Respiratory · General Medicine
Sarcoidosis
Also known as Sarcoidosis · Non-caseating granulomatous disease · Lofgren syndrome · Scadding stages · Heerfordt syndrome · Boeck sarcoid
Sarcoidosis is a multisystem granulomatous disease of unknown cause characterised by non-caseating (hard) granulomas in one or more organs. It most often affects the lungs and intrathoracic lymph nodes of young and middle-aged adults (peak age 20 to 40 years), with a striking predilection for African ancestry (about three times the incidence of white Americans) and Scandinavians. Presentation ranges from an incidental finding of bilateral hilar lymphadenopathy through acute Löfgren syndrome (erythema nodosum plus bilateral hilar lymphadenopathy plus ankle arthritis, with fever) to chronic multisystem disease involving the skin (lupus pernio), eyes (uveitis), heart (arrhythmia, sudden death), nervous system (cranial nerve VII palsy), liver, spleen, bone and kidney (hypercalcaemia). Diagnosis requires a compatible clinical and radiological picture plus histological non-caseating granulomas, after exclusion of mimics (tuberculosis, fungal infection, berylliosis, lymphoma). The Scadding chest radiograph stages 0 to IV grade pulmonary involvement. Sixty to seventy per cent of cases resolve spontaneously; corticosteroids are first-line for significant disease; methotrexate, azathioprine and infliximab are steroid-sparing for chronic or refractory disease.
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags
Overview & Definition
Sarcoidosis is a multisystem granulomatous disorder of unknown cause defined pathologically by the presence of non-caseating (hard) granulomas — organised aggregates of epithelioid macrophages, multinucleated Langhans giant cells and a rim of CD4+ T-lymphocytes — in one or more organs, in the absence of any infection or malignancy that could explain them. The cause is unknown, but the dominant model is an exaggerated antigen-driven Th1 immune response in a genetically susceptible host: an as-yet-unidentified antigen (microbial or environmental) is presented and fails to be cleared, sustained macrophage and T-cell activation drives granuloma formation, and the resulting inflammatory mass distorts or destroys the tissue it occupies. Although any organ can be involved, the lungs and intrathoracic lymph nodes are affected in over ninety per cent of patients, and the disease is therefore classified and staged principally on its thoracic manifestations.[3][7]
The clinical spectrum is exceptionally broad. At one end is the patient with incidental bilateral hilar lymphadenopathy on a chest X-ray taken for another reason, entirely well, who may never need treatment. At the other is the young patient who presents in complete heart block, ventricular tachycardia, or with rapidly progressive pulmonary fibrosis. Between these extremes lies every conceivable combination of respiratory, cutaneous, ocular, cardiac, neurological, hepatic, renal and constitutional disease. The disease is named for the skin — Boeck's sarkoid (flesh-like) lesions — and it was Jonathan Hutchinson who first described cutaneous sarcoid in 1877, but its modern identity rests on the multisystem granuloma and on the exclusion of its mimics, above all tuberculosis.[7]
Two clinical syndromes anchor the diagnosis at the bedside. Löfgren syndrome, the acute presentation, is the triad of bilateral hilar lymphadenopathy, erythema nodosum and ankle arthritis (periarthritis) with fever, and carries an excellent prognosis — most patients resolve within two years without specific therapy, and biopsy is often unnecessary when the picture is classical. Heerfordt syndrome (uveoparotid fever) is the combination of uveitis, parotid enlargement, fever and cranial nerve VII palsy, and signals more extensive disease requiring corticosteroids. Beyond these syndromes, the diagnosis is built methodically: a compatible clinical-radiological picture, histological non-caseating granulomas from the most accessible involved tissue, and exclusion of tuberculosis, fungal infection, berylliosis and lymphoma.[1][11]

Classification
Sarcoidosis is classified along three axes that the examiner will probe: tempo (acute versus chronic, split at two years), radiographic stage (the Scadding chest X-ray stages 0 to IV), and organ involvement. The first two are objective and reproducible; the third drives management, because certain organs (heart, nerves, eyes, calcium) demand treatment regardless of radiographic stage.[1][7]
Acute sarcoidosis (under two years) presents abruptly, often as Löfgren syndrome or a febrile illness with erythema nodosum, and is the form most likely to resolve spontaneously. Chronic sarcoidosis (over two years) is insidious, often pulmonary or multisystem, tends to fibrose, and is the form most likely to need long-term immunosuppression. The acute-chronic split correlates with the host's genetics — HLA-DRB1*03 in Löfgren syndrome predicts resolution, whereas HLA-DRB1*15 predicts chronicity.[4]
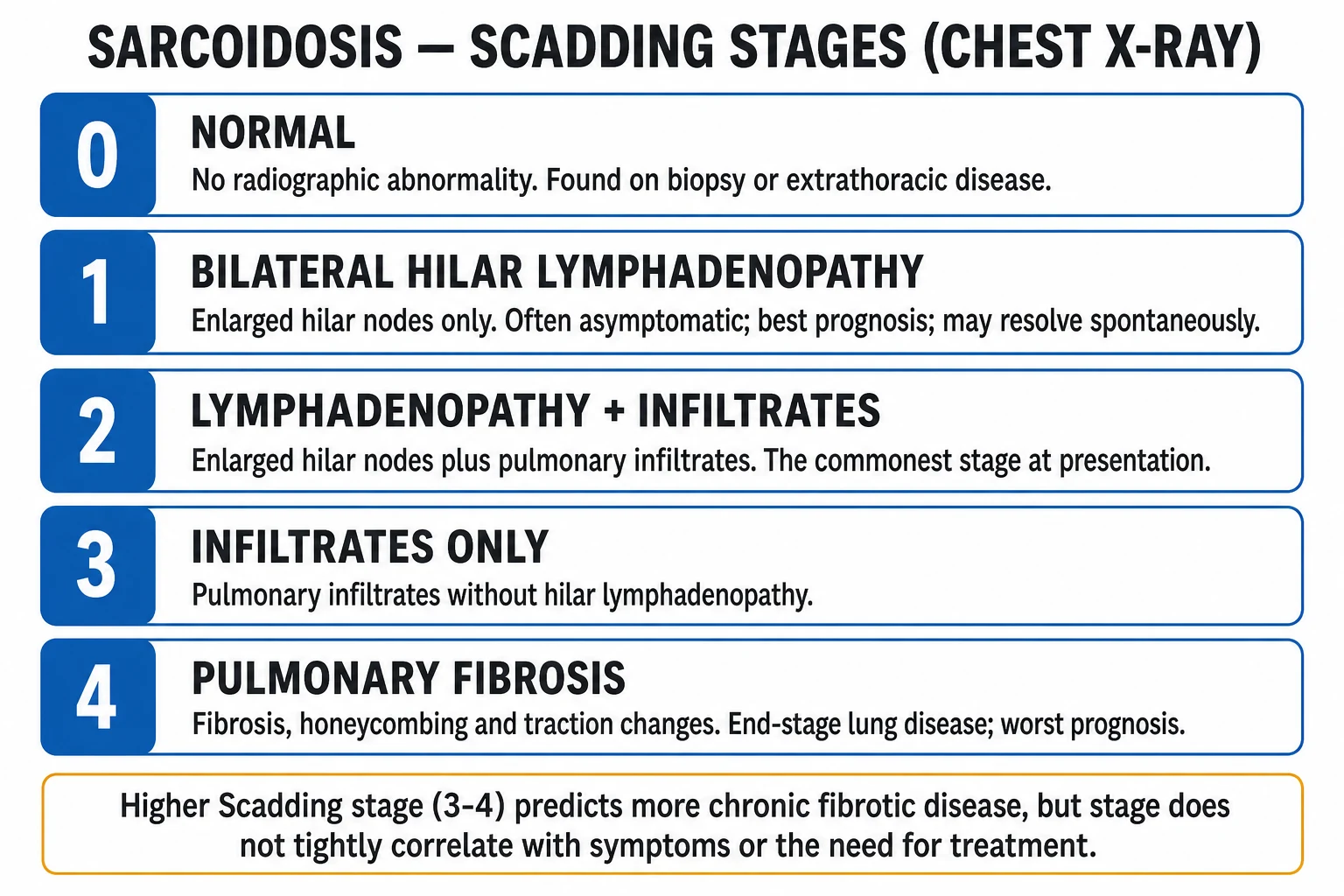
The Scadding radiographic staging system (1961) is the universal language of pulmonary sarcoidosis and is reproduced verbatim below. Stage does not tightly track symptoms or the need for treatment — a stage II patient may be asymptomatic and a stage I patient may have disabling uveitis — but it does predict prognosis: stages I and II resolve more often than stages III and IV.[7]
Acute (under 2 yr)
- Abrupt onset, often Löfgren syndrome
- Erythema nodosum, fever, arthralgia
- HLA-DRB1*03 — resolves spontaneously
- Often no biopsy, no steroids needed
Chronic (over 2 yr)
- Insidious onset, pulmonary fibrosis
- Lupus pernio, multisystem disease
- HLA-DRB1*15 — persistent
- Long-term steroids plus steroid-sparing agents
Organ-based
- Pulmonary over 90 per cent
- Skin: erythema nodosum, lupus pernio
- Eyes: uveitis; Heart: arrhythmia
- Neuro: CN VII; Renal: hypercalcaemia
Sarcoidosis — the numbers that decide a question
Epidemiology & Risk Factors
Sarcoidosis is a worldwide disease with a striking age, sex, racial and geographic distribution. The peak incidence is in adults under 40, with a second smaller peak in women over 50 (often presenting with Löfgren syndrome). Lifetime risk is about 0.85 per cent in white Americans but approaches 2.4 per cent in African-Americans, who also have more severe, more chronic and more extrathoracic disease. Scandinavians, Irish and African ancestry populations are most affected; the disease is uncommon in Indians and East Asians, although Japanese patients have a notably high rate of cardiac sarcoidosis despite a low overall incidence.[4]
The cause is unknown, but epidemiological and molecular work points to a gene-environment interaction. The landmark ACCESS (A Case Control Etiologic Study of Sarcoidosis) enrolled over 700 matched pairs and found modest environmental and occupational associations: exposure to mould and mildew, insecticides, and agricultural employment were modestly over-represented in cases, while tobacco smoking was inversely associated (smokers are less likely to develop sarcoidosis, a paradox the examiner may test). No single infectious agent was confirmed, and the study concluded that sarcoidosis is likely the common endpoint of several environmental triggers in a susceptible host. Subsequent work has revived the infectious trigger hypothesis: Cutibacterium (formerly Propionibacterium) acnes has been detected in granulomas, and mycobacterial antigens (particularly the Mycobacterium tuberculosis catalase-peroxidase KatG) elicit T-cell responses in sarcoid patients, though no organism is cultured from the lesions. Beryllium exposure produces a clinically and histologically indistinguishable disease (chronic beryllium disease), proving that an inorganic metal antigen can drive non-caseating granuloma formation in a susceptible host.[5]
The genetic architecture is increasingly mapped. The strongest associations are within the major histocompatibility complex on chromosome 6: HLA-DRB1*03 defines Löfgren syndrome and a resolving course; HLA-DRB1*15 predicts chronicity. BTNL2 (butyrophilin-like 2), Annexin A11 (ANXA11) and CC-chemokine receptor 2 (CCR2) variants each modestly raise susceptibility. These alleles are distributed differently across populations, helping to explain the ethnic variation in incidence and phenotype.[4]
Pathophysiology
The granuloma is the unit of sarcoidosis. A non-caseating granuloma is a tightly organised collection of activated epithelioid macrophages (large cells with abundant eosinophilic cytoplasm), multinucleated Langhans giant cells (formed by macrophage fusion, with peripheral horseshoe nuclei), surrounded by a cuff of CD4+ T-helper-1 lymphocytes and a thin outer rim of CD8+ cells and B cells. Unlike the granuloma of tuberculosis, there is no central caseous necrosis — hence "non-caseating" or "hard" — although fibrinoid necrosis may occasionally occur in the centre (necrotising sarcoid granulomatosis is a related vasculitic variant).[7]
The cellular and molecular cascade begins when an antigen-presenting cell (tissue macrophage or dendritic cell) ingests an antigen that it cannot fully degrade, processes it onto an MHC class II molecule, and presents it to a naïve CD4+ T cell in the context of co-stimulatory signals. A Th1 polarisation follows, driven by interleukin-12 and interleukin-18, producing CD4+ T cells that secrete interleukin-2, interferon-gamma and tumour necrosis factor-alpha. Interferon-gamma further activates macrophages; TNF-alpha is the master cytokine sustaining the granuloma and is the rationale for anti-TNF (infliximab) therapy in refractory disease. Interleukin-2 drives clonal T-cell expansion, producing the lymphocytosis and elevated CD4:CD8 ratio seen in bronchoalveolar lavage. The activated macrophages also secrete angiotensin-converting enzyme (ACE) — explaining the modest rise in serum ACE used to follow disease activity, though not to diagnose it.[3][7]

Two downstream consequences generate much of the morbidity and the examinable biochemistry. First, activated granuloma macrophages express the 25-hydroxyvitamin D-1-alpha-hydroxylase (CYP27B1), the same enzyme normally confined to the kidney proximal tubule. They therefore convert 25-hydroxyvitamin D into active 1,25-dihydroxyvitamin D (calcitriol) in an unregulated, substrate-driven way. The result is increased intestinal calcium absorption and bone resorption, producing hypercalcaemia and (more often) hypercalciuria, with the attendant risks of nephrocalcinosis, nephrolithiasis and chronic kidney disease. This explains the bedside pearl that a sarcoid patient's calcium rises in summer (sunlight generates vitamin D substrate) and falls with corticosteroids (which suppress granuloma activity).[15]
Second, granulomas may follow one of two fates. They may resolve by apoptosis, leaving no scar — the favourable outcome of Löfgren syndrome. Or they may persist and drive fibrosis, particularly under the influence of transforming growth factor-beta and fibroblast activation, producing the upper-lobe-predominant pulmonary fibrosis of Scadding stage IV with traction bronchiectasis, honeycombing, volume loss and, eventually, pulmonary hypertension and cor pulmonale. Why some patients resolve and others fibrose is the central unsolved question of the disease and is partly genetic (HLA-DRB1*15) and partly environmental.[3]
Clinical Presentation
Sarcoidosis can present through almost any organ, and the presentation reflects the tempo (acute versus chronic) and the organs involved. A focused multisystem history and examination is therefore mandatory at the first encounter, not a respiratory review alone.[3]
Pulmonary disease (over 90 per cent of patients) is often detected incidentally as bilateral hilar lymphadenopathy on a chest X-ray. Symptomatic pulmonary disease produces a dry cough, exertional dyspnoea and vague retrosternal chest discomfort; pleuritic pain and haemoptysis are uncommon and suggest an alternative or a complication (aspergilloma in a fibrotic cavity). Constitutional symptoms — fatigue, weight loss, low-grade fever and night sweats — are common and may dominate the picture, occasionally mimicking lymphoma or tuberculosis.[3][7]
Cutaneous disease is the most visible and examinable extrathoracic manifestation, and the skin lesion often predicts prognosis. Erythema nodosum — tender, erythematous subcutaneous nodules on the shins — is a non-specific (reactive) lesion that is the hallmark of acute sarcoidosis and Löfgren syndrome and signals a good prognosis.[13] Lupus pernio — chronic, violaceous, indurated plaques on the nose, cheeks, ears and lips — is a specific (granulomatous) lesion that signals chronic disease, upper-respiratory involvement, bone cysts and pulmonary fibrosis, and carries a poor prognosis. Other specific lesions include scar sarcoidosis (granulomatous infiltration of old scars, tattoos or venepuncture sites), papular sarcoidosis (small translucent papules on the face and trunk, common in African ancestry) and, less often, granuloma annulare and erythema multiforme.[12][14]
Ocular sarcoidosis affects about a quarter of patients and may precede other manifestations by years. Anterior uveitis presents with a painful red eye, photophobia and blurred vision; posterior uveitis with floaters and visual loss; keratoconjunctivitis sicca with dry gritty eyes. Untreated uveitis causes synechiae, glaucoma, cataract and blindness, so every patient with suspected sarcoidosis needs a slit-lamp examination by an ophthalmologist. Heerfordt syndrome (uveoparotid fever) is the combination of uveitis, parotid enlargement, fever and cranial nerve VII palsy.[3]
Cardiac sarcoidosis is the leading cause of sarcoid-related death. It presents with palpitations, syncope, conduction disease (bundle branch block, atrioventricular block, complete heart block), ventricular arrhythmia, cardiomyopathy and heart failure, and may declare itself as sudden cardiac death. Any cardiac symptom in a patient with known or suspected sarcoidosis mandates an ECG, echocardiogram, 24-hour Holter and (when suspicion is high) cardiac MRI or FDG-PET.[9]
Neurosarcoidosis occurs in 5 to 15 per cent and most often involves the cranial nerves (especially the facial nerve, which may be bilateral), the leptomeninges (chronic meningitis with headache and lymphocytic CSF), the hypothalamic-pituitary axis (diabetes insipidus, hyperprolactinaemia, panhypopituitarism), the spinal cord (myelopathy) and the cerebral parenchyma (seizures, mass lesions). Heerfordt syndrome with its facial palsy is the classic neurocutaneous presentation.[3]
Other organ involvement includes hepatic and splenic granulomas (often asymptomatic, with mildly deranged liver enzymes and hepatosplenomegaly), bone cysts (especially the phalanges of the hands, with dactylitis), bone marrow infiltration (anaemia, leukopenia), renal disease (granulomatous interstitial nephritis, nephrocalcinosis and nephrolithiasis from hypercalciuria), parotid and salivary enlargement, and skeletal muscle involvement (myopathy or nodular masses). Hypercalcaemia itself causes polyuria, polydipsia, constipation, abdominal pain and, at high levels, confusion and arrhythmia.[15]
In the elderly, sarcoidosis is more insidious and cardiac disease is more common than pulmonary; hypercalcaemia may be occult and mistaken for malignancy. In pregnancy, disease usually improves (the immunosuppressive shift of pregnancy) but may flare postpartum. In immunocompromised patients and on immune checkpoint inhibitors, a sarcoid-like reaction can appear and must be distinguished from infection. Blau syndrome (NOD2 mutation) is a familial paediatric granulomatous triad of arthritis, uveitis and dermatitis that mimics sarcoidosis but is monogenic and non-pulmonary.[3]
Differential Diagnosis
The differential of sarcoidosis is the differential of the granulomatous diseases and of bilateral hilar lymphadenopathy. The examiner expects you to list the mimics and, crucially, the features that distinguish each.[1][7]
Tuberculosis is the central mimic, especially in TB-endemic regions. Both produce granulomatous lymphadenitis and pulmonary infiltrates, both cause constitutional symptoms, and both may coexist. The distinguishing feature is the histology: TB produces caseating granulomas with central necrosis, and acid-fast bacilli are demonstrable on Ziehl-Neelsen stain, Xpert MTB/RIF NAAT or mycobacterial culture. Sarcoid granulomas are non-caseating and culture-negative. The principle "exclude TB before starting steroids" is non-negotiable, because corticosteroids can accelerate undiagnosed TB.[1]
Fungal infections — histoplasmosis, coccidioidomycosis, blastomycosis and cryptococcosis — produce granulomatous disease with pulmonary infiltrates and lymphadenopathy that can be radiographically indistinguishable. They are distinguished by endemic exposure history, fungal serology, antigen testing (Histoplasma galactomannan, Cryptococcus antigen) and culture or special stains (Grocott methenamine silver, PAS) of biopsy tissue.[1]
Chronic beryllium disease (berylliosis) produces non-caseating granulomas that are histologically identical to sarcoidosis; the only way to distinguish them is the occupational exposure history (aerospace, nuclear, electronics, dental industries) and a positive beryllium lymphocyte proliferation test (BeLPT) on blood or bronchoalveolar lavage. This is the disease that proves an inorganic metal antigen can generate a sarcoid-like granuloma.[5]
Lymphoma (Hodgkin and non-Hodgkin) and metastatic malignancy cause mediastinal lymphadenopathy and B-symptoms; biopsy shows malignant cells rather than granulomas. Hypersensitivity pneumonitis produces centrilobular nodules and lymphocytic interstitial infiltrates but is distinguished by exposure history (bird fancier's, farmer's lung) and loose, poorly formed non-necrotising granulomas around bronchioles. Idiopathic pulmonary fibrosis causes basal fibrosis without significant adenopathy. Granulomatosis with polyangiitis is a necrotising granulomatous vasculitis with upper-airway and renal disease; Crohn disease is granulomatous but confined to the gut. A sarcoid-like reaction in draining lymph nodes adjacent to a malignancy (or triggered by immune checkpoint inhibitors) can falsely suggest systemic sarcoidosis.[3]
Sarcoidosis
- Non-caseating granuloma
- Bilateral hilar lymphadenopathy
- AFB and fungal stains/cultures negative
- Elevated ACE and BAL CD4:CD8 over 3.5
- Multiorgan; Löfgren syndrome; lupus pernio
Tuberculosis
- Caseating granuloma with central necrosis
- AFB stain or Xpert MTB/RIF positive
- Apical cavitation on chest X-ray
- Caseating lymph node; scrofula
- Exclude before any steroids
Fungal
- Granuloma +/- necrosis; organisms visible
- Endemic exposure (histoplasmosis, cocci)
- Positive serology, antigen, special stains
- May mimic miliary or nodular disease
- Treat with antifungals, not steroids
Berylliosis
- Histologically IDENTICAL non-caseating granuloma
- Occupational beryllium exposure
- Positive beryllium lymphocyte proliferation test
- Aerospace, nuclear, dental industries
- Treat by removal of exposure + steroids
For Löfgren syndrome, the differential is that of erythema nodosum itself: streptococcal pharyngitis, drugs (oral contraceptives, sulfonamides), inflammatory bowel disease, pregnancy, histoplasmosis and coccidioidomycosis. The bilateral hilar lymphadenopathy of Löfgren syndrome, when classical, allows a clinical diagnosis without biopsy.[13]
Clinical & Bedside Assessment
The bedside examination of a patient with suspected sarcoidosis is deliberately multisystem. Begin with the respiratory system: inspect for cachexia and cyanosis, measure respiratory rate and oxygen saturation, and auscultate for fine inspiratory crackles over the mid and upper zones (the fibrotic stage IV). Signs of pulmonary hypertension (loud P2, right ventricular heave, raised JVP, peripheral oedema) indicate advanced disease. The skin is examined next with the lights bright: erythema nodosum on the shins (tender erythematous nodules), lupus pernio on the nose, cheeks and ears (chronic violaceous indurated plaques), and scar sarcoidosis (induration of old scars, tattoos or venepuncture tracks). Examine the eyes for a red anterior chamber and photophobia (uveitis) and lacrimal and parotid glands for enlargement. Palpate all peripheral lymph node groups (cervical, axillary, epitrochlear, inguinal) and examine the abdomen for hepatosplenomegaly. The hands deserve attention — dactylitis and bony swelling of the phalanges (Jüngling disease, radiolucent bone cysts). The neurological examination must include all cranial nerves, with particular attention to the facial nerve (VII), which may be affected bilaterally and is the commonest cranial neuropathy.[3][7]
The cardiovascular examination is mandatory and must be supplemented by a 12-lead ECG on every patient, because cardiac sarcoidosis can present with subclinical conduction disease — a first-degree or higher AV block, bundle branch block or ventricular ectopy — that is invisible at the bedside but visible on the ECG. Any conduction abnormality, palpitation or syncope mandates echocardiography and ambulatory monitoring.[9]
Bedside observations flag the dangerous presentations: hypoxia (respiratory compromise), bradycardia or runs of ventricular ectopy (cardiac sarcoid), polyuria, polydipsia and confusion (hypercalcaemia), and new facial weakness or visual change (neuro-ocular disease). The reflex in any of these is to escalate organ-specific evaluation immediately — they are not variants of benign pulmonary disease.[3]
Investigations
The diagnosis of sarcoidosis rests on three pillars: a compatible clinical and radiological picture, histological non-caseating granulomas in at least one tissue, and exclusion of the mimics (tuberculosis, fungal infection, berylliosis, lymphoma). The ATS 2020 guideline and the 1999 WASOG/ATS/ERS statement both codify this principle, and Löfgren syndrome is the principal exception where biopsy is not required when the triad is classical.[1][7]
Imaging. The chest X-ray is the entry test and stages the disease (Scadding 0 to IV). The classical appearance is bilateral hilar and right paratracheal lymphadenopathy (the "1-2-3 sign" or garland triad), often with middle and upper zone infiltrates. The high-resolution CT is more sensitive and shows perilymphatic nodules (along bronchovascular bundles, fissures and subpleural regions), bilateral hilar and mediastinal lymphadenopathy (often with calcification in chronic disease), the galaxy sign (a large nodule with surrounding small satellite nodules), and the reversed-halo sign (atoll sign). In stage IV disease there is upper-lobe-predominant fibrosis with traction bronchiectasis, volume loss and honeycombing. Whole-body FDG-PET can map active inflammatory disease, including occult cardiac, neural and osseous involvement, and guides biopsy.[1]
Pulmonary function tests. Pulmonary sarcoidosis produces a restrictive defect with reduced vital capacity (FVC) and total lung capacity and a disproportionately reduced diffusion capacity (DLCO). The FVC is the single best measure to follow over time and to gauge response to corticosteroids; a fall of 10 per cent or more in FVC or a fall of 15 per cent or more in DLCO is a recognised threshold to initiate or escalate therapy.[8]
Histology. The goal is to demonstrate non-caseating granulomas from the most accessible involved tissue, balancing diagnostic yield against procedural risk. The order of preference is: skin lesion (highest yield, lowest risk, fully ambulatory); transbronchial lung biopsy (yield over 60 per cent in stage I to II); endobronchial ultrasound-guided transbronchial needle aspiration (EBUS-TBNA) of mediastinal and hilar nodes (yield 80 to 90 per cent, the modern preferred bronchoscopic technique); conjunctival or labial salivary gland biopsy; and, if other approaches fail, surgical (VATS) lung or lymph-node biopsy. All biopsy material must be stained for acid-fast bacilli (Ziehl-Neelsen) and fungi (Grocott methenamine silver, PAS), and submitted for mycobacterial and fungal culture, to exclude the mimics.[1]
Bloods. A full blood count may show anaemia, leukopenia or lymphopenia. Serum angiotensin-converting enzyme (ACE) is elevated in about 60 per cent of active cases but is non-specific (also raised in TB, lymphoma, asbestosis, hepatitis, hyperthyroidism and diabetes) and non-diagnostic; its main role is in following disease activity over time in an individual patient. Serum and 24-hour urinary calcium are essential — hypercalciuria often precedes and is more common than hypercalcaemia. Liver function tests, renal function and electrolytes screen for hepatic and renal involvement. Serum ACE genotype has limited clinical utility. Soluble interleukin-2 receptor is an emerging marker. Tuberculin skin test is typically anergic (negative) in active sarcoidosis, a curiosity the examiner may probe.[1]
Bronchoalveolar lavage. BAL shows a lymphocytic alveolitis with a raised CD4:CD8 ratio, typically over 3.5 (and often over 4.0). A ratio over 3.5 has a sensitivity and specificity of about 90 per cent for sarcoidosis in the right clinical context, supporting the diagnosis, though it is not used in isolation.[7]
Organ-specific tests. Every patient should have a 12-lead ECG; if cardiac sarcoid is suspected, add echocardiography (regional wall motion abnormality, reduced ejection fraction), 24-hour Holter monitoring (conduction disease, ventricular arrhythmia), and cardiac MRI with late gadolinium enhancement (the typical pattern is basal, subepicardial or mid-myocardial enhancement of the lateral or septal wall) or FDG-PET (focal uptake indicating active inflammation). Slit-lamp examination by an ophthalmologist screens for uveitis. MRI of the brain and spine with gadolinium and CSF analysis (lymphocytic pleocytosis, raised protein, normal glucose, oligoclonal bands) are reserved for suspected neurosarcoidosis. Liver ultrasound or biopsy is reserved for significant hepatomegaly or derangement.[1][9]
SARCOID
Management — Resuscitation

Most patients with sarcoidosis are not acutely unwell and are managed as outpatients, but a small number present with time-critical, life-threatening disease in which the resuscitation reflex must precede the immunosuppressive ladder. The three dangerous scenarios are complete heart block or sustained ventricular arrhythmia, severe hypercalcaemia, and acute respiratory failure.[9][15]
Cardiac sarcoid with conduction disease or arrhythmia is the leading cause of sudden death. The immediate priorities are continuous cardiac monitoring, transcutaneous or transvenous pacing for symptomatic high-grade AV block, and intravenous corticosteroids (methylprednisolone 0.5 to 1 g daily for one to three days, then oral prednisolone) to suppress active inflammation. Sustained ventricular tachycardia or fibrillation is managed with advanced life support, an implantable cardioverter-defibrillator (ICD), and referral for electrophysiology evaluation.[9]
Severe hypercalcaemia (corrected calcium over 3.4 mmol/L, or symptomatic) is managed with aggressive intravenous isotonic saline (3 to 6 L in the first 24 hours), intravenous bisphosphonate (zoledronic acid 4 mg or pamidronate 60 to 90 mg) to inhibit bone resorption, subcutaneous calcitonin (4 IU/kg every 12 hours for a rapid but transient effect), and corticosteroids (prednisolone 20 to 40 mg per day) to suppress the underlying granulomatous 1-alpha-hydroxylase activity. The calcium falls because the granuloma stops converting 25-OH-D to active calcitriol.[15]
Acute respiratory failure from extensive alveolitis or superimposed infection (pneumothorax in fibrotic disease, haemorrhage) is managed with supplemental oxygen, non-invasive or invasive ventilation as needed, treatment of any precipitant, and intravenous corticosteroids for active alveolitis. Status epilepticus or rapidly progressive myelopathy from neurosarcoidosis is managed with standard anticonvulsant protocols and intravenous methylprednisolone (1 g daily for three to five days).[2]
Management — Definitive & Stepwise
Definitive management follows a clear ladder that the examiner will probe in detail: observe, corticosteroid, steroid-sparing, anti-TNF for refractory disease, and organ-specific measures. The thresholds for stepping up the ladder are as important as the drugs themselves.[2][8]
Step 1 — Observe. Asymptomatic pulmonary sarcoidosis (Scadding stage 0 to II with stable lung function and no dangerous organ involvement) is observed without therapy, because the disease resolves spontaneously in the majority. A reasonable surveillance schedule is clinical and spirometric review every 3 to 6 months for the first two years. Asymptomatic stage III disease is observed similarly; stage IV fibrosis is treated symptomatically.[2]
Step 2 — Corticosteroids (first-line when treatment is needed). Oral prednisolone 20 to 40 mg per day (roughly 0.5 mg/kg/day) is started for symptomatic pulmonary disease (cough, dyspnoea, declining lung function), ocular disease not controlled by topical therapy, cardiac, neurologic or renal involvement, symptomatic or persistent hypercalcaemia, and disfiguring cutaneous disease. The ERS 2021 guideline recommends an initial dose of 0.3 to 0.6 mg/kg/day for 4 to 6 weeks, then a taper over 6 to 24 months to the lowest effective maintenance dose (5 to 15 mg/day), recognising that relapse is common as steroids are tapered. Löfgren syndrome usually needs only NSAIDs; steroids are reserved for severe arthritis or symptomatic disease. In cardiac and neurosarcoidosis, the dose is higher — prednisolone 0.5 to 1 mg/kg/day — and is maintained for longer. Bone protection (calcium and vitamin D, a bisphosphonate) and Pneumocystis prophylaxis (co-trimoxazole 480 mg daily or three times weekly) are considered on long courses.[2][6]
[2]Step 3 — Steroid-sparing agents are added when disease is chronic or relapsing on steroid taper, when steroid toxicity is unacceptable, or for specific organs. The ERS 2021 guideline names methotrexate as the first steroid-sparing agent of choice. Methotrexate 10 to 25 mg once weekly (titrated from 7.5 to 10 mg weekly up) with daily folic acid 5 mg once weekly (or 1 mg daily) is the workhorse of chronic sarcoid; baseline and monitoring bloods include FBC, liver function and renal function every 4 to 8 weeks; chest X-ray is monitored for pneumonitis. Azathioprine 1 to 3 mg/kg/day (check thiopurine methyltransferase (TPMT) activity first to avoid fatal myelosuppression) is a near-equivalent alternative. Mycophenolate mofetil 1 to 2 g/day is preferred for neurologic and cutaneous sarcoidosis. Leflunomide 10 to 20 mg/day is an alternative. Hydroxychloroquine 200 to 400 mg/day (5 mg/kg lean body weight) is used principally for cutaneous disease (lupus pernio), hypercalcaemia and joint disease; baseline and annual eye (OCT) screening for maculopathy is mandatory. Chloroquine 250 to 500 mg/day is an alternative antimalarial with the same eye-risk caveat.[2][6][15]
Step 4 — Anti-TNF and refractory disease. For refractory pulmonary, cardiac or neurosarcoidosis unresponsive to corticosteroids and two steroid-sparing agents, infliximab is the agent with the best evidence. The Baughman 2006 randomised placebo-controlled trial (the INFLEX trial) showed that infliximab 3 to 5 mg/kg at weeks 0, 2 and 6, then every 8 weeks produced a modest but significant improvement in FVC in chronic pulmonary sarcoidosis. Adalimumab (40 mg subcutaneously every other week, with an induction of 80 mg then 40 mg at week 1) is an alternative fully human monoclonal. Screening for latent tuberculosis (IGRA ± chest X-ray) before anti-TNF is mandatory, as is hepatitis B screening. Rituximab and JAK inhibitors (tofacitinib) have emerging evidence for highly refractory disease.[2][10]
Organ-specific measures. In cardiac sarcoidosis, prednisolone 0.5 to 1 mg/kg/day is combined with a steroid-sparing agent early; a permanent pacemaker is indicated for symptomatic AV block, and an implantable cardioverter-defibrillator (ICD) is indicated for sustained ventricular arrhythmia, prior cardiac arrest, or an ejection fraction under 35 per cent despite optimised therapy. Catheter ablation of refractory ventricular tachycardia, heart failure pharmacotherapy (beta-blocker, ACE inhibitor, MRA), and heart transplantation for end-stage disease complete the cardiac toolkit. In ocular sarcoidosis, topical and periocular corticosteroids with cycloplegia are used for anterior uveitis; posterior uveitis requires systemic steroids and steroid-sparing agents. In hypercalcaemia, in addition to corticosteroids, the patient is advised to avoid excess sunlight and vitamin D supplementation and to maintain hydration. In stage IV fibrosis, long-term oxygen therapy for hypoxaemia (PaO2 under 7.3 kPa or 55 mmHg), pulmonary rehabilitation, treatment of pulmonary hypertension (targeted therapy if mean PAP over 35 mmHg), and lung transplantation for selected end-stage patients complete the ladder.[2][9]
Prednisolone (1st line)
- 20 to 40 mg PO once daily (0.5 mg/kg/day)
- Taper over 6 to 24 months to lowest effective dose
- Indications: symptomatic lung, cardiac, neuro, ocular, hypercalcaemia
- Bone protection + PJP prophylaxis on long courses
Methotrexate (steroid-sparing)
- 10 to 25 mg PO/SC once weekly with folic acid 5 mg weekly
- First-line steroid-sparing agent (ERS 2021)
- Monitor FBC, LFT, creatinine every 4 to 8 weeks
- Contraindicated in pregnancy and liver disease
Azathioprine
- 1 to 3 mg/kg/day PO
- Check TPMT activity before starting
- Monitor FBC, LFT weekly then monthly
- Pregnancy-safe (relative); alternative to methotrexate
Infliximab (refractory)
- 3 to 5 mg/kg IV at weeks 0, 2, 6 then every 8 weeks
- Refractory pulmonary, cardiac, neuro sarcoid
- Screen for latent TB and hepatitis B first
- Baughman 2006 RCT showed modest FVC gain
Specific Subtypes & Scenarios
Löfgren syndrome is the acute presentation and is treated as a clinical diagnosis when the triad is classical. It comprises bilateral hilar lymphadenopathy, erythema nodosum and bilateral ankle arthritis (periarthritis) with fever. The prognosis is excellent — 80 to 90 per cent of patients resolve within two years without specific therapy. Management is NSAIDs and rest; a short course of corticosteroids is reserved for severe arthritis or persistent symptoms. Biopsy is not required when the picture is classical (the ATS 2020 guideline endorses this), but the patient must be reviewed to exclude mimics (streptococcal infection, drugs, inflammatory bowel disease). HLA-DRB1*03 positivity predicts resolution.[1][11][13]
Heerfordt syndrome (uveoparotid fever) is the combination of uveitis, parotid enlargement, fever and cranial nerve VII (facial) palsy. It is a multisystem acute sarcoid that requires systemic corticosteroids and close ophthalmology input. The facial palsy often resolves with treatment.[3]
Cardiac sarcoidosis deserves its own paragraph because it kills. The presenting rhythm disturbances range from first-degree AV block, bifascicular block and complete heart block through sustained monomorphic ventricular tachycardia to sudden cardiac death. Diagnosis rests on cardiac MRI (basal, subepicardial or mid-wall late gadolinium enhancement of the septum or lateral wall, often with thinning) or FDG-PET (focal uptake), with the Heart Rhythm Society 2014 criteria providing the diagnostic framework. Treatment combines corticosteroids (0.5 to 1 mg/kg/day) to suppress active inflammation, an early steroid-sparing agent, and device therapy (pacemaker for AV block, ICD for secondary prevention of ventricular arrhythmia, and increasingly for primary prevention in selected patients).[9]
Neurosarcoidosis most often involves the cranial nerves (especially the facial nerve, often bilateral and sometimes recurrent), the basal leptomeninges (chronic meningitis), the hypothalamic-pituitary axis (diabetes insipidus, hyperprolactinaemia), the spinal cord (myelopathy) and the cerebral parenchyma (seizures, mass lesions). MRI with gadolinium shows leptomeningeal enhancement, often basal, and nodular parenchymal lesions; CSF shows lymphocytic pleocytosis with raised protein and normal or low glucose. Treatment is high-dose corticosteroids followed by early steroid-sparing (methotrexate, mycophenolate) because neurosarcoid is the most steroid-dependent and relapse-prone form.[3]
Cutaneous sarcoidosis divides into the acute form (erythema nodosum — NSAIDs, good prognosis) and the chronic form (lupus pernio, papular sarcoid, scar sarcoidosis — hydroxychloroquine 200 to 400 mg/day, methotrexate, and for refractory lupus pernio, anti-TNF). Intralesional corticosteroids (triamcinolone) help localised lesions. Lupus pernio is refractory and disfiguring, and is associated with upper-respiratory-tract and bone involvement.[12][14]
Stage IV (fibrotic) pulmonary sarcoid is the end-stage, characterised by upper-lobe fibrosis, traction bronchiectasis, volume loss, honeycombing and frequently pulmonary hypertension and chronic respiratory failure. Corticosteroids have limited effect on established fibrosis; management is supportive — long-term oxygen, pulmonary rehabilitation, treatment of pulmonary hypertension, vaccination, and lung transplantation in selected patients (sarcoid recurs in the graft but rarely causes graft failure).[2]
Complications & Pitfalls
The complications of sarcoidosis are organ-specific and follow the pattern of the disease. Pulmonary complications include progressive fibrosis with traction bronchiectasis and honeycombing, respiratory failure, pulmonary hypertension (mean PAP over 25 mmHg), cor pulmonale, secondary aspergilloma colonising an upper-lobe cavity (which may cause massive haemoptysis), pneumothorax from ruptured bullae, and opportunistic infection in the immunosuppressed patient. Cardiac complications — sudden cardiac death, heart failure, conduction disease requiring pacing, and ventricular arrhythmia requiring ICD — are the leading cause of sarcoid-related mortality and the principal reason to screen every patient with an ECG.[9]
Renal complications arise from hypercalcaemia and hypercalciuria (nephrocalcinosis, nephrolithiasis, obstructive uropathy) and from granulomatous interstitial nephritis; untreated, these progress to chronic kidney disease. Ocular complications — synechiae, glaucoma, cataract and blindness — are the risk of untreated uveitis and the reason every patient needs slit-lamp screening. Neurologic complications include permanent cranial neuropathy, chronic meningitis, hydrocephalus, myelopathy and treatment-resistant seizures. Endocrine disturbance includes hypercalcaemia, diabetes insipidus, and (from corticosteroids) hyperglycaemia, osteoporosis and adrenal suppression.[3][15]
The classic pitfalls are examinable and recurring. The first is missing cardiac sarcoidosis in a young patient with a conduction abnormality — every newly diagnosed sarcoid patient needs an ECG, and any conduction disease mandates echocardiography and cardiac MRI. The second is failing to exclude tuberculosis and fungal infection before starting corticosteroids — caseating granuloma on histology, a positive AFB stain, Xpert MTB/RIF or fungal culture must redirect the diagnosis. The third is over-treating asymptomatic disease with corticosteroids when observation would have sufficed. The fourth is ignoring hypercalcaemia until the patient presents with renal failure from nephrocalcinosis — a 24-hour urinary calcium is part of the initial work-up. The fifth is neglecting drug toxicities: methotrexate hepatotoxicity and pneumonitis, azathioprine myelosuppression (especially in TPMT-deficient patients), hydroxychloroquine maculopathy, and infliximab's risk of reactivating latent tuberculosis. The sixth is misattributing lupus pernio (a specific, chronic, poor-prognosis lesion) to a benign dermatosis.[2][6]
Exam application bank (NEET-PG / INICET)
One-line answer
Sarcoidosis is a multisystem granulomatous disease of unknown cause characterised by non-caseating (hard) granulomas in one or more organs. It most often affects the lungs and intrathoracic lymph nodes of young and middle-aged adults (peak age 20 to 40 years), with a striking predilection for African ancestry (about three times the incidence of white Americans) and Scandinavians. Presentation ranges from an incidental finding of bilateral hilar lymphadenopathy through acute Löfgren syndrome (erythema nodosum plus bilateral hilar lymphadenopathy plus ankle arthritis, with fever) to chronic multisystem disease involving the skin (lupus pernio), eyes (uveitis), heart (arrhythmia, sudden death), nervous system (cranial nerve VII palsy), liver, spleen, bone and kidney (hypercalcaemia). Diagnosis requires a compatible clinical and radiological picture plus histological non-caseating granulomas
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard. [1]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes. [1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change. [1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each. [1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory. [1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Sarcoidosis.
Prognosis & Disposition
The prognosis of sarcoidosis is good for the majority and grave for the minority, and the job of the clinician is to identify which patient sits at which end of the curve. Overall, 60 to 70 per cent of patients experience spontaneous or treatment-induced resolution within two years. Löfgren syndrome has the best outlook — 80 to 90 per cent resolve within two years, especially HLA-DRB1*03-positive patients, and the disease does not recur. Acute presentation, erythema nodosum, Scadding stage I and short symptom duration all predict resolution.[1][4][11]
The poor-prognosis features are: lupus pernio; Scadding stage III and especially stage IV (fibrosis); chronic uveitis; hypercalcaemia; African ancestry (more chronic and severe disease); age of onset over 40; nasal mucosal involvement; bone cysts; and neurologic, cardiac or renal involvement. These patients are the ones who will need long-term corticosteroids and steroid-sparing agents, and who carry the disease-related mortality.[4][15]
The leading causes of sarcoid-related death are respiratory failure from pulmonary fibrosis, sudden cardiac death, and neurologic disease. Mortality is reported at 1 to 5 per cent in specialty cohorts, though population data suggest a smaller figure. Relapse after withdrawal of therapy occurs in about a third to a half of patients with chronic disease, particularly within the first six months of tapering.[2]
Disposition. Most patients are managed as outpatients with respiratory or general medicine follow-up and the appropriate organ specialists (cardiology, ophthalmology, neurology, dermatology, nephrology) as the phenotype demands. Admission is reserved for suspected or confirmed cardiac sarcoid with arrhythmia, severe symptomatic hypercalcaemia, acute respiratory failure, and rapidly progressive neurosarcoidosis. Every patient needs an explicit safety-net and follow-up plan — pulmonary function tests and chest imaging at regular intervals, ECG at every visit, slit-lamp annually, and clear advice to return urgently with palpitations, syncope, visual change or polyuria.[1]
Special Populations
Sarcoidosis in pregnancy usually improves during the third trimester as the maternal immune system shifts toward Th2 (immunosuppressive), and may flare in the postpartum period. Corticosteroids are safe in pregnancy and remain first-line; azathioprine is relatively safe with specialist input. Methotrexate and mycophenolate mofetil are absolutely contraindicated (teratogenic) and must be stopped before conception. Breastfeeding is generally acceptable on low-dose prednisolone.[6]
African ancestry patients have about three times the incidence of white Americans, more often present with extrathoracic disease (cardiac, cutaneous lupus pernio, bone cysts, ocular), and have more chronic, more severe and more steroid-dependent disease. The female preponderance is more marked. These patients warrant closer surveillance and earlier steroid-sparing therapy.[4]
Elderly patients present more insidiously; cardiac sarcoidosis is more common than in the young, the constitutional features may be mistaken for malignancy, and hypercalcaemia may be occult. The threshold for cardiac MRI and Holter monitoring should be lower. Comorbidity (chronic kidney disease, diabetes, osteoporosis) complicates corticosteroid use and pushes toward early steroid-sparing agents.[3]
Children are rarely affected; paediatric sarcoidosis is most often multisystem and paucibacillary, with ocular and joint involvement prominent. Blau syndrome — an autosomal dominant NOD2 (CARD15) mutation — produces a familial granulomatous triad of arthritis, uveitis and dermatitis in young children and is the principal differential; it lacks the pulmonary disease of classic sarcoidosis.[3]
Immunocompromised patients and those on immune checkpoint inhibitors (anti-PD-1, anti-CTLA-4) may develop a sarcoid-like granulomatous reaction that mimics idiopathic sarcoidosis; biopsy and exclusion of infection remain essential, and the reaction often responds to corticosteroids or to cessation of the trigger.[3]
Evidence, Guidelines & Regional Differences
The two governing modern guidelines are the American Thoracic Society (ATS) 2020 Clinical Practice Guideline on Diagnosis and Detection of Sarcoidosis (Crouser et al.) and the European Respiratory Society (ERS) 2021 Clinical Practice Guideline on Treatment of Sarcoidosis (Baughman et al.). The ATS guideline endorses tissue confirmation of non-caseating granulomas in most cases but accepts a clinical diagnosis of Löfgren syndrome without biopsy when the triad is classical; it supports EBUS-TBNA as the preferred bronchoscopic diagnostic technique (high yield, low complication rate) over blind transbronchial biopsy. The ERS guideline codifies corticosteroids as first-line therapy, methotrexate as the first steroid-sparing agent, and infliximab for refractory disease; it explicitly recommends against routine antibiotics and against first-line anti-TNF in treatment-naïve patients.[1][2]
The Delphi consensus treatment algorithm (Rahaghi et al., 2020) operationalises the ladder for pulmonary sarcoidosis: observe asymptomatic disease; start prednisolone 20 to 40 mg/day for symptomatic or worsening disease; add methotrexate at steroid taper or relapse; switch to or add infliximab for refractory disease; consider transplant for end-stage fibrosis.[8]
Landmark evidence includes the ACCESS study (Newman 2004), which mapped the modest environmental associations and the gene-environment model; the Baughman 2006 randomised controlled trial of infliximab (the first positive RCT of anti-TNF in sarcoidosis, showing a modest FVC gain); and a series of methotrexate observational cohorts establishing steroid-sparing efficacy. The Heart Rhythm Society 2014 expert consensus provides the criteria for the diagnosis of cardiac sarcoidosis.[5][9][10]
Controversies and grey zones include: when exactly to start corticosteroids in asymptomatic stage II disease (the ERS recommends treatment for worsening lung function or significant symptoms); whether to screen all patients for cardiac sarcoidosis with MRI/PET or only those with symptoms or ECG abnormality (most centres screen selectively, although Japanese guidelines recommend routine cardiac screening because of the high cardiac incidence); the utility of serum ACE (low specificity limits its diagnostic role but it remains useful for tracking activity); the optimal duration of therapy (relapse is common within six months of tapering); and the role of newer agents — JAK inhibitors (tofacitinib), phosphodiesterase-4 inhibitors (apremilast), and rituximab — which remain investigational.[2][4]
In TB-endemic regions (India, sub-Saharan Africa, parts of South-East Asia), sarcoidosis is a diagnosis of exclusion with a particular obligation to exclude tuberculosis by AFB stain, Xpert MTB/RIF NAAT and mycobacterial culture of every biopsy before starting corticosteroids — a sarcoid-like granuloma in a high-TB-burden country is TB until proven otherwise. Japanese guidelines emphasise routine cardiac screening because cardiac sarcoidosis is over-represented in the Japanese population. The ATS and ERS guidelines are the international reference standards; Indian (NTEP-aligned) and NICE practice adds the TB-exclusion reflex by default.[1][4]
Exam Pearls
The examiner will reward precise one-liners and a handful of discriminating facts. The following are the high-yield minutiae that decide a sarcoidosis question.[1][3][11]
The definition. Sarcoidosis is a multisystem non-caseating granulomatous disease of unknown cause affecting the lungs and intrathoracic lymph nodes in young adults (peak 20 to 40 years). The granuloma is non-caseating — the single word that distinguishes it from tuberculosis.[3]
The epidemiology. African ancestry (3 times the incidence and more severe), Scandinavian and Northern European, slight female predominance, and inverse association with smoking (smokers are less likely to develop sarcoidosis — a counterintuitive fact worth remembering).[4]
The syndromes. Löfgren syndrome = bilateral hilar lymphadenopathy plus erythema nodosum plus ankle arthritis with fever, acute and benign, often no biopsy, NSAIDs. Heerfordt syndrome (uveoparotid fever) = uveitis plus parotid enlargement plus fever plus cranial nerve VII palsy.[11]
The Scadding stages. 0 normal; I bilateral hilar lymphadenopathy; II lymphadenopathy plus infiltrates; III infiltrates only; IV fibrosis. I to II best prognosis, IV worst.[7]
The investigations. Compatible clinical-radiological picture plus non-caseating granulomas plus exclusion of TB and fungal. Chest X-ray shows bilateral hilar and right paratracheal lymphadenopathy (1-2-3 sign); CT shows perilymphatic nodules; BAL shows CD4:CD8 ratio over 3.5; ACE is supportive, not diagnostic; 24-hour urinary calcium detects hypercalciuria.[1]
The treatment. Observe asymptomatic disease; prednisolone 20 to 40 mg/day, taper over 6 to 24 months for symptomatic disease or organ-threatening involvement; methotrexate 10 to 25 mg weekly as first steroid-sparing agent; infliximab 3 to 5 mg/kg for refractory disease; hydroxychloroquine for cutaneous; NSAIDs for Löfgren.[2]
The dangerous organs. Heart (arrhythmia, sudden death, steroids plus ICD), nerves (cranial VII, hypothalamus, seizures), eyes (uveitis, blindness), calcium (hypercalcaemia, nephrocalcinosis). The lung is the commonest organ but not the most dangerous.[9]
The prognosis. 60 to 70 per cent resolve; Löfgren 80 to 90 per cent in two years; lupus pernio, stage IV, hypercalcaemia, cardiac, neuro and renal disease predict a chronic, steroid-dependent course. Death is from respiratory failure, sudden cardiac death or neurologic disease.[4][15]
References
- [1]Crouser ED, Maier LA, Wilson KC, et al. Diagnosis and Detection of Sarcoidosis. An Official American Thoracic Society Clinical Practice Guideline Am J Respir Crit Care Med, 2020.PMID 32293205
- [2]Baughman RP, Valeyre D, Korsten P, et al. ERS clinical practice guidelines on treatment of sarcoidosis Eur Respir J, 2021.PMID 34140301
- [3]Seve P, Pacheco Y, Durupt F, et al. Sarcoidosis: A Clinical Overview from Symptoms to Diagnosis Cells, 2021.PMID 33807303
- [4]Rossides M, Darlington P, Kullberg S, et al. Sarcoidosis: Epidemiology and clinical insights J Intern Med, 2023.PMID 36872840
- [5]Newman LS, Rose CS, Bresnitz EA, et al. A case control etiologic study of sarcoidosis: environmental and occupational risk factors Am J Respir Crit Care Med, 2004.PMID 15347561
- [6]Gerke AK. Treatment of Sarcoidosis: A Multidisciplinary Approach Front Immunol, 2020.PMID 33329511
- [7]Hunninghake GW, Costabel U, Ando M, et al. Statement on sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999 Am J Respir Crit Care Med, 1999.PMID 10430755
- [8]Rahaghi FF, Baughman RP, Saketkoo LA, et al. Delphi consensus recommendations for a treatment algorithm in pulmonary sarcoidosis Eur Respir Rev, 2020.PMID 32198218
- [9]Mankad P, Sequeira J, Reddy VN, et al. Cardiac Sarcoidosis Curr Cardiol Rep, 2019.PMID 31768666
- [10]Baughman RP, Drent M, Kavuru M, et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement Am J Respir Crit Care Med, 2006.PMID 16840744
- [11]Chapa-Rodriguez A, Bhatt SP, Gill RR, et al. Lofgren Syndrome 2026.PMID 29493940
- [12]Ezeh N, Caplan A, Rosenbach M, et al. Cutaneous Sarcoidosis Dermatol Clin, 2023.PMID 37236714
- [13]Perez-Garza DM, Chavez-Alvarez S, Ocampo-Candiani J, et al. Erythema Nodosum: A Practical Approach and Diagnostic Algorithm Am J Clin Dermatol, 2021.PMID 33683567
- [14]Abdelghaffar M, Hwang E, Damsky W. Cutaneous Sarcoidosis Clin Chest Med, 2024.PMID 38245372
- [15]Baughman RP, Culver DA, Judson MA. Treatment of Sarcoidosis Clin Rev Allergy Immunol, 2015.PMID 25989728