further cycles or bone marrow transplant (BMT) for high-risk
- APL specifically: ATRA + arsenic trioxide + ATRA-based chemo (different from other AML)
Complications of Treatment
- Tumour lysis syndrome: hyperuricaemia, hyperkalaemia, hyperphosphataemia → renal failure. Prevention: rasburicase, allopurinol, aggressive hydration
- Infection: febrile neutropenia → urgent broad-spectrum IV antibiotics (piperacillin-tazobactam ± gentamicin ± vancomycin)
- Pancytopenia: transfusion support (RBC, platelets)
- Mucositis, nausea, alopecia: supportive care
- Late effects: cardiotoxicity (anthracyclines), growth impairment, secondary malignancies, infertility
ALL TREATMENT
TUMOUR LYSIS
Epidemiology and Risk Factors
Childhood leukaemia accounts for 30% of all paediatric malignancies, with an annual incidence of approximately 40-45 cases per million children under 15 years. ALL is the single most common paediatric cancer, roughly 4-5 times more common than AML in children (the reverse ratio of adults).[2]
Key epidemiological features: [1]
- Peak age: 2-5 years for B-cell precursor ALL, reflecting the delayed consequence of a prenatal leukaemic translocation plus a postnatal second hit
- Sex: slight male predominance (M:F approximately 1.2:1 for ALL)
- Geography: incidence highest in high-income countries (likely surveillance bias); ALL accounts for a higher proportion of childhood cancer in affluent populations [1]
Predisposing genetic conditions: [1]
Environmental associations (weaker evidence): high birth weight, maternal X-ray exposure during pregnancy, paternal preconception smoking, benzene or solvent exposure. There is no convincing evidence that electromagnetic fields, routine vaccinations, or common childhood infections directly cause leukaemia.[2]
Pathophysiology
Childhood leukaemia arises from clonal expansion of haematopoietic precursor cells arrested at an early stage of differentiation. The immature blasts proliferate uncontrollably, fail to undergo apoptosis, and accumulate in the bone marrow, progressively displacing normal haematopoiesis. [1]
The two-hit hypothesis (adapted from Knudson):[1]
- First hit (prenatal): a chromosomal translocation occurs in utero in a haematopoietic stem cell or early progenitor. This has been proven by Guthrie card (neonatal blood spot) backtracking studies — leukaemic fusion genes such as ETV6-RUNX1 are detectable at birth in children who later develop ALL, yet only about 1% of newborns carrying the translocation actually develop leukaemia.
- Second hit (postnatal): additional genetic event(s) in the pre-leukaemic clone trigger overt leukaemia. Common second hits include deletion of CDKN2A/B, PAX5, ETV6 or IKZF1, and activating mutations in RAS pathway genes (KRAS, NRAS). [1]
Consequences of blast infiltration: [1]
- Marrow failure: blasts crowd out normal precursors, causing anaemia, neutropenia and thrombocytopenia
- Organ infiltration: liver, spleen, lymph nodes, CNS meninges, testes, bones and skin (leukaemia cutis)
- Metabolic derangement: rapid cell turnover causes tumour lysis syndrome with hyperuricaemia, hyperkalaemia and hyperphosphataemia [1]
Key fusion oncoproteins: ETV6-RUNX1 blocks lymphoid differentiation; PML-RARA blocks myeloid maturation at the promyelocyte stage (APL); BCR-ABL1 provides constitutive tyrosine kinase signalling (Philadelphia-positive ALL, targetable with imatinib and dasatinib).[1]
Differential Diagnosis
The presentation of childhood leukaemia — fever, pallor, bruising, organomegaly, bone pain — overlaps with several common paediatric conditions. A FBC and blood film is the critical first investigation in every child with unexplained cytopenia or systemic symptoms. [1]
Key differentials to exclude: [1]
- Immune thrombocytopenia (ITP): isolated thrombocytopenia with normal haemoglobin and neutrophil count, no blasts on film, otherwise well child — the most important distinction at the bedside
- Aplastic anaemia: pancytopenia with hypocellular marrow and no blasts — the key distinguishing feature from leukaemia on bone marrow biopsy
- Infectious mononucleosis (EBV): atypical lymphocytes may mimic blasts; heterophile antibody and EBV serology confirm; usually no significant pancytopenia
- Juvenile idiopathic arthritis (JIA): bone and joint pain with fever, but FBC is normal; systemic JIA (Still disease) may cause cytopenias and ferritin elevation mimicking HLH
- Neuroblastoma: retrobulbar ecchymosis (raccoon eyes), abdominal mass, opsoclonus-myoclonus; diagnose with elevated urinary VMA and HVA, tissue biopsy
- Osteomyelitis or septic arthritis: localised bone pain with fever; blood count usually normal
- Haemophagocytic lymphohistiocytosis (HLH): persistent fever, splenomegaly, cytopenias, very high ferritin, high soluble IL-2 receptor [1]
Treatment Protocols: UKALL and AML Regimens
ALL — UKALL Protocol (phases and drug dosing)
The UKALL protocol (currently UKALL 2019) is the risk-stratified backbone of ALL treatment in the UK and Ireland. Total duration is 2 to 3.5 years (boys are treated longer than girls due to testicular sanctuary and higher relapse risk).[2]
Phase 1 — Induction (4-6 weeks): [1]
| Drug | Dose | Route | Duration |
|---|---|---|---|
| Dexamethasone | 6 mg/m²/day | PO | Days 1-28 |
| Vincristine | 1.5 mg/m² (max 2 mg per dose) | IV weekly | Days 1, 8, 15, 22 |
| PEG-asparaginase | 1000-2500 IU/m² | IM or IV | Days 4, 18 |
| Daunorubicin | 25 mg/m² | IV weekly | Days 1, 8, 15 (standard and high-risk only) |
| Intrathecal methotrexate | Age-adjusted (see below) | IT | Days 1, 8, 15, 22 |
- Dexamethasone is preferred over prednisolone — better CNS penetration, lower CNS relapse rate, improved event-free survival (UKALL 97/99 evidence)
- Intrathecal methotrexate age-adjusted doses: under 1 year = 6 mg; 1-2 years = 8 mg; 2-3 years = 10 mg; 3 years and over = 12 mg
- Asparaginase allergy or pancreatitis: switch from PEG-asparaginase to Erwinia asparaginase (alternative preparation) [1]
Phase 2 — Consolidation and intensification (4-8 weeks): [1]
- High-dose methotrexate: 2-5 g/m² IV over 24 hours with folinic acid (leucovorin) rescue — provides CNS sanctuary penetration
- 6-Mercaptopurine: 50-75 mg/m²/day PO throughout consolidation
- Cytarabine and cyclophosphamide in some intensification blocks (UKALL includes two delayed intensification blocks) [1]
Phase 3 — CNS-directed therapy: [1]
- Intrathecal methotrexate throughout treatment (CNS is a sanctuary site where systemic drugs penetrate poorly)
- Cranial radiotherapy (12-18 Gy): now reserved for high-risk CNS disease only (CNS3 status at diagnosis or CNS relapse); avoided in standard-risk patients due to late neurocognitive effects, secondary brain tumours and endocrinopathy [1]
Phase 4 — Maintenance (continuous, 18-30 months): [1]
| Drug | Dose | Frequency |
|---|---|---|
| 6-Mercaptopurine | 50-75 mg/m²/day | Daily PO (titrate to ANC 1-2 × 10⁹/L) |
| Methotrexate | 20 mg/m² | Weekly PO |
| Vincristine and dexamethasone pulses | As per induction doses | Every 4-12 weeks |
- TPMT (thiopurine methyltransferase) testing before starting 6-mercaptopurine: homozygous-deficient patients need approximately 10% of standard dose due to risk of fatal myelosuppression [1]
Philadelphia chromosome-positive (Ph+) ALL: add tyrosine kinase inhibitor — imatinib 300-340 mg/m²/day or dasatinib 60-80 mg/m²/day from induction onwards; stem cell transplant in first complete remission was historical standard but is now less frequent with TKI integration.[2]
AML — intensive chemotherapy
AML treatment is shorter but far more intensive than ALL, with prolonged inpatient stays, severe myelosuppression and high transfusion and infection burden.[3]
| Phase | Drugs | Details |
|---|---|---|
| Induction (2 cycles) | Cytarabine 100 mg/m²/day CI × 10 days + Daunorubicin 50 mg/m²/day × 3 days + Etoposide 100 mg/m²/day × 5 days (ADE 10+3+5) | APL excluded |
| Consolidation (3-4 cycles) | High-dose cytarabine 3 g/m² q12h × 4-6 doses ± anthracycline or etoposide | Risk-adapted |
| Stem cell transplant | Matched unrelated donor or matched sibling donor | High-risk cytogenetics, MRD-positive, relapse |
APL (M3) — specific regimen: [1]
- ATRA (all-trans retinoic acid): 25 mg/m²/day PO until remission (induces differentiation of the leukaemic promyelocytes)
- Arsenic trioxide: 0.15 mg/kg/day IV (differentiation and pro-apoptotic therapy)
- Plus ATRA-based chemotherapy (different from other AML subtypes)
- Differentiation syndrome (formerly APL differentiation or retinoic acid syndrome): fever, respiratory distress, weight gain, pleural and pericardial effusions, renal failure — treat with dexamethasone 10 mg IV twice daily and hold ATRA if severe [1]
DEXA OVER PRED
Tumour Lysis Syndrome
Tumour lysis syndrome (TLS) is an oncological emergency caused by the massive release of intracellular contents when tumour cells are rapidly lysed — typically at the start of chemotherapy in patients with high tumour burden (WCC over 100 × 10⁹/L, bulky disease, Burkitt leukaemia or lymphoma, high-grade AML).[4]
[1]Clinical features: oliguria or anuria, haematuria, cardiac arrhythmias (from hyperkalaemia), muscle cramps, paraesthesiae and seizures (from hypocalcaemia), lethargy, nausea and potentially cardiac arrest. [1]
Prevention protocol (start BEFORE chemotherapy): [1]
| Strategy | Details |
|---|---|
| Hydration | IV fluids at 3 L/m²/day (or 2-3 mL/kg/hr) without added potassium. Maintain urine output above 100 mL/m²/hr. Do NOT alkalinise the urine — alkalinisation worsens calcium-phosphate precipitation and xanthine crystallisation |
| Allopurinol (low to intermediate risk) | 10 mg/kg/day PO in 2-3 divided doses (max 300 mg/day), or 200-300 mg/m²/day. Xanthine oxidase inhibitor — prevents formation of new uric acid |
| Rasburicase (high risk or established TLS) | 0.15 to 0.2 mg/kg IV as a single dose (can repeat at 0.15 mg/kg after 24 hr if needed, up to 5 days). Recombinant urate oxidase — degrades existing uric acid to allantoin (unlike allopurinol which only prevents new formation). Contraindicated in G6PD deficiency (causes severe haemolysis) |
Monitoring during the TLS risk period: [1]
- U&E, urate, phosphate, calcium and creatinine every 4-6 hours for the first 48-72 hours
- Continuous cardiac monitoring if potassium above 6 mmol/L
- Treat hyperkalaemia: calcium gluconate 0.5 mL/kg of 10% IV (cardioprotection), insulin-dextrose infusion, nebulised salbutamol, and calcium resonium or sodium polystyrene sulfonate [1]
HIGH PHOS HIGH K LOW CA
Febrile Neutropenia Management
Febrile neutropenia is a medical emergency requiring empirical broad-spectrum IV antibiotics within 1 hour of presentation (the "door-to-needle" time). It is the leading cause of treatment-related mortality in paediatric oncology. [1]
Definitions: [1]
- Fever: single oral or axillary temperature at or above 38.5°C, or at or above 38.0°C sustained for at least 1 hour
- Neutropenia: absolute neutrophil count (ANC) below 0.5 × 10⁹/L (or below 1.0 × 10⁹/L and expected to fall further) [1]
Empirical antibiotic algorithm (NICE CG151 and UK paediatric oncology guidance): [1]
| Step | Indication | Antibiotic regimen |
|---|---|---|
| 1. First-line monotherapy | Standard-risk febrile neutropenia | Piperacillin-tazobactam 80 mg/kg (of piperacillin component) IV every 8 hours (max 4.5 g per dose) OR Ceftazidime 50 mg/kg IV every 8 hours (max 2 g per dose) |
| 2. Add aminoglycoside | Clinically unstable, severe sepsis, suspected resistant organism | Gentamicin 6-7 mg/kg IV once daily (monitor trough levels) |
| 3. Add glycopeptide | Suspected line infection, severe mucositis, MRSA colonisation, haemodynamic instability | Vancomycin 15 mg/kg IV every 8 hours (max 1 g per dose; monitor levels) or Teicoplanin 10 mg/kg IV every 12 hours for 3 doses then daily |
| 4. Add antifungal | Persistent fever beyond 96 hours despite broad-spectrum antibiotics | Caspofungin 70 mg/m² loading then 50 mg/m²/day OR Liposomal amphotericin B 3 mg/kg/day |
| 5. Add antiviral | Mucocutaneous HSV or VZV reactivation, severe mucositis | Aciclovir 250-500 mg/m² IV every 8 hours |
Risk stratification: [1]
Duration of antibiotics: [1]
- If a microbiological source is identified: complete the appropriate targeted course (typically 7-14 days)
- If no source is found and the child is afebrile: continue until afebrile for at least 48 hours AND the ANC is recovering (above 0.5 × 10⁹/L) — do NOT stop antibiotics while still neutropenic in high-risk patients
- If fever persists beyond 96 hours despite broad-spectrum cover: reassess — repeat cultures, perform imaging (CT sinuses and chest for invasive fungal infection) and escalate antifungal therapy [1]
Long-term Follow-up and Late Effects
With 5-year survival now exceeding 90% for ALL, the growing cohort of childhood leukaemia survivors makes late effects of treatment a major clinical priority. The Childhood Cancer Survivor Study (CCSS) has shown that by age 45, over 95% of survivors have at least one chronic health condition and nearly 80% have a severe or life-threatening condition.[5]
Key late effects and surveillance: [1]
| Late effect | Cause | Surveillance |
|---|---|---|
| Cardiotoxicity (cardiomyopathy, arrhythmia, heart failure) | Anthracyclines (daunorubicin, doxorubicin) — cumulative dose-dependent. Risk rises sharply above 250 mg/m² | Echocardiogram annually (or biennially if low dose), ECG, BNP or pro-BNP |
| Growth impairment and short stature | Cranial irradiation causing growth hormone deficiency; spinal irradiation causing direct spinal growth arrest | Height and weight every 6-12 months; GH stimulation testing; consider GH replacement |
| Endocrine dysfunction | Craniospinal irradiation causing hypothyroidism, premature ovarian failure, obesity and metabolic syndrome | Annual TFTs, gonadotrophins and sex steroids, BMI, fasting glucose and lipids |
| Secondary malignancies | Alkylating agents (cyclophosphamide) causing AML or MDS; topoisomerase II inhibitors (etoposide) causing AML; cranial radiotherapy causing brain tumours (meningioma, glioma) | Lifelong surveillance; counselling on symptom awareness; MRI if symptomatic |
| Infertility and gonadal damage | Alkylating agents (cyclophosphamide, ifosfamide) depleting germ cells; cranial radiotherapy disrupting hypothalamic-pituitary-gonadal axis | Baseline and periodic FSH, LH, oestradiol or testosterone; semen analysis in males; AMH in females |
| Neurocognitive deficits | Cranial radiotherapy plus intrathecal methotrexate causing IQ decline, attention and working memory deficits, executive dysfunction | Neuropsychological assessment at end of treatment and periodically; educational support |
| Bone health | Glucocorticoids and methotrexate causing osteoporosis and avascular necrosis (femoral head) | DEXA scan; calcium and vitamin D supplementation |
Fertility preservation
Fertility preservation must be discussed before treatment starts, because gonadotoxic agents cause irreversible germ cell loss: [1]
[1]Follow-up schedule: [1]
- Year 1-2 off therapy: review every 3-6 months (FBC, clinical examination, endocrine and cardiac surveillance)
- Year 3-5: annual review
- Year 5 onwards (long-term survivorship clinic): annual comprehensive review with risk-adapted surveillance (echocardiogram, endocrine panels, DEXA, psychosocial screening) [1]
Psychological Support for Family
A diagnosis of childhood leukaemia has a profound and lasting psychosocial impact on the child, parents, siblings and extended family. The multidisciplinary team (MDT) is central to holistic care, recognising that psychological wellbeing is inseparable from physical treatment outcomes. [1]
Impact on the child: [1]
- Diagnosis disclosure should be honest, age-appropriate and incremental — children sense parental distress and concealment increases anxiety. Use play specialists and age-adapted materials to explain the illness and treatment.
- Treatment-related distress: needle phobia, procedural anxiety, body image changes (alopecia, steroid-induced weight gain and mood swings), and separation from peers during prolonged hospitalisations
- School absence and reintegration: plan early with educational liaison; gradual return to school; individualised education plan (IEP); awareness training for teachers and classmates
- Long-term psychological sequelae: PTSD (up to 20% of survivors), depression, anxiety and neurocognitive deficits affecting academic achievement and employment [1]
Impact on parents and caregivers: [1]
- Acute grief reaction, anxiety, depression and financial strain (time off work, travel, accommodation near the treatment centre)
- Post-traumatic stress symptoms in up to 40% of mothers
- Relationship strain and marital difficulties
- Need for clear, consistent communication; written information; a documented care plan; and a named key worker (usually the clinical nurse specialist) [1]
Impact on siblings (often overlooked): [1]
- Jealousy, guilt, loneliness, behavioural regression and academic decline are common
- Sibling support programmes and dedicated psychology input are essential [1]
Support services: [1]
| Service | Role |
|---|---|
| Clinical nurse specialist (CNS) | Key worker; coordinates care; first point of contact 24/7 |
| Play specialist or therapist | Procedural preparation, distraction, therapeutic play, coping strategies |
| Psychologist or psychiatrist | Assessment and treatment of anxiety, depression, PTSD; CBT; family therapy |
| Social worker | Financial support, housing, community resources, benefits advice |
| School liaison teacher | Educational planning, hospital school, reintegration support |
| Youth support and charity | Practical, emotional and financial support; peer networks |
PARENTS NEED TOO
Prognosis and Survival
ALL prognostic factors: [1]
Key survival milestones: [1]
- ALL: 5-year event-free survival approximately 85-90% in standard-risk patients; overall survival over 95% at 5 years in the best-risk groups. Most relapses occur within 3 years of diagnosis; late relapse (off therapy) is uncommon.
- AML: 5-year event-free survival approximately 60-70% with intensive chemotherapy plus stem cell transplant for high-risk disease. Early deaths (within 4 weeks of diagnosis) from bleeding or infection remain approximately 3-5%.
- APL: historically the most fatal AML subtype because of DIC-related haemorrhage — now the most curable with ATRA plus arsenic trioxide, achieving over 80% cure.[3]
Relapse management: [1]
- Most common sites: bone marrow (over 80%), CNS (5-10%), testes (in boys)
- Treatment: re-induction chemotherapy followed by allogeneic stem cell transplant for most relapses
- CAR-T cell therapy (tisagenlecleucel, brand Kymriah): CD19-directed chimeric antigen receptor T-cell therapy, approved for refractory or relapsed B-cell ALL in patients aged 25 years and under; produces remission rates of approximately 80% in heavily pre-treated disease
- Blinatumomab: a bispecific CD19/CD3 T-cell engager antibody — another immunotherapy option for relapsed or refractory B-ALL [1]
High-Risk Subgroups and Targeted Therapy
The subgroups below carry intrinsically worse prognosis and are managed with dedicated, intensified or biologically targeted strategies layered onto (or replacing) standard backbone chemotherapy. They are high-yield exam territory because they test knowledge of cytogenetics, novel immunotherapy and transplant indications. [1]
Philadelphia Chromosome-Positive ALL
The Philadelphia chromosome — t(9;22)(q34;q11) generating the BCR-ABL1 fusion gene — encodes a constitutively active tyrosine kinase that drives leukaemic proliferation. It accounts for only around 3% of childhood ALL but was historically its worst-risk cytogenetic subset, with event-free survival under 20% on chemotherapy alone.[6]
Management revolution — tyrosine kinase inhibitors (TKIs): [1]
- TKIs are added to conventional ALL chemotherapy from induction onwards and are continued throughout all phases, including maintenance
- Imatinib (first-generation, 300 to 340 mg/m²/day): the original TKI, transformed outcomes when combined with intensive chemotherapy in COG trial AALL0031
- Dasatinib (second-generation, 60 to 100 mg/m²/day): preferred at many centres — better CNS penetration, broader kinase inhibition including Src-family kinases; demonstrated excellent activity in COG AALL0622 and is increasingly first-line in Ph+ paediatric ALL[6]
Role of bone marrow transplant (allogeneic HSCT): [1]
[1]Infant Leukaemia
Leukaemia in infants under 12 months is biologically and clinically distinct and carries the poorest prognosis of all childhood ALL subgroups, with 4-year event-free survival around 50 to 60%. It is uncommon (roughly 2 to 4% of childhood ALL) but disproportionately lethal. [1]
Molecular hallmark — KMT2A (MLL) rearrangement: [1]
- Rearrangement of the KMT2A gene at 11q23 (formerly MLL) is found in approximately 80% of infant ALL — the single dominant driver
- The commonest partner is AFF1 (AF4) at t(4;11), producing the KMT2A-AFF1 fusion
- KMT2A-rearranged blasts arrest at an immature pro-B stage (CD19 positive, CD10 negative) with frequent aberrant myeloid co-expression (CD15, CD33)
- Mechanism: KMT2A fusion proteins are aberrant histone methyltransferases that drive HOX-independent leukaemogenesis — a distinct biology from older children's ALL [1]
Clinical features of infant leukaemia: [1]
- Presentation in the first months of life (often under 6 months), with very high WCC (frequently over 300 × 10⁹/L), massive hepatosplenomegaly, and a high rate of central nervous system and extramedullary (skin, chloroma) involvement
- Hyperleucocytosis and tumour lysis syndrome at presentation are common [1]
Different treatment protocol (Interfant): [1]
- Infants are not treated on standard UKALL-type regimens; they are enrolled on dedicated Interfant protocols (Interfant-06; the current Interfant-iALL backbone), which are steroid-poor and built on cytarabine and anthracycline-containing blocks rather than the lymphoid backbone used in older children
- Cranial radiotherapy is avoided — infants are exquisitely vulnerable to devastating neurotoxicity and developmental harm
- Blinatumomab added to the Interfant backbone (Interfant-iALL 009m) improved event-free survival in KMT2A-rearranged infants and is being integrated as standard — a landmark advance for this historically dismal disease[7]
- Allogeneic BMT is considered for the highest-risk infants — particularly those with KMT2A-AFF1 and a slow minimal residual disease response or refractory disease after induction
INFANT DIFFERENCES
Relapsed Acute Lymphoblastic Leukaemia
Despite overall cure rates above 90%, 10 to 15% of children with ALL relapse, and relapse remains the leading cause of leukaemia death in children. Relapse is biologically and prognostically heterogeneous — management is dictated by timing, site and MRD response, and it is the principal setting in which novel immunotherapies and transplant are deployed. [1]
Relapse risk stratification (timing and site): [1]
| Relapse category | Definition | Prognosis |
|---|---|---|
| Very early | Within 18 months of diagnosis | Poor |
| Early | 18 to 36 months from diagnosis, or within 6 months off therapy | Intermediate |
| Late | Beyond 36 months, or more than 6 months off therapy | Favourable |
| Isolated extramedullary (CNS, testis) | No marrow disease | Better than marrow relapse |
| Bone marrow relapse | Over 5% blasts in marrow | Worse, especially if combined with CNS/testis |
Re-induction (salvage) chemotherapy: [1]
- All relapsed patients receive re-induction therapy to achieve a second complete remission — the prerequisite for any curative strategy
- Regimens incorporate mitoxantrone, fludarabine, cytarabine, clofarabine and often asparaginase; the goal is MRD negativity before proceeding to transplant or immunotherapy
- Achieving MRD negativity after re-induction is the strongest predictor of post-relapse survival [1]
Novel immunotherapy for relapsed or refractory B-ALL: [1]
- Blinatumomab — a bispecific T-cell engager (BiTE) antibody linking CD19 (on the B-leukaemic blast) to CD3 (on cytotoxic T cells), activating polyclonal T-cell killing. Administered as a continuous intravenous infusion over 28 days per cycle. Licensed for MRD-positive and relapsed or refractory B-ALL in children and adults; high response rates even in heavily pre-treated disease and effective at eradicating MRD[8]
- Tisagenlecleucel (Kymriah) — a CD19-directed chimeric antigen receptor (CAR) T-cell therapy using a 4-1BB costimulatory domain. The patient's own T cells are genetically engineered to express an anti-CD19 receptor, expanded and reinfused. Approved for refractory, relapsed or second-relapse B-cell ALL up to age 25 years; the ELISA global trial reported a complete remission rate of approximately 81% within three months of infusion[9]
- Inotuzumab ozogamicin — an anti-CD22 antibody-drug conjugate, another option for relapsed or refractory B-ALL
Exam application bank (NEET-PG / INICET)
One-line answer
Childhood leukaemia is the most common childhood malignancy (30% of all childhood cancers). Acute lymphoblastic leukaemia (ALL) accounts for 75-80%; acute myeloid leukaemia (AML) 15-20%. Peak age: 2-5 years. Presentation: bone marrow failure (anaemia, infection, bleeding), organ infiltration (hepatosplenomegaly, lymphadenopathy, CNS). Diagnosis: FBC + blood film + bone marrow aspirate (morphology, flow cytometry, cytogenetics). Treatment: risk-stratified chemotherapy. 5-year survival ALL: 90%, AML: 65-70%.
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard. [1]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes. [1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change. [1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each. [1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory. [1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Childhood Leukaemia.
[1]Bone marrow transplant (allogeneic HSCT) in relapse: [1]
- Indicated for most bone marrow relapses (except very-low-risk late isolated relapses), and for very early or early relapse with poor MRD response after re-induction
- Donor sources: matched sibling donor (preferred), matched unrelated donor, haploidentical or umbilical cord blood
- Delivered in second complete remission, after cytoreductive re-induction or as consolidation following CAR-T/blinatumomab-induced remission
- Curative intent, but carries substantial morbidity: graft-versus-host disease, infertility, second malignancy and transplant-related mortality [1]
Supportive Care
Modern cure rates depend as much on supportive care as on cytotoxic therapy — infection remains the leading cause of treatment-related death in paediatric oncology, and late effects dominate survivorship. A systematic, protocol-driven approach is mandatory and is frequently examined. [1]
Infection prophylaxis
- Pneumocystis jirovecii pneumonia (PJP) prophylaxis — co-trimoxazole (trimethoprim-sulfamethoxazole) is the standard, given throughout chemotherapy whenever the child is on steroids or immunosuppressed, and continued for several months after therapy. PJP pneumonitis carries high mortality in immunocompromised children and is almost entirely preventable with co-trimoxazole.
- Antifungal prophylaxis — posaconazole (or micafungin/fluconazole) during periods of prolonged profound neutropenia (notably AML induction and re-induction). Posaconazole is preferred for its mould-active cover against Aspergillus, the dominant invasive fungal pathogen in this setting.
- Antiviral prophylaxis — aciclovir for HSV and VZV prophylaxis, particularly in seropositive children during induction.
- Antibacterial — penicillin V after splenectomy or in functional asplenia; fluoroquinolone prophylaxis during severe neutropenia in some protocols.
- G-CSF to shorten neutropenia and reduce infection in intensive (especially AML) cycles. [1]
Viral monitoring
- CMV monitoring is critical, especially around allogeneic stem cell transplant: weekly PCR surveillance in at-risk (donor-positive or recipient-positive) pairs, with pre-emptive therapy (ganciclovir, foscarnet or letermovir) triggered by rising viral load, to prevent CMV disease (pneumonitis, colitis). Reactivation also occurs with prolonged lymphodepletion after CAR-T and blinatumomab.
- EBV monitoring for post-transplant lymphoproliferative disorder (PTLD), and adenovirus surveillance in heavily immunosuppressed patients. [1]
Transfusion and supportive measures
- Irradiated blood products to prevent transfusion-associated graft-versus-host disease (ta-GvHD) in immunocompromised patients, and CMV-negative or leucodepleted components as required
- Mucositis prevention (palifermin in selected regimens), oral and dental care, and nutritional support via enteral feeding or parenteral nutrition
- Vaccination: inactivated vaccines are safe during therapy; live vaccines (MMR, varicella, rotavirus) are deferred until immune reconstitution after treatment ends — revaccination is often required post-therapy [1]
Late-effect supportive care
- Growth hormone (recombinant somatropin) for growth hormone deficiency and short stature following cranial irradiation (CNS-directed therapy), once leukaemia is in remission and the height deficit confirmed on growth charts and provocation testing — earlier is better, as responsiveness declines with age
- Endocrine surveillance (thyroid, pubertal timing, bone density) for hypothalamic-pituitary effects of cranial RT and steroid exposure
- Psychosocial, educational and neurocognitive support, with formal neuropsychological assessment for children who received CNS-directed therapy [1]
SUPPORTIVE ABCs
References
- [1]Hunger SP, Mullighan CG. Acute Lymphoblastic Leukemia in Children N Engl J Med, 2015.PMID 26465987
- [2]Pui CH, Evans WE. A 50-year journey to cure childhood acute lymphoblastic leukemia Semin Hematol, 2013.PMID 23953334
- [3]Creutzig U, Kutny MA, Barr R, et al. Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel Blood, 2012.PMID 22879540
- [4]Coiffier B, Altman A, Pui CH, et al. Guidelines for the management of pediatric and adult tumor lysis syndrome: an evidence-based review J Clin Oncol, 2008.PMID 18509186
- [5]Oeffinger KC, Mertens AC, Sklar CA, et al. Chronic health conditions in adult survivors of childhood cancer N Engl J Med, 2006.PMID 17035650
- [6]Slayton WB, Schultz KR, Kairalla JA, et al. Dasatinib Plus Intensive Chemotherapy in Children, Adolescents, and Young Adults With Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: Results of Children's Oncology Group Trial AALL0622 J Clin Oncol, 2018.PMID 29812996
- [7]van der Sluis IM, de Lorenzo P, Kotecha RS, et al. Blinatumomab Added to Chemotherapy in Infant Lymphoblastic Leukemia N Engl J Med, 2023.PMID 37099340
- [8]von Stackelberg A, Locatelli F, Zugmaier G, et al. Phase I/Phase II Study of Blinatumomab in Pediatric Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia J Clin Oncol, 2016.PMID 27998223
- [9]Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia N Engl J Med, 2018.PMID 29385370