Psychiatry · General Medicine
Autism Spectrum Disorder
Also known as Autism spectrum disorder · ASD · Autism · Asperger syndrome · Pervasive developmental disorder
Autism spectrum disorder (ASD) is a neurodevelopmental disorder defined in DSM-5 and ICD-11 by persistent deficits in social communication and social interaction across multiple contexts (the three sub-domains of social-emotional reciprocity, nonverbal communicative behaviour, and relationships) PLUS restricted, repetitive patterns of behaviour, interests or activities (at least 2 of 4: stereotyped/repetitive movements, speech or object use; insistence on sameness/rituals; restricted interests of abnormal intensity; and hyper- or hypo-reactive sensory behaviour). Onset is in the early developmental period, the symptoms are present across contexts, and they cause clinically significant functional impairment. Global prevalence is about 1 in 100 children and the most recent US CDC ADDM surveillance reports 1 in 36 (2.8 percent) among 8-year-olds; the male-to-female ratio is about 3 to 4:1; heritability is approximately 80 percent. There is no medication that treats the core ASD — management is early intensive behavioural and developmental intervention, multidisciplinary support, and symptom-targeted pharmacotherapy for comorbidities (risperidone and aripiprazole are the two FDA-approved drugs for irritability associated with paediatric ASD).
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Exam tags
Red flags
Overview & Definition
Autism spectrum disorder (ASD) is a lifelong neurodevelopmental condition whose expression ranges from a non-verbal child with severe intellectual disability and self-injurious behaviour to a verbally gifted, employed adult with a satisfying career and family. The clinical task is the same across that spectrum: (1) recognise the early signs, (2) confirm the diagnosis against DSM-5 criteria through multidisciplinary assessment, (3) start intensive intervention in the most plastic developmental window, (4) identify and treat comorbidities, and (5) support the individual and family across the life course.[1]
The unifying clinical thread is that social communication and the flexibility of behaviour/thought develop atypically — not that the child "cannot" relate, but that relating happens in a different sensory, cognitive and emotional register. ASD is a spectrum in two senses: severity (DSM-5 Levels 1 to 3) and profile (a verbally able child with intense special interests differs more in kind than in degree from a non-verbal child with severe ID). The diagnostic unifier is the two-symptom-domain structure.[9]

Autism spectrum disorder — the headline numbers
Classification
ASD is now a single diagnostic category under both DSM-5 and ICD-11, replacing the historical DSM-IV pervasive developmental disorders (PDD) — autistic disorder, Asperger disorder, PDD-NOS, childhood disintegrative disorder, and Rett disorder. The rationale was that the historical subcategories were applied inconsistently, did not predict outcome or treatment response, and shared the same two-symptom-domain structure differing mainly in degree.[9]
ASD vs its closest neighbours — the discriminating feature in each
Social (pragmatic) communication disorder
- Identical social-communication deficits
- NO restricted/repetitive behaviours or sensory features
- Single discriminator: the RRB/sensory domain is absent in SCD
- DSM-5 introduced SCD to capture children who would previously have been labelled PDD-NOS without RRBs
Intellectual disability without ASD
- Social skills are COMMENSURATE with overall developmental level
- Child interacts appropriately for their mental age
- ASD is added only when social communication is MORE impaired than expected for the ID (DSM-5 criterion E)
- Commonly co-exists with ASD (~30-50 percent)
Developmental language disorder
- Language delayed but social-communicative INTENT is preserved
- Strong gesture, eye contact, joint attention, pretend play
- Child actively tries to communicate (pulls hand, points)
- Audiology must be assessed and hearing impairment excluded
ADHD
- Social difficulties arise from inattention and impulsivity
- No restricted/repetitive behaviours or sensory core
- Frequently CO-EXISTS with ASD (40-50 percent) — both diagnoses are made
- Treat the ADHD with stimulants/atomoxetine (slightly lower response in ASD)
Selective mutism
- Child speaks normally at home, mute in specific settings
- Normal nonverbal communication and joint attention
- Onset after age 5; an anxiety-spectrum disorder
- No restricted/repetitive behaviours or sensory features
Reactive attachment disorder
- Severe early NEGLECT or institutional care is the cause
- Persistent social withdrawal and emotional disturbance
- Distinctive history; not a developmental trajectory
- Recovery is possible with stable caregiving
DSM-5 specifiers and severity levels
Every ASD diagnosis is annotated with specifiers that capture the individual profile and shape intervention: [1]
- WITH or WITHOUT accompanying intellectual impairment (and the level of impairment);
- WITH or WITHOUT accompanying language impairment (and if present, the current level — none, some words, phrase speech, fluent);
- Associated with a known medical or genetic condition or environmental factor (e.g. fragile X syndrome, tuberous sclerosis complex, Rett syndrome, prenatal valproate exposure);
- Associated with another neurodevelopmental, mental or behavioural disorder;
- WITH catatonia (a recognised but under-diagnosed specifier). [1]
DSM-5 severity levels — applied SEPARATELY to social-communication and restricted/repetitive-behaviour domains
Level 1 — requiring support
- Social: initiates interactions but atypically; responds to direct approaches; reduced interest; difficulty with one-to-one
- RRB: difficulty switching activities; organisation problems interfere with independence
- Support required for successful participation
Level 2 — requiring substantial support
- Social: marked deficits in verbal and nonverbal social-communication; limited initiation; reduced/atypical response even with support
- RRB: inflexibility of behaviour, frequent distress at change, multiple repetitive behaviours obvious to casual observer
- Substantial support required even with accommodation
Level 3 — requiring very substantial support
- Social: severe deficits in verbal/nonverbal social-communication; very few initiations; minimal response to others
- RRB: preoccupation with restricted interests; extreme distress at change; markedly restricted repertoire
- Very substantial support required for daily functioning
Subtypes within the spectrum (clinical, not DSM-5 categories)
- ASD with intellectual impairment (~30-50 percent; IQ below 70) — more severe language delay, more epilepsy, more self-injurious behaviour; genetic workup higher yield.
- ASD without intellectual impairment (the former "Asperger" profile) — preserved cognitive milestones, fluent but odd/pedantic speech with impaired pragmatics, intense circumscribed interests, normal self-help; diagnosis often delayed into school-age or adulthood.
- Regressive ASD (~15-25 percent) — apparently normal development for 15 to 24 months followed by loss of words, eye contact and social engagement.
- Syndromic ASD — fragile X syndrome (commonest inherited single-gene association), tuberous sclerosis complex (commonest neurocutaneous cause; ~25-50 percent of TSC cases meet ASD criteria), Rett syndrome (MECP2), PTEN hamartoma tumour syndrome (with macrocephaly), 15q11-q13 duplication, 16p11.2 deletion/duplication, 22q11.2 deletion, CHARGE, Sotos.
- Female phenotype — better superficial social skills, special interests in socially acceptable domains, more internalising/masking, diagnosis delayed to adulthood.
- Adult-diagnosed ASD — anxiety, depression, burnout, employment and relationship difficulty as the presenting complaint; diagnosis from a careful developmental history. [1]
Epidemiology & Risk Factors
Prevalence has risen steadily over four decades — a consequence largely of better recognition, broader criteria, earlier identification and improved service access rather than a true surge in incidence. The CDC ADDM 2023 surveillance (Shaw et al., MMWR; 8-year-old US children in 2018) reports 1 in 36 (2.8 percent)[4], up from 1 in 54 in the 2021 report (Shaw et al.)[3]; the WHO global estimate is approximately 1 in 100 children. The male-to-female ratio is reported as 3 to 4:1 but overstates the true sex difference because girls are systematically under-diagnosed.
The epidemiology that examiners test
Established risk factors:
- Genetic and familial (dominant): heritability ~80 percent (Sandin 2017 JAMA Swedish cohort — 83 percent; Bai/Sandin 2019 five-country cohort — 80 percent)[11][10]; monozygotic twin concordance 60-90 percent vs 0-20 percent for dizygotic twins; sibling recurrence risk 10-20 percent rising to 30-50 percent with two affected children — drives universal sibling screening.
- Advanced parental age — paternal over 40 and maternal over 35 (de-novo mutation hypothesis).
- Perinatal factors — prematurity and very low birth weight, fetal growth restriction, hypoxic-ischaemic injury, multiple gestation, short inter-pregnancy interval.
- Maternal exposures in pregnancy — sodium valproate (the strongest single medication signal), thalidomide, certain SSRIs, maternal infection/fever, gestational diabetes, autoimmune disease.
- Socioeconomic — very low socioeconomic status is associated in some cohorts; access-driven ascertainment plays a role.
What does NOT cause autism (high-yield counselling point):
- Vaccines — including the measles-mumps-rubella (MMR) vaccine and thimerosal-containing vaccines — do NOT cause ASD. The 1998 Wakefield Lancet paper was retracted as fraudulent; the largest cohort studies (e.g. Hviid et al. 2019, Annals of Internal Medicine, Danish cohort of over 657 000 children) show no association. This remains a high-yield counselling point.
- Parenting style ("refrigerator mother") — abandoned as baseless decades ago. [1]
Pathophysiology
ASD is best understood as altered brain wiring beginning in the late first and second trimester, driven predominantly by polygenic variation with contributions from rare high-effect variants, prenatal environmental exposures and immune/inflammatory processes. There is no single causal lesion — a useful framing is altered excitation/inhibition balance and atypical connectivity. [1]
Genetic architecture. ASD is among the most heritable of psychiatric disorders — heritability estimates of 60-90 percent[11][10]. The genetic architecture is heterogeneous:
- Common polygenic variants (SNPs of small individual effect, polygenic risk scores) account for the majority of liability.
- Rare de-novo and inherited copy-number variants (CNVs) explain ~10-20 percent — clinically relevant examples are 16p11.2 deletion/duplication, 15q11-q13 duplication, 22q11.2 deletion, 1q21.1, 7q11.23 (Williams syndrome reciprocal).
- Single-gene (syndromic) associations — fragile X syndrome (FMR1, the commonest inherited cause of intellectual disability and the commonest single-gene ASD association), tuberous sclerosis complex (TSC1/TSC2 — the commonest neurocutaneous cause), Rett syndrome (MECP2), PTEN hamartoma tumour syndrome (present with macrocephaly), CHD8, SHANK3, NLGN3/4, NRXN1. [1]
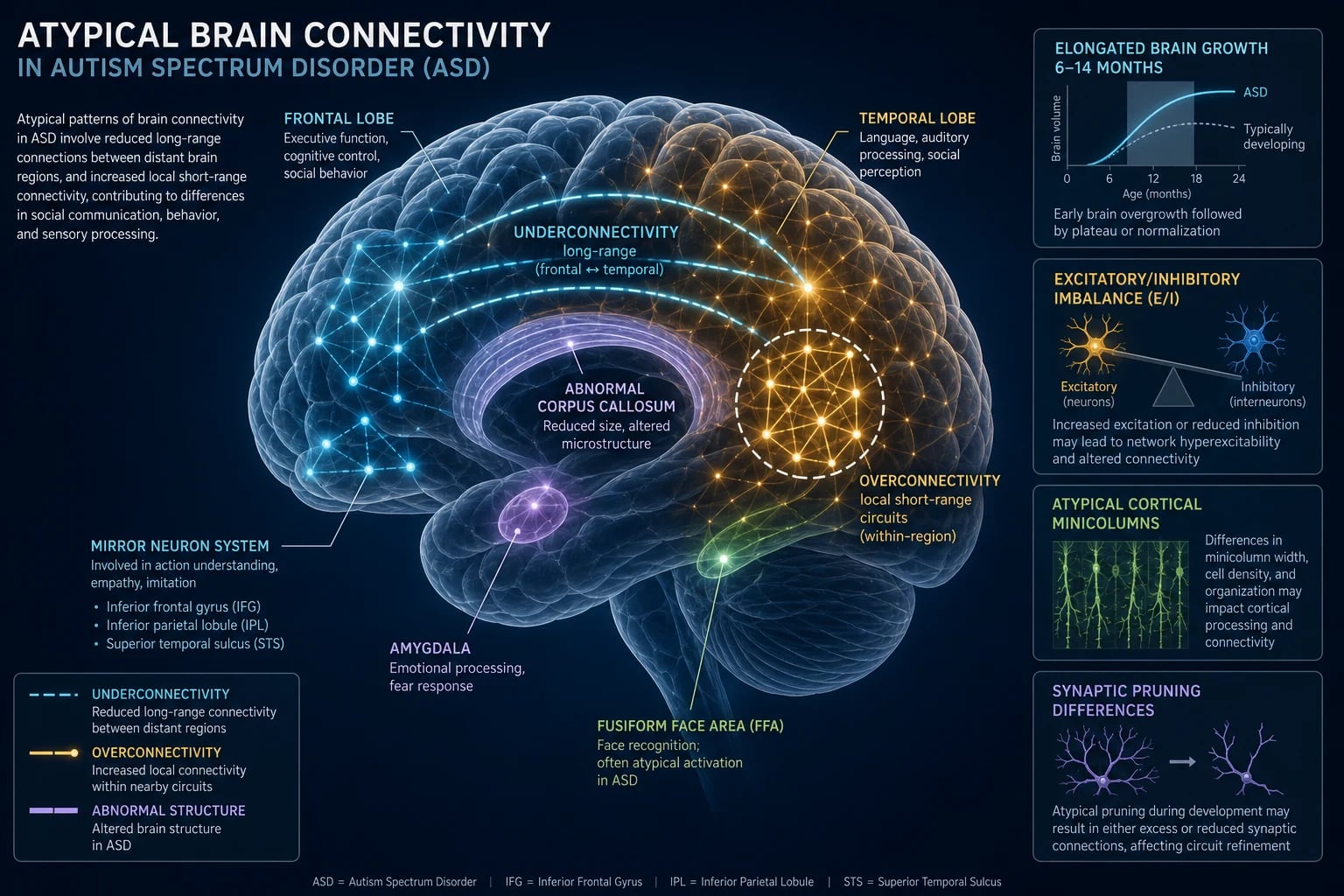
Macroscopic brain trajectory. Early brain overgrowth in the first two years — frontal and temporal cortices enlarged, with increased total brain volume in roughly 15 percent — followed by plateau and a near-normal adult brain size. The mechanism is thought to involve excess cortical precursor proliferation, abnormal synaptic pruning and altered white-matter trajectory. [1]
Microscopic (histological) signature. Post-mortem studies (Bailey, Courchesne and others) show abnormal minicolumn architecture (more numerous, narrower, less well-defined minicolumns), reduced GABAergic (inhibitory) interneuron markers (GAD67, GABA-A receptor binding), and patchy heterotopic neurons in the molecular layer — a fingerprint of late prenatal migrational error.[9]
White-matter connectivity. Two complementary findings explain the cognitive profile: under-connectivity of long-range tracts (fronto-temporal, corpus callosum) producing weak global/integrative processing, and over-connectivity of local short-range circuits producing detail-focused processing and sometimes savant abilities. This is the biological substrate of weak central coherence and the Enhancing Perceptual Performance (EPF) model. [1]
Key brain regions. Prefrontal cortex (social cognition, executive function), superior temporal sulcus and temporoparietal junction (biological motion, theory of mind), fusiform face area (hypo-active when viewing faces — explains gaze avoidance), amygdala (emotional salence and anxiety; enlarged in early childhood), mirror-neuron system at the inferior frontal gyrus (action understanding, imitation), and cerebellum (motor coordination and affective/cognitive processing; Purkinje-cell loss is one of the most consistent findings). [1]
Neurochemistry. Hyperserotonemia of autism — blood (platelet) serotonin is elevated in 25-30 percent (a biomarker, not a causal explanation, since platelet serotonin does not cross the blood-brain barrier). GABAergic dysfunction and glutamatergic dysregulation underpin the excitation/inhibition (E/I) imbalance hypothesis. Oxytocin and vasopressin pathways are implicated in social affiliation (and form the basis of experimental therapeutic trials). Dopaminergic reward circuitry is implicated in restricted interests. [1]
Immune and neuroinflammatory contributions. Maternal immune activation (maternal infection, fever, or autoantibodies against fetal brain proteins) and microglial activation in the autistic brain support an immune component — the valproate-exposure animal model recapitulates many features. [1]
Embryological timing. The histological and brain-volume data converge on a disturbance in late first and second trimester (8 to 24 weeks gestation) — the period of cortical neurogenesis, neuronal migration and early synaptogenesis. [1]
Clinical Presentation
The two symptom domains (social communication; restricted/repetitive behaviour) produce a recognisable phenotype that evolves with age. The earliest detectable signs appear between 12 and 24 months, and recognition at this age is the single highest-yield clinical skill. [1]
[1]By age band. In pre-school children one sees delayed or echolalic speech, pronoun reversal, monotone or unusual prosody, absence of pretend play (preference for repetitive sensorimotor play like lining up objects or spinning wheels), intense attachment to unusual objects, insistence on sameness with distress at small changes, and restricted interests. School-age children show difficulty with peers and group play, literal interpretation of language with failure to grasp sarcasm/jokes/idioms, pedantic overly-formal speech ("little professor"), intense circumscribed interests (e.g. trains, dates, timetables, dinosaurs), school refusal, anxiety and uneven academic profiles (often with strong rote memory and weak comprehension). Adolescents and adults present with social anxiety, depression, burnout from chronic masking, vulnerability to exploitation, and difficulties with intimate relationships and employment.[9]
The four restricted/repetitive-behaviour domains (DSM-5) — know all four:
- Stereotyped or repetitive motor movements, speech or object use — hand-flapping, rocking, spinning, toe-walking; echolalia (immediate or delayed), verbal rituals, scripting from films; lining up, spinning objects.
- Insistence on sameness — distress at small changes, need to take the same route, eat the same food, rigid adherence to rules and rituals.
- Highly restricted, fixated interests of abnormal intensity — trains, dinosaurs, timetables, vacuum cleaners, a single pop star or fictional character.
- Hyper- or hypo-reactivity to sensory input — covering ears, avoiding bright lights, food-texture aversion (hyper); high pain threshold, seeking deep pressure, under-reacting to name (hypo). [1]
Atypical presentations. [1]
- Girls and women — under-diagnosed. Better superficial social skills, stronger language, special interests in socially acceptable domains (animals, celebrities, friendship rather than objects), more camouflaging/masking, more internalising (anxiety, eating disorders, self-harm, chronic fatigue). The female phenotype is recognised late, often in adulthood.[9]
- Verbal children and adults with average/above IQ — present with anxiety, depression, burnout, relationship and employment difficulty; diagnosis only emerges when a careful developmental history is taken.
- Regressive ASD (15-25 percent) — apparently normal development for 15 to 24 months followed by loss of words, eye contact and social engagement; mandates urgent referral and exclusion of Landau-Kleffner syndrome (acquired epileptic aphasia with epileptiform EEG), metabolic and neurodegenerative disease.
- ASD with intellectual disability (30-50 percent, IQ below 70) — more severe language delay, more epilepsy, more self-injury; ASD is added when social communication is more impaired than expected for the developmental level (DSM-5 criterion E).
- ASD with co-occurring epilepsy (20-30 percent overall, up to 35-40 percent with severe ID) — seizure types include infantile spasms (especially in tuberous sclerosis), focal seizures, generalised tonic-clonic and atypical absence; EEG abnormalities without clinical seizures are common; onset peaks in early childhood and again in adolescence.
Differential Diagnosis
The differential is broad because delayed language, social withdrawal and repetitive behaviours are non-specific. The discriminating feature in each case is shown below.[1][9]
Distinguishing ASD from its closest differentials
Social (pragmatic) communication disorder (DSM-5)
- Same social-communication deficits
- NO restricted/repetitive behaviours or sensory features
- The single discriminator: presence or absence of the RRB/sensory domain
Hearing impairment
- MUST be excluded in EVERY child with delayed language
- Strong nonverbal communication, joint attention and social interest once the channel is restored
- Audiology / auditory brainstem response before confirming ASD
Global developmental delay / intellectual disability without ASD
- Social skills COMMENSURATE with overall developmental level
- Child interacts appropriately for their mental age
- ASD is added only when social communication is more impaired than expected
Selective mutism
- Speaks normally at home, mute in specific social settings
- Normal nonverbal communication and joint attention
- An anxiety-spectrum disorder; onset after age 5
ADHD
- Social difficulty arises from inattention and impulsivity, not from core social-communication deficit
- No restricted/repetitive behaviours or sensory core
- Frequently CO-EXISTS (40-50 percent) — both diagnoses are made
Stereotypic movement disorder
- Repetitive movements (head-banging, body-rocking, hand-biting) WITHOUT the ASD social-communication deficit
- Common in severe ID and in sensory deprivation
- Treat the movement; ASD criteria are not met
Fragile X syndrome
- Long face, large ears, post-pubertal macro-orchidism, intellectual disability
- Social anxiety with hypersociability and gaze aversion
- FMR1 testing mandatory in any child with global developmental delay and ASD features
Tuberous sclerosis complex
- Cortical tubers, facial angiofibromas, ash-leaf macules, infantile spasms, renal/cardiac lesions
- The commonest neurocutaneous cause of ASD (~25-50 percent of TSC cases meet ASD criteria)
- TSC1/TSC2 testing; clinical genetics referral
Rett syndrome (MECP2, girls)
- Apparently normal for 6-18 months, then regression with loss of purposeful hand use replaced by hand-wringing
- Deceleration of head growth, gait apraxia, breathing irregularities
- Now coded separately from ASD, but may meet ASD criteria during the regression phase
Childhood disintegrative disorder / Heller syndrome
- Normal development for at least 2 years, then severe regression in language, social and self-help skills
- DSM-5 subsumes into ASD but mandates exclusion of metabolic/neurodegenerative disease
- Prognosis more guarded than non-regressive ASD
Landau-Kleffner syndrome (acquired epileptic aphasia)
- Loss of language with epileptiform EEG (especially electrical status epilepticus of slow sleep)
- An important TREATABLE mimic of regressive autism
- Steroids and anti-seizure medication may restore language
Reactive attachment disorder (RAD) / disinhibited social engagement disorder (DSED)
- Severe early neglect or institutional care
- RAD — withdrawn and unresponsive; DSED — over-familiar with strangers
- Recovery possible with stable caregiving
Clinical & Bedside Assessment
Developmental surveillance is performed at every well-child visit (AAP recommends 9, 18, 24 and 30 months) by eliciting parent concerns, observing social-communication behaviours and screening with a validated tool.[1]
Universal screening — the M-CHAT-R/F (Modified Checklist for Autism in Toddlers, Revised with Follow-up; Robins 2014 validation in a US primary-care sample of over 18 000 toddlers).[5] The AAP recommends an autism-specific screen at the 18- AND 24-month visits. The M-CHAT-R/F is:
- a 20-item parent questionnaire (free, translated into many languages);
- a child screens positive on the initial screen with a total score of 3 or more, OR with any two of the seven critical items;
- a positive initial screen triggers the structured Follow-Up Interview, which uses a standard script to confirm or refute each endorsed item;
- if the child remains positive on the Follow-Up (score of 2 or more), the positive predictive value is ~47.5 percent for ASD and ~95 percent for ANY developmental concern — the child is referred immediately for diagnostic evaluation AND for early intervention (do not wait for the diagnostic confirmation to begin services). [1]
Other screening tools: the STAT (Screening Tool for Autism in Toddlers — interactive, second-level), the SCQ (Social Communication Questionnaire — parent report, ages 4+), the SRS-2 (Social Responsiveness Scale — dimensional severity, 2.5-18 years), the CAST (Childhood Autism Spectrum Test) and the AQ / AQ-10 / AQ-50 (Autism Spectrum Quotient — older adolescents and adults). [1]
Diagnostic assessment is multidisciplinary (paediatrician/child psychiatrist, clinical psychologist, speech-language therapist, occupational therapist, special educator, social worker) and uses the gold-standard instruments:[1]
- ADOS-2 (Autism Diagnostic Observation Schedule, 2nd edition) — a semi-structured, standardised, interactive assessment with modules chosen by age and language level (Toddler Module, Modules 1 to 4).
- ADI-R (Autism Diagnostic Interview-Revised) — a standardised, semi-structured parent interview covering the developmental history. Both require specific training and are used in specialist clinics and research. [1]
Diagnostic history-taking — key elements:
- Pregnancy, perinatal and neonatal history; family history of ASD, ADHD, language delay, learning difficulty, intellectual disability, psychiatric illness.
- Early developmental milestones — social smile, eye contact, babbling, first words, pointing, joint attention, pretend play, walking, self-help.
- Any regression — and its exact timing (15 to 24 months typical of ASD; later or more catastrophic regression mandates exclusion of Landau-Kleffner, metabolic and neurodegenerative disease).
- Routines, rituals, restricted interests, sensory behaviours, sleep, feeding, GI function. [1]
Direct clinical observation looks for: eye contact, joint attention, response to name, social smile, pointing, imitation, pretend play, language (echolalia, pronoun reversal, prosody), the child's response to the examiner's overtures, restricted/repetitive behaviours and sensory behaviours. [1]
Functional impact and risk assessment — assess school, friendships, family life, self-care and safety; assess risk of self-injurious behaviour, absconding/elopement, pica, suicidality in adolescents (significantly elevated in verbally able adults), and abuse/exploitation in adults. [1]
Investigations
ASD is a clinical diagnosis against DSM-5 criteria; there is no blood test, EEG or imaging study that confirms it. Investigations are directed at excluding mimics, identifying associated (syndromic) conditions, and characterising comorbidities.[1]
Mandatory first-line:
- Audiology assessment (audiometry and/or auditory brainstem response) in EVERY child with delayed language and EVERY suspected ASD referral, before the diagnosis is confirmed.
- Formal vision assessment. [1]
Genetic testing — NICE CG128 and the AAP recommend chromosomal microarray (CMA, array CGH or SNP array) and fragile X (FMR1) testing for ALL children with confirmed ASD; add MECP2 if Rett features, PTEN if macrocephaly, TSC1/TSC2 if tuberous sclerosis features; whole-exome sequencing is increasingly used in specialist genetics. [1]
Metabolic workup — only when indicated by regression, severe/profound ID, dysmorphism, organomegaly or developmental plateau: plasma amino acids, urinary organic acids, serum lactate, creatine metabolites, very-long-chain fatty acids, uric acid, mucopolysaccharide screen, transferrin isoelectric focusing (congenital disorders of glycosylation), 7-dehydrocholesterol (Smith-Lemli-Opitz). [1]
Targeted tests:
- Blood lead level in children with pica or developmental delay in older housing.
- Thyroid function (TSH, free T4) with any regression or signs of hypothyroidism.
- EEG when there is a history of seizures, regression (especially language regression — consider Landau-Kleffner), staring spells, or significant cognitive/behavioural regression. The EEG is NOT routine in ASD without seizures (AAN/AAP).
- MRI brain is NOT routine; reserve for focal neurological signs, abnormal head size (above 2.5 SD or below -2.5 SD), regression, midline facial defects, neurocutaneous stigmata, or an abnormal neurological examination. [1]
Reproduced named tools and criteria (used for diagnosis, severity grading and tracking):
- DSM-5 criteria A-E (see Classification) — the diagnostic standard.
- DSM-5 severity Levels 1 to 3 — applied separately to social-communication and RRB domains.
- M-CHAT-R/F scoring — 20 items; initial positive at total score 3 or more, or any two of seven critical items; Follow-Up administered if initial positive; persistent score of 2 or more on the Follow-Up warrants immediate referral.[5]
- Vineland-3 — adaptive behaviour scales with four domains (Communication; Daily Living Skills; Socialisation; Motor Skills — optional), yielding an Adaptive Behaviour Composite standard score (mean 100, SD 15). Used for baseline and longitudinal adaptive-functioning measurement.
- CARS-2 (Childhood Autism Rating Scale, 2nd edition) — a 15-item clinician-rated behaviour scale; the total score classifies minimal-to-no symptoms (15-29.5), mild-moderate (30-36.5) and severe (37-60) ASD.
Management — Resuscitation

ASD is rarely a medical emergency, but several scenarios demand immediate recognition and management: [1]
The acute presentations of ASD and their first-line response
Acute severe behavioural disturbance / aggression / self-injury
- Ensure safety of patient and others; reduce sensory/environmental triggers (quiet low-stimulation room, dim lights, remove unnecessary staff, allow a trusted caregiver)
- De-escalation with familiar objects, visual supports and predictable language
- Pharmacological only as a last resort: lorazepam 0.05 mg/kg PO/IM (max 2 mg) OR low-dose antipsychotic (risperidone 0.25-0.5 mg PO/IM, olanzapine 5 mg orodispersible) when there is imminent risk
Acute regression with neurological signs
- Treat as possible status epilepticus / Landau-Kleffner / neurodegenerative or metabolic emergency
- Urgent EEG, glucose, electrolytes, ammonia, lactate, toxicology, neuroimaging
- Paediatric neurology referral
First convulsion / status epilepticus
- ABC, recovery position, IV lorazepam 0.1 mg/kg (buccal or intranasal midazolam if no IV access)
- EEG and neurology referral; do not over-diagnose ASD stereotypies as seizures — they are distinguishable
Severe pica with ingestion
- Identify substance (lead, button battery, caustic, foreign body)
- Bloods including lead level and FBC with basophilic stippling; abdominal X-ray
- Poison centre and surgical input
Elopement / absconding
- Immediate coordinated search (police, hospital security, community)
- Water-drowning risk alert — autistic wanderers are drawn to water
- Wearable ID/tracking devices; preventive planning with door alarms
Catatonia in ASD
- Mute, posturing, negativism, waxy flexibility, stereotypy, refusal to eat or drink
- Lorazepam challenge and urgent psychiatry; ECT for refractory cases
Acute dystonic reaction to antipsychotic
- IM or IV procyclidine 5 mg or benztropine 1-2 mg
- Review antipsychotic choice/dose
Neuroleptic malignant syndrome
- STOP the antipsychotic
- IV fluids, cooling, dantrolene 1 mg/kg IV and/or bromocriptine; ICU
Management — Definitive & Stepwise
There is no medication that treats the CORE social-communication or cognitive deficits of ASD. The evidence base is for early, intensive, structured behavioural and developmental intervention; medications target comorbid symptoms only.[1][9]
Principles: start early; make the plan individualised and multidisciplinary; build on the child's strengths; involve the family as partners; coordinate with school; review and revise periodically; and minimise polypharmacy. [1]
Step 1 — Behavioural and educational intervention (FIRST-LINE, the core of management):
- Applied Behaviour Analysis (ABA) and discrete-trial teaching — structured, intensive.
- Early Start Denver Model (ESDM) — naturalistic, play-based, for children 12 to 48 months. Dawson et al. 2010 RCT showed significant gains in IQ, receptive and expressive language, and adaptive behaviour in toddlers receiving ESDM versus community care.[6]
- Early Intensive Behavioural Intervention (EIBI) — the Lovaas model; 20 to 40 hours per week.
- Pivotal Response Training (PRT) — targets pivotal skills (motivation, self-initiation).
- TEACCH — structured teaching with visual supports.
- PECS (Picture Exchange Communication System) and AAC (augmentative and alternative communication) devices for non-verbal children.
- Social-skills training groups.
Step 2 — Communication: speech and language therapy targeting receptive, expressive and pragmatic language; AAC for the non-verbal; prosody and conversation skills for verbally able children. [1]
Step 3 — Occupational therapy: sensory integration therapy (evidence mixed but widely used), motor-coordination, activities of daily living, environmental modification, visual schedules. [1]
Step 4 — Education: an individualised education plan (IEP / EHCP / 504 plan) with appropriate placement (mainstream with support, resource room, autism unit, specialist school), structured classroom, visual timetables, predictable routines, movement/sensory breaks, and modifications for communication and behavioural needs. [1]
Step 5 — Family-centred support: parent-mediated intervention and parent training (Hanen, Triple P, Stepping Stones), sibling support, respite care, financial/benefits advice, signposting to parent-led organisations. [1]
Step 6 — Symptom-targeted pharmacotherapy (NEVER first-line for core ASD): [1]
Symptom-targeted drug therapy in ASD — agent, dose, route, rationale, monitoring
Severe irritability, aggression, self-injury (FDA-APPROVED)
- Risperidone 0.25-3 mg/day PO (start 0.25-0.5 mg, titrate by 0.5 mg every 1-2 weeks; McCracken 2002 NEJM RUPP trial)
- Aripiprazole 5-15 mg/day PO (start 1-2 mg; Marcus 2011)
- Both approved for ages 5-16 (risperidone) / 6-17 (aripiprazole)
- Monitor weight, BMI, waist, fasting glucose and lipids, prolactin, EPSE/AIMS, ECG
ADHD comorbidity
- Methylphenidate (slightly lower response and more adverse effects than in primary ADHD)
- Atomoxetine 0.5-1.2 mg/kg/day; guanfacine extended-release
- Caution: may transiently worsen stereotypies/irritability
Anxiety, depression, OCD
- SSRI — fluoxetine first; START LOW, GO SLOW
- Monitor for behavioural activation and worsened sleep
- CBT adapted for ASD alongside medication
Sleep disturbance
- Behavioural and sleep-hygiene measures FIRST
- Melatonin 2-6 mg PO 30-60 minutes before bedtime (evidence-based, CARI trial)
- Avoid long-term sedating antipsychotics for sleep
Epilepsy
- Per seizure type — levetiracetam, lamotrigine, sodium valproate
- AVOID VALPROATE in girls of childbearing potential (teratogenic)
- Vagus-nerve stimulation and dietary therapies for refractory cases
ASD pharmacotherapy — FDA-approved vs the rest
Treatments NOT recommended (no evidence, several harmful): chelation, hyperbaric oxygen, secretin, megadose vitamins, restrictive diets (gluten-free/casein-free), facilitated communication, rapid prompting, stem-cell therapy, and MMS/chlorine dioxide. The AAP and NICE explicitly list these as "do not use".[1]
Escalation triggers for specialist referral: regression (urgent paediatric neurology + EEG + metabolic workup); uncontrolled epilepsy; severe self-injurious or aggressive behaviour refractory to behavioural and pharmacological measures (inpatient psychiatry); suspicion of an underlying syndromic/genetic cause (clinical genetics); suspected comorbid psychiatric illness. [1]
Transition planning starts at age 14 (per NICE and AAP), with explicit transition to adult mental-health/ID services, supported employment, independent-living support, mental-health care, social opportunities and benefits — a known weak point in services. [1]
Specific Subtypes & Scenarios
- ASD with intellectual impairment (~30-50 percent) — more severe language delay, more epilepsy, more self-injury; higher genetic-workup yield; prognosis driven by the ID. Use AAC aggressively; behavioural supports and respite central.
- ASD without intellectual impairment (Asperger-equivalent) — preserved cognitive milestones, fluent but odd speech with impaired pragmatics, intense circumscribed interests, normal self-help; diagnosis often delayed. High rates of anxiety, depression, bullying. Psychoeducation, CBT adapted for ASD, workplace adjustments.
- Regressive ASD (~15-25 percent) — loss of words and social engagement at 15 to 24 months after apparently normal development; exclude Landau-Kleffner, metabolic and neurodegenerative disease; begin intensive intervention immediately.
- Syndromic ASD — fragile X, tuberous sclerosis complex, Rett (MECP2), PTEN hamartoma tumour syndrome (macrocephaly), 15q duplication, 16p11.2 deletion/duplication, 22q11.2 deletion (DiGeorge), CHARGE, Sotos; manage the underlying syndrome alongside the ASD.
- ASD with catatonia — mute, posturing, negativism, immobility, refusal to eat/drink; managed with lorazepam and, if refractory, ECT.
- Female phenotype — under-diagnosed; anxiety, eating disorders, self-harm, burnout; female-aware diagnostic pathways (AQ-10/AQ-50, RAADS-R); strengths-based framing.
- Adult-diagnosed ASD — anxiety, depression, burnout, employment and relationship difficulty as the presenting complaint; emphasise self-understanding, sensory environment, routines, workplace adjustments, mental-health support; neurodiversity-affirming framing.
- Severity Level 3 (requiring very substantial support) — non-verbal or minimal speech, minimal response to others, extreme distress at change, severely restricted repertoire; lifelong supported living is the norm. [1]
Complications & Pitfalls
- Psychiatric — anxiety disorders 40 percent (GAD, social anxiety, OCD), depression 25-30 percent (peaks in adolescence/adulthood), ADHD 40-50 percent, oppositional defiant and conduct disorders, eating disorders (especially ARFID — avoidant/restrictive food intake disorder driven by sensory aversion, and anorexia in females), and significantly elevated suicidality (especially in verbally able adults).
- Neurological — epilepsy 20-30 percent overall (up to 40 percent with severe ID); epileptiform EEG without clinical seizures in up to 30 percent; catatonia; sleep disorders.
- Behavioural — severe self-injurious behaviour (hand-biting, head-banging, eye-poking), aggression, severe temper tantrums, elopement/wandering, pica (and lead poisoning), property destruction; physical injury from stereotypies.
- Medical — gastrointestinal disorders (constipation, diarrhoea, abdominal pain in 25-40 percent), feeding problems/ARFID, sleep disorders (50-80 percent), dental problems, obesity (especially with antipsychotics).
- Psychosocial — bullying and peer victimisation, social isolation, school refusal/exclusion, family stress and parent burnout, financial strain, reduced employment and independent living, vulnerability to exploitation and abuse.
- Diagnostic overshadowing — a major source of morbidity: medical and psychiatric symptoms attributed to "the autism" rather than investigated and treated (e.g. dental pain causing behavioural deterioration).
- Medication-related — metabolic syndrome, type 2 diabetes, weight gain (risperidone more than aripiprazole), hyperprolactinaemia/gynaecomastia, EPSE/akathisia/tardive dyskinesia, QT prolongation, sedation, behavioural activation with SSRIs.
- Premature mortality — life expectancy reduced by an estimated 16 to 30 years in those with ID and/or epilepsy; leading causes include epilepsy, suffocation, accidents/injury, suicide (especially in those without ID) and comorbid medical illness. [1]
Prognosis & Disposition
Prognosis is highly variable and shaped by baseline cognitive and language ability, severity of symptoms, age and intensity of intervention, presence of ID/epilepsy, and the family/community environment. The same diagnosis spans non-verbal-with-severe-ID through to gifted-and-independent. [1]
Strongest favourable predictors: non-verbal IQ above 70 (especially above 85); functional phrase speech by age 5 (the single best predictor of adult independence and social outcome); early intensive intervention before age 4 to 5; family involvement; milder social-communication deficits; absence of epilepsy; and a supportive educational environment. [1]
Adverse predictors: persistent absence of spoken language at age 5; IQ below 50; comorbid epilepsy (especially infantile spasms); severe ID; dysmorphic features or a syndromic/genetic cause; marked regression; and severe self-injurious behaviour. [1]
Outcome trajectories (approximate): about 15-25 percent lose the diagnosis over time and function independently; about 30 percent achieve reasonable independence with support; about 30-50 percent remain significantly dependent throughout life requiring family, residential or supported care. [1]
ASD is lifelong — the core neurocognitive style persists even when symptoms improve with intervention; ongoing needs include adult mental-health support, employment with reasonable adjustments, social and relationship support, and management of comorbid conditions. Most children are managed in the community via multidisciplinary services with school input; inpatient admission is reserved for severe behavioural disturbance, catatonia, or assessment of acute regression/medical complications; transition to adult services starts at age 14. [1]
Special Populations
- Girls and women — under-diagnosed; present with anxiety, eating disorders, self-harm, burnout, sensory overload, missed until adulthood. Female-aware diagnostic pathways; counselling must address masking/burnout and provide strengths-based framing.[9]
- Intellectually able / "twice-exceptional" — present with anxiety, depression, exhaustion from camouflaging, sensory overload, employment and relationship difficulties; need workplace adjustments, psychoeducation, and mental-health support rather than deficit-focused intervention.
- Adults — diagnosis increasingly made in adulthood; assessment via specialist adult ASD services; emphasise self-understanding, sensory environment and routines, accommodations, support for co-occurring mental health; strengths-based neurodiversity-affirming framing.
- Severe intellectual disability / minimally verbal — emphasise functional communication (AAC), behavioural support, sensory and environmental modification, medical review for GI/dental/sleep causes of distress, family support and respite, and a genetics workup.
- Pregnancy and motherhood in autistic women — heightened sensory overload, anxiety, comorbid mental illness; tailored perinatal mental-health support, clear communication, sensory-friendly maternity environment; medication review (SSRIs and antipsychotics weighed against teratogenicity and breastfeeding risks; valproate is CONTRAINDICATED).
- Comorbid epilepsy — manage per seizure type (levetiracetam, lamotrigine; AVOID valproate in girls of childbearing potential); review seizure control, monitor for side effects and interactions; vagus-nerve stimulation and dietary therapies for refractory cases.
- Elderly/ageing — emerging population; care needs include sensory decline, dementia (limited data), loss of routine/caregivers, mental health; few bespoke services.
- Indigenous, low-income, rural and migrant populations — marked inequity in diagnosis and services; culturally adapted screening tools, interpreter use, family/community-based intervention models.
- Inpatient and acute mental-health settings — predictable low-stimulus environment, consistent staff, visual supports, allow AAC and trusted items, minimise restraints, multidisciplinary autism-trained input, family liaison.
Evidence, Guidelines & Regional Differences
Landmark papers and guidelines to know:[1][9]
- AAP Clinical Report 2020 (Hyman SL et al., Pediatrics; PMID 31843864) — the authoritative paediatric guidance on identification, evaluation and management; mandates universal M-CHAT-R/F screening at 18 and 24 months and multidisciplinary assessment.
- CDC ADDM surveillance 2023 (Shaw KA et al., MMWR; PMID 36952289) — current US prevalence ~1 in 36 (2.8 percent) among 8-year-olds.[4] The 2021 report (PMID 34855727) reported 1 in 54.[3]
- Robins DL et al. 2014 (Pediatrics; PMID 24366990) — M-CHAT-R/F validation in a large US primary-care sample; established the 3+/2+ thresholds and a PPV of ~47.5 percent for ASD.[5]
- Dawson G et al. 2010 (Pediatrics; PMID 19948568) — the ESDM randomised controlled trial; significant gains in IQ, receptive and expressive language, and adaptive behaviour in toddlers; landmark evidence for early naturalistic developmental behavioural intervention.[6]
- McCracken JT et al. 2002 (NEJM; PMID 12151468) — the RUPP Autism Network trial establishing risperidone efficacy for severe irritability/aggression/self-injury in ASD, with significant weight gain.[8]
- Marcus RN et al. 2011 (J Clin Psychiatry; PMID 21813076) — aripiprazole efficacy and tolerability for irritability in paediatric ASD; basis of FDA approval.[7]
- Sandin S et al. 2017 (JAMA; PMID 28973605) — Swedish population twin/family study estimating ASD heritability at ~83 percent, with minimal shared-environment contribution.[11]
- Bai D et al. 2019 (JAMA Psychiatry; PMID 31314057) — five-country cohort; heritability ~80 percent; environmental factors contribute but familial/genetic factors dominate.[10]
- Lai MC et al. 2014 (Lancet; PMID 24074734) — comprehensive clinical and biological review of autism.[9]
- Okoye C et al. 2023 (Cureus; PMID 37692637) — review of early diagnosis, risks and benefits.[2]
- NICE CG128 (2011, updated 2021) — UK guidance on autism diagnosis in under-19s; recommends against routine biological tests, for multidisciplinary diagnostic assessment, and against unsupported biomedical interventions.
- WHO 2023 — comprehensive and integrated services for autism and other developmental disorders; rights-based, community, family-centred framing.
Controversies:
- Rising prevalence — better recognition vs true increase; the consensus is that the rise is largely ascertainment.
- Universal vs targeted screening — the AAP/CDC mandate universal M-CHAT-R/F screening at 18 and 24 months; the UK NSC currently recommends AGAINST universal population screening on grounds of test accuracy and intervention evidence. A clear UK-US regional delta to carry.
- Neurodiversity movement — reframes autism as difference rather than disorder, opposing cure-focused and some behavioural interventions; influences how clinicians frame diagnosis and treatment.
- The (retracted, fraudulent) Wakefield 1998 Lancet MMR paper drove a vaccine-confidence crisis that persists; the largest published studies (Hviid et al. 2019, Annals of Internal Medicine, Danish cohort of over 657 000 children) show NO link between MMR and autism. [1]
Regional delta summary: [1]
Screening policy — the UK-US delta you must carry
US (AAP / CDC)
- Universal M-CHAT-R/F screening at 18 AND 24 months
- All positive screens trigger the Follow-Up Interview and referral to early intervention
- Backed by the AAP Clinical Report 2020
UK (NICE / National Screening Committee)
- Developmental surveillance in primary care with TARGETED referral
- National Screening Committee currently recommends AGAINST universal population screening
- Concerns: test accuracy, natural history, intervention-evidence base
India (Indian Academy of Pediatrics)
- Developmental surveillance with M-CHAT-R at 18 and 24 months
- Adapted to local services and resources
- Emphasis on early referral to developmental services
Exam Pearls
- DSM-5 has TWO domains, NOT the old DSM-IV triad — (A) social-communication deficits in all three sub-domains + (B) at least 2 of 4 restricted/repetitive-behaviour domains; symptoms present early; cause impairment; not better explained by ID alone.
- The three DSM-5 social-communication sub-domains (learn verbatim): social-emotional reciprocity; nonverbal communicative behaviours; developing/maintaining/understanding relationships.
- The four DSM-5 restricted/repetitive-behaviour domains (need 2 or more): stereotyped/repetitive movements/speech/object use; insistence on sameness/rituals; restricted interests of abnormal intensity; hyper-/hypo-reactivity to sensory input.
- Severity levels — Level 1 needing support, Level 2 needing substantial support, Level 3 needing very substantial support — applied SEPARATELY to the social-communication and RRB domains.
- ASD vs Social (Pragmatic) Communication Disorder — the ONLY difference is the presence of restricted/repetitive behaviours/sensory issues (present in ASD, absent in SCD).
- M-CHAT-R/F — 20-item parent screen; positive at 3 or more; Follow-Up at 2 or more; universal at 18 and 24 months per AAP.
- Two FDA-approved drugs for irritability associated with paediatric ASD: risperidone (ages 5-16) and aripiprazole (ages 6-17). NO drug treats core ASD.
- The numbers — prevalence 1 in 36 (CDC 2023); male-to-female 3-4:1; heritability ~80 percent; sibling recurrence 10-20 percent; epilepsy 20-30 percent; ADHD 40-50 percent; ID 30-50 percent; sleep problems 50-80 percent.
- Commonest genetic associations — fragile X (FMR1, commonest inherited cause of ID), tuberous sclerosis (TSC1/TSC2, commonest neurocutaneous ASD cause), 15q duplication, 16p11.2 deletion, MECP2 (Rett), PTEN (with macrocephaly), CHD8.
- Regression in 15-25 percent at 15 to 24 months — ALWAYS exclude Landau-Kleffner (acquired epileptic aphasia with epileptiform EEG), metabolic and neurodegenerative disease.
- MMR does NOT cause autism — the Wakefield 1998 paper was retracted and fraudulent; counsel parents with the Danish 2019 cohort evidence.
- Melatonin (2-6 mg PO at bedtime) is evidence-based for sleep in ASD (CARI trial) — a frequently tested fact.
- Diagnostic workup of every confirmed case — audiology + chromosomal microarray + fragile X testing (NICE/AAP); add EEG if seizures/regression; MRI only if focal signs or abnormal head size.
- Do NOT use unsupported biomedical treatments (chelation, secretin, HBOT, GFCF diet, facilitated communication, MMS) — no evidence, several harmful.
- Intellectual disability is added as a diagnosis when social communication is even MORE impaired than expected for the overall developmental level — DSM-5 criterion E.
- Diagnostic overshadowing is the single most important pitfall — never attribute new medical/psychiatric symptoms to "the autism" without investigation. [1]
Exam application bank (NEET-PG / INICET)
One-line answer
Autism spectrum disorder (ASD) is a neurodevelopmental disorder defined in DSM-5 and ICD-11 by persistent deficits in social communication and social interaction across multiple contexts (the three sub-domains of social-emotional reciprocity, nonverbal communicative behaviour, and relationships) PLUS restricted, repetitive patterns of behaviour, interests or activities (at least 2 of 4: stereotyped/repetitive movements, speech or object use; insistence on sameness/rituals; restricted interests of abnormal intensity; and hyper- or hypo-reactive sensory behaviour). Onset is in the early developmental period, the symptoms are present across contexts, and they cause clinically significant functional impairment. Global prevalence is about 1 in 100 children and the most recent US CDC ADDM surveillance reports 1 in 36 (2.8 percent) among 8-year-olds; the male-to-female ratio is about 3 to 4:1;
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard. [1]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes. [1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change. [1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each. [1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory. [1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Autism Spectrum Disorder.
References
- [1]Hyman SL, Levy SE, Myers SM, Council on Children with Disabilities, Section on Developmental and Behavioral Pediatrics. Identification, Evaluation, and Management of Children With Autism Spectrum Disorder Pediatrics, 2020.PMID 31843864
- [2]Okoye C, Obialo-Ibeawuchi CM, Obajeun OA. Early Diagnosis of Autism Spectrum Disorder: A Review and Analysis of the Risks and Benefits Cureus, 2023.PMID 37692637
- [3]Shaw KA, Maenner MJ, Bakian AV, et al. Early Identification of Autism Spectrum Disorder Among Children Aged 4 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2018 MMWR Surveill Summ, 2021.PMID 34855727
- [4]Shaw KA, Bakian AV, Bilder DA, et al. Early Identification of Autism Spectrum Disorder Among Children Aged 4 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2020 MMWR Surveill Summ, 2023.PMID 36952289
- [5]Robins DL, Casagrande K, Barton M, Chen CM, Dumont-Mathieu T, Fein D. Validation of the modified checklist for Autism in toddlers, revised with follow-up (M-CHAT-R/F) Pediatrics, 2014.PMID 24366990
- [6]Dawson G, Rogers S, Munson J, et al. Randomized, controlled trial of an intervention for toddlers with autism: the Early Start Denver Model Pediatrics, 2010.PMID 19948568
- [7]Marcus RN, Owen R, Kamen L, et al. Safety and tolerability of aripiprazole for irritability in pediatric patients with autistic disorder: a 52-week, open-label, multicenter study J Clin Psychiatry, 2011.PMID 21813076
- [8]McCracken JT, McGough J, Shah B, et al., Research Units on Pediatric Psychopharmacology Autism Network. Risperidone in children with autism and serious behavioral problems N Engl J Med, 2002.PMID 12151468
- [9]Lai MC, Lombardo MV, Baron-Cohen S. Autism Lancet, 2014.PMID 24074734
- [10]Bai D, Yip BHK, Windham GC, et al. Association of Genetic and Environmental Factors With Autism in a 5-Country Cohort JAMA Psychiatry, 2019.PMID 31314057
- [11]Sandin S, Lichtenstein P, Kuja-Halkola R, Hultman CM, Larsson H, Reichenberg A. The Heritability of Autism Spectrum Disorder JAMA, 2017.PMID 28973605