Rheumatology · General Medicine
ANCA-Associated Vasculitis (GPA, MPA & EGPA)
Also known as ANCA-associated vasculitis · AAV · Granulomatosis with polyangiitis · GPA · Wegener granulomatosis · Microscopic polyangiitis · MPA · Eosinophilic granulomatosis with polyangiitis · EGPA · Churg-Strauss
ANCA-associated vasculitis (AAV) comprises three necrotising small-vessel vasculitides associated with anti-neutrophil cytoplasmic antibodies (ANCA): granulomatosis with polyangiitis (GPA, formerly Wegener) — PR3-ANCA (c-ANCA), ENT (epistaxis, saddle nose, deafness) plus respiratory (sinusitis, lung nodules/cavitation) plus renal (pauci-immune glomerulonephritis); microscopic polyangiitis (MPA) — MPO-ANCA (p-ANCA), renal plus pulmonary without granulomatous ENT; eosinophilic granulomatosis with polyangiitis (EGPA, Churg-Strauss) — asthma, eosinophilia, sinus/nasal polyposis then vasculitic phase (neuropathy, cardiomyopathy, renal). The feared pulmonary-renal syndrome (alveolar haemorrhage plus rapidly progressive glomerulonephritis) is a medical emergency. Diagnosis combines ANCA (immunofluorescence plus PR3/MPO ELISA), urinalysis, renal biopsy (pauci-immune necrotising crescentic GN), and CT chest. Induction is high-dose glucocorticoids plus rituximab or cyclophosphamide, with plasma exchange for severe renal/pulmonary disease; maintenance is rituximab or azathioprine; mepolizumab (anti-IL-5) for EGPA.
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags
Overview & Definition
ANCA-associated vasculitis (AAV) is the archetype of the pulmonary-renal syndrome — a group of disorders where small-vessel inflammation causes simultaneous alveolar haemorrhage and rapidly progressive glomerulonephritis (RPGN). The 2012 Revised International Chapel Hill Consensus Conference (CHCC) nomenclature defines AAV as a necrotising vasculitis, predominantly affecting small vessels (capillaries, venules, arterioles and small arteries), with few or no immune deposits (pauci-immune), associated with ANCA.[3] The three AAV subtypes are granulomatosis with polyangiitis (GPA, formerly Wegener granulomatosis), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA, Churg-Strauss). A fourth phenotype, renal-limited vasculitis (RLV), describes pauci-immune necrotising crescentic GN with ANCA positivity but no extrarenal disease.
The clinical skill is recognising the multi-system pattern — ENT plus lung plus renal equals GPA; renal plus pulmonary without ENT equals MPA; asthma plus eosinophilia plus neuropathy equals EGPA — confirming with ANCA (PR3/MPO) and biopsy, and treating aggressively. Untreated severe AAV is fatal within months, but with modern immunosuppression (high-dose glucocorticoids plus rituximab or cyclophosphamide, with selective plasma exchange) the 5-year survival now approaches 75 to 90 percent.[1] Two therapeutic revolutions have reshaped the field: rituximab (anti-CD20 B-cell depletion) has replaced cyclophosphamide in many induction regimens — particularly in relapsing and PR3-positive disease — and mepolizumab (anti-IL-5) targets the eosinophilic biology of EGPA. The alternative complement pathway is now an established therapeutic target, with avacopan (C5a receptor blocker) licensed for steroid-sparing induction.[1]
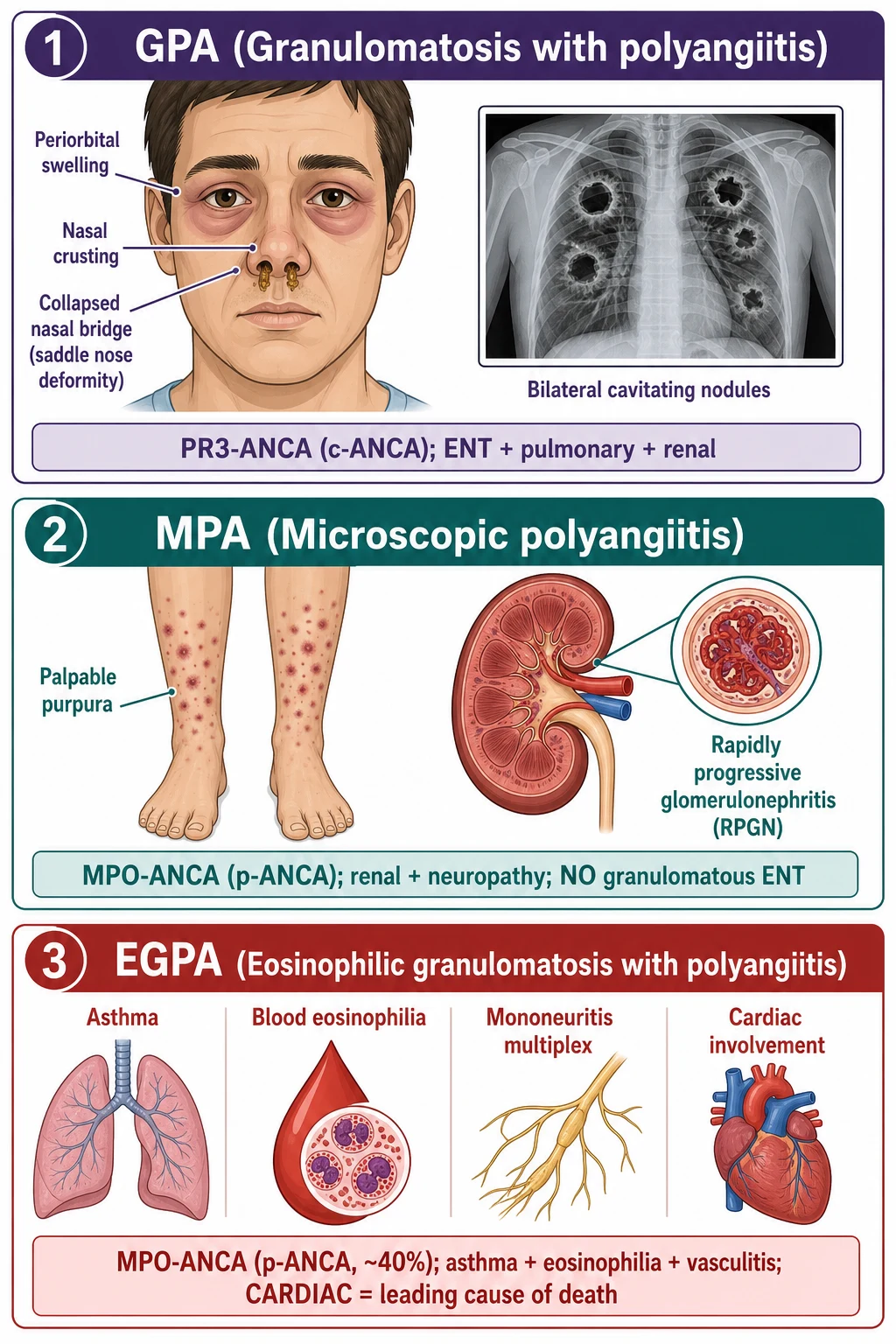
Classification
The 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides (Jennette et al.) is the foundational classification system, organising vasculitis by dominant vessel size (large, medium, small, variable) and by pathological and serological features.[3] AAV sits within the small-vessel vasculitis group, alongside immune-complex small-vessel vasculitides (IgA vasculitis, cryoglobulinaemic vasculitis, anti-GBM disease, hypocomplementaemic urticarial vasculitis). The 2021 ACR/VF (American College of Rheumatology/Vasculitis Foundation) classification criteria and the 2022 EULAR recommendations for AAV management complement the CHCC nomenclature, with growing emphasis on a PR3-AAV vs MPO-AAV serological split that better predicts relapse and therapeutic response than the clinical phenotype.[1]
GPA (Wegener)
- PR3-ANCA (c-ANCA) in approximately 75 to 90 percent
- Necrotising granulomatous inflammation of upper AND lower respiratory tract
- Pauci-immune necrotising glomerulonephritis
- ENT: epistaxis, sinusitis, saddle-nose, subglottic stenosis, conductive deafness, orbital pseudotumour
- Lungs: nodules (often cavitating), infiltrates, alveolar haemorrhage
- Most relapsing subtype; rituximab preferred for induction and maintenance
MPA
- MPO-ANCA (p-ANCA) in 40 to 80 percent
- Necrotising small-vessel vasculitis with renal and pulmonary capillaritis
- NO granulomatous inflammation, NO destructive ENT disease
- Idiopathic RPGN, pulmonary haemorrhage, neuropathy, palpable purpura
- More renal-limited than GPA; less relapsing
- Treated with steroids + rituximab or cyclophosphamide; PLEX if severe renal
EGPA (Churg-Strauss)
- MPO-ANCA in approximately 40 percent (ANCA-positive phenotype more vasculitic/renal)
- Three phases: allergic (asthma, allergic rhinitis, nasal polyposis), eosinophilic (eosinophilia over 10 percent, pulmonary infiltrates), vasculitic (mononeuritis multiplex, cardiomyopathy)
- Cardiomyopathy is the leading cause of death
- Mepolizumab (anti-IL-5) for eosinophilic phenotype
- Cyclophosphamide for severe organ-threatening (cardiac, neuropathy, renal)
Renal-limited vasculitis
- Pauci-immune necrotising crescentic GN with ANCA positivity
- No extrarenal disease (no ENT, pulmonary, neuropathic features)
- Treated as MPA-equivalent: steroids plus rituximab or cyclophosphamide
- Outcomes depend on creatinine at presentation and biopsy chronicity
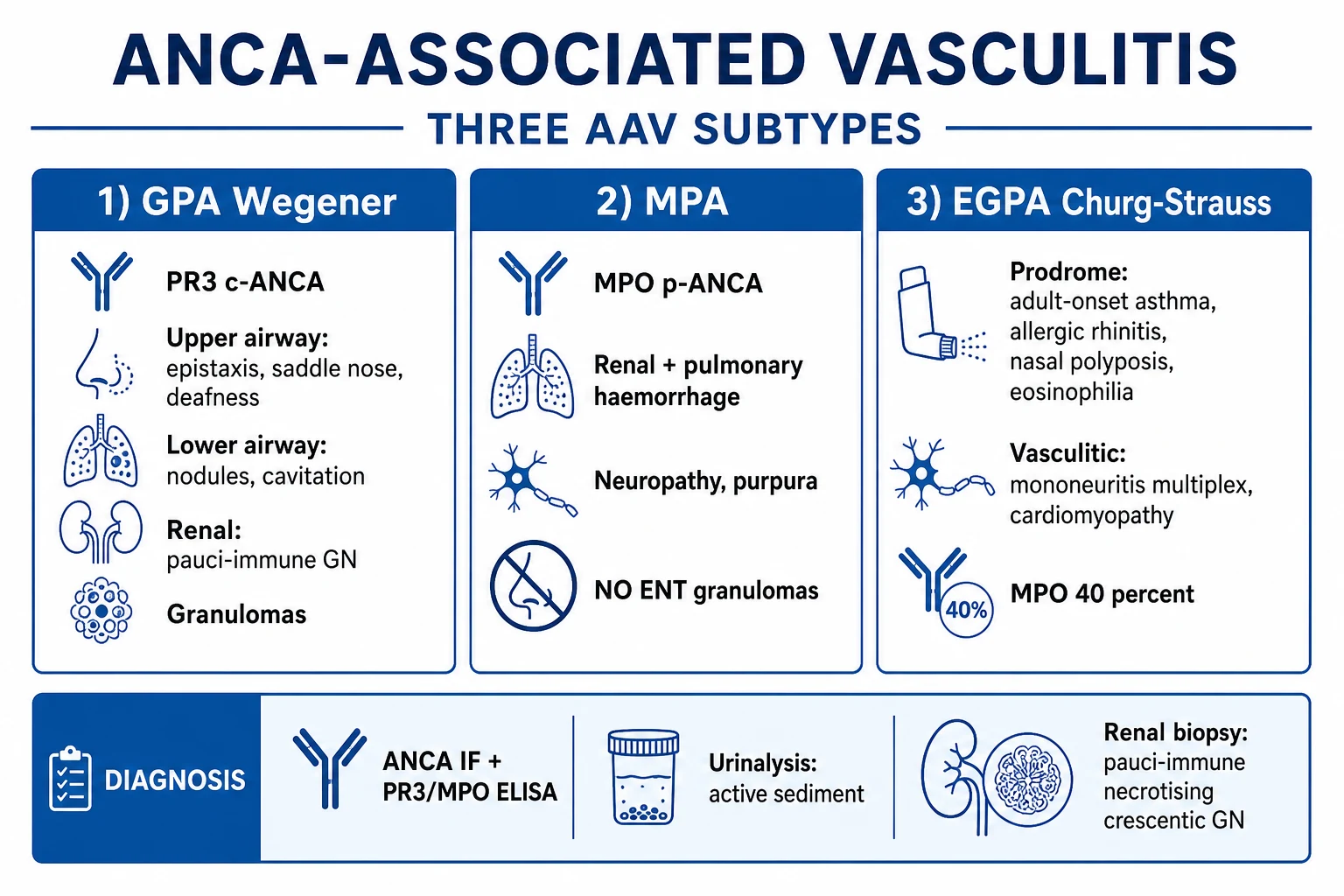
Epidemiology & Risk Factors
AAV is uncommon but not rare. The annual incidence is approximately 10 to 20 per million population per year, with a prevalence of 100 to 200 per million; in Northern European populations figures are at the higher end.[1] Incidence rises with age, peaking at 65 to 75 years, although EGPA often presents slightly younger (40 to 55 years). The sex ratio is roughly equal in elderly-onset disease, with a slight female predominance in younger-onset MPA and EGPA.
A striking geographic and ethnic gradient is recognised: PR3-ANCA and GPA are commoner in Northern European and Caucasian populations, while MPO-ANCA and MPA predominate in East Asian populations (Japan, China) and in people of African ancestry. The reasons are partly genetic — HLA-DP variants associate with PR3/GPA and HLA-DQ and SERPINA1 (alpha-1-antitrypsin) with MPO/MPA — and partly environmental.[1]
Environmental and occupational risk factors include silica exposure (mining, farming, construction, sandblasting), occupational solvents, drugs (propylthiouracil, hydralazine, levamisole-adulterated cocaine, anti-TNF agents), chronic Staphylococcus aureus nasal carriage (a relapse cofactor in GPA), and smoking. The dual-hit hypothesis proposes that genetic predisposition plus an environmental insult triggers autoantigen exposure and ANCA formation. [1]
DRUGS that trigger AAV — the contemporary causes
DRUGS
less common but reported drug-induced AAV
high-titre p-ANCA plus PR3, ear, retiform purpura
classic cause; p-ANCA, often renal-limited
p-ANCA plus ANA, anti-histone; can be renal-threatening
remission usually follows drug withdrawal
Pathophysiology
AAV is the prototypic antibody-driven autoimmune vasculitis. The central mechanism is ANCA-mediated neutrophil activation at the vessel wall. ANCA are IgG antibodies directed against neutrophil granule proteins — most commonly proteinase 3 (PR3) and myeloperoxidase (MPO). The pathogenic sequence is: [1]
- Priming. Cytokines (IL-1, TNF-alpha) from infection or inflammation prime neutrophils, translocating PR3 and MPO from intracellular granules to the cell surface.
- Binding. Circulating ANCA bind surface PR3/MPO and cross-link Fc-gamma receptors, generating a strong activation signal.
- Degranulation and ROS release. Activated neutrophils release proteases (PR3, elastase), reactive oxygen species, and pro-inflammatory cytokines that injure the endothelium.
- NETosis. Neutrophils extrude neutrophil extracellular traps (NETs) — webs of DNA decorated with PR3, MPO, histones and antimicrobial peptides — which propagate ANCA formation, deposit in glomeruli, and trigger thrombosis and tissue injury. Impaired NET clearance (e.g. by DNase I) perpetuates autoantigen exposure.
- Alternative complement pathway amplification. ANCA-activated neutrophils release C3 and factor B activators that fuel the alternative pathway amplification loop, generating C5a. C5a, acting via the C5a receptor (CD88) on neutrophils, recruits and primes yet more neutrophils, completing a self-sustaining inflammatory circuit. This loop explains why biopsy shows a pauci-immune pattern (little/no IgG/complement deposition) despite intense complement activation — the alternative pathway amplifies inflammation locally without forming detectable immune complexes. Avacopan (C5aR antagonist) targets this loop and is licensed for steroid-sparing induction.[1]
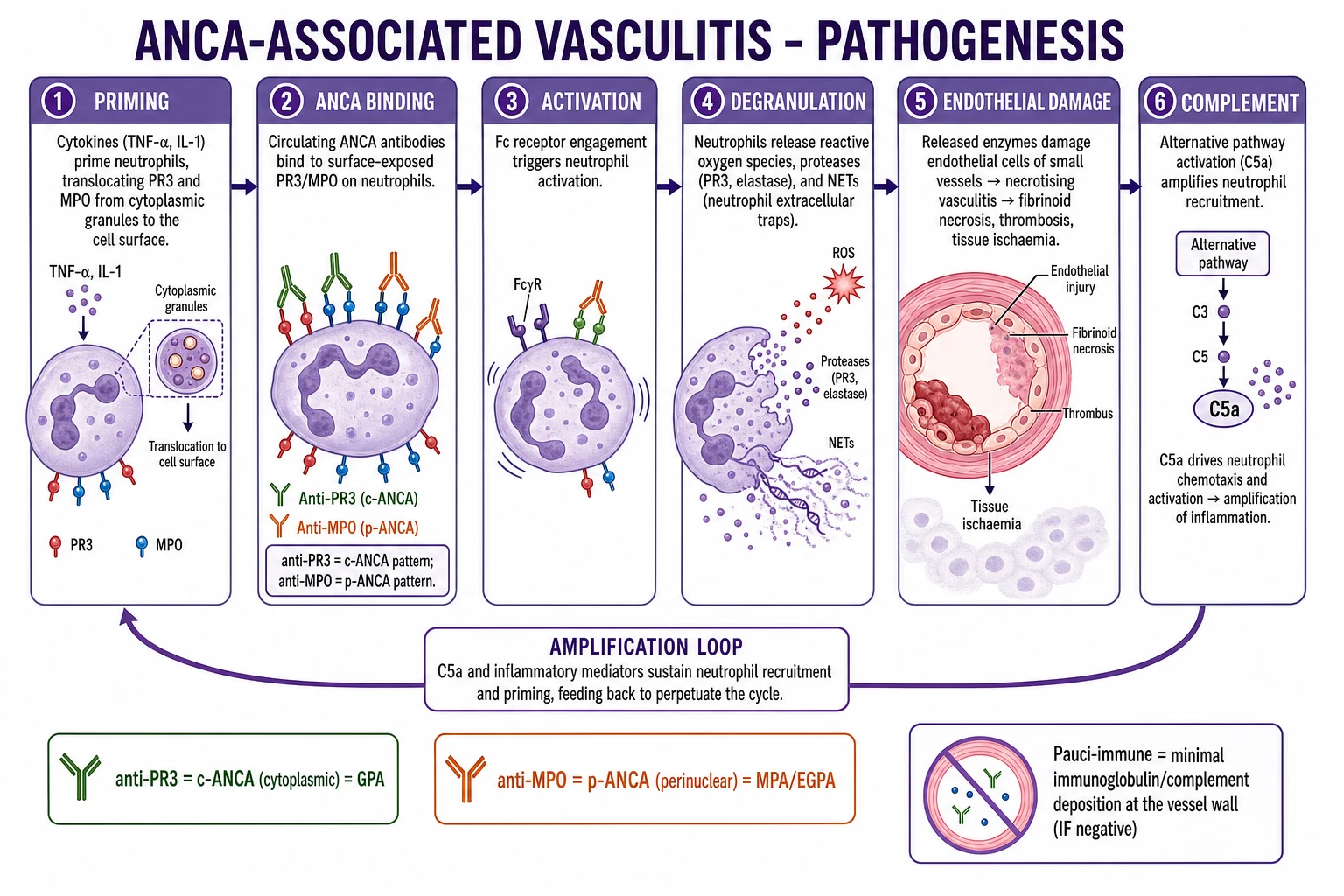
Phenotypic differences mirror the antigen: PR3-AAV shows more granulomatous, ENT and respiratory disease with a relapsing course, while MPO-AAV is more renal-limited and indolent but less relapsing. EGPA is pathogenically distinct — a Th2-skewed process with IL-5-driven eosinophil survival and activation, eosinophilic tissue infiltration, IgE elevation (especially in the ANCA-negative eosinophilic phenotype), granuloma formation (often with an eosinophilic necrotic core), and necrotising vasculitis. The eosinophilic phenotype often lacks vasculitic features and behaves more like severe asthma with eosinophilic organ inflammation; the ANCA-positive MPO phenotype behaves more like MPA with renal and neuropathic involvement.[2]
Clinical Presentation
AAV presents across multiple systems simultaneously or sequentially, and the clinical pattern often points to the subtype. Constitutional features (fever, fatigue, weight loss, myalgia, arthralgia) are common to all subtypes and may precede organ-specific disease by weeks or months. [1]
Granulomatosis with polyangiitis (GPA)
GPA classically involves the ENT, the lower respiratory tract, and the kidneys, in any order and combination. [1]
- ENT (90 percent at presentation, eventually nearly 100 percent): refractory sinusitis, epistaxis (often bilateral, with nasal crusting), oral and nasal ulcers (painless, unlike the painful ulcers of Behcet), saddle-nose deformity (collapse of the nasal bridge from cartilage destruction), subglottic tracheal stenosis (stridor, dyspnoea, requires dilatation), conductive or sensorineural hearing loss (serous otitis media from Eustachian tube dysfunction, or cochlear vasculitis), orbital pseudotumour (proptosis, diplopia, chemosis), and recurrent salivary gland swelling.
- Lower respiratory tract: cough, dyspnoea, haemoptysis (the cardinal sign of alveolar haemorrhage), pleuritic chest pain. Imaging shows multiple pulmonary nodules (often cavitating), consolidations, ground-glass infiltrates (diffuse alveolar haemorrhage), pleural effusion, and rarely tracheobronchial involvement.
- Renal: rapidly progressive glomerulonephritis — rising creatinine, microscopic haematuria, dysmorphic red cells, red cell casts, sub-nephrotic proteinuria, oedema, hypertension. Without treatment, evolves to ESKD within weeks.
- Other: peripheral neuropathy (mononeuritis multiplex), palpable purpura, scleritis/episcleritis, pericarditis, mesenteric vasculitis, prostatitis. [1]
Microscopic polyangiitis (MPA)
MPA is the archetype of pauci-immune necrotising small-vessel vasculitis without granulomatous inflammation. [1]
- Renal (the commonest organ involved): idiopathic rapidly progressive pauci-immune GN — the dominant presentation. Active urinary sediment with red cell casts; rising creatinine.
- Pulmonary: pulmonary capillaritis causing diffuse alveolar haemorrhage (DAH) — haemoptysis, dyspnoea, hypoxaemia, falling haemoglobin, rising carbon monoxide transfer factor (KCO).
- Other: peripheral neuropathy (mononeuritis multiplex), palpable purpura, scleritis, constitutional symptoms, arthralgia.
- No destructive ENT disease — the cardinal distinction from GPA. [1]
Eosinophilic granulomatosis with polyangiitis (EGPA, Churg-Strauss)
EGPA evolves through three sequential phases, often over years: [1]
- Prodromal (allergic) phase: adult-onset asthma (often severe, late-onset, often resistant to standard therapy), allergic rhinitis, nasal polyposis — frequently present years before vasculitis emerges.
- Eosinophilic phase: marked peripheral eosinophilia (over 10 percent or 1.5 times 10⁹/L), tissue eosinophilia, Loeffler-like fleeting pulmonary infiltrates, gastrointestinal eosinophilia (abdominal pain, diarrhoea).
- Vasculitic phase: necrotising vasculitis — mononeuritis multiplex (the commonest vasculitic manifestation; wrist drop, foot drop, sensory loss), eosinophilic cardiomyopathy (heart failure, arrhythmia, eosinophilic endomyocardial fibrosis — the leading cause of death), mesenteric vasculitis, glomerulonephritis (more often in the ANCA-positive phenotype), cutaneous purpura and nodules. [1]
AAV — key numbers that decide an answer
Atypical presentations
- Elderly onset: more renal-predominant and pulmonary haemorrhage, less ENT/granulomatous; worse tolerance of cyclophosphamide and high-dose steroids (favour rituximab and reduced-dose steroids, per PEXIVAS); age over 65 is a Five Factor Score point.
- Drug-induced AAV (propylthiouracil, hydralazine, levamisole-cocaine): high-titre p-ANCA plus PR3, cutaneous purpura (especially levamisole — retiform, ear, necrotic), arthralgia, often renal-limited; remits on stopping the drug, with immunosuppression if organ-threatening.
- Limited GPA (seronegative, ENT only): ANCA may be negative; biopsy and imaging required; treated with local measures, methotrexate or rituximab.
- Double-positive ANCA plus anti-GBM (Goodpasture overlap): pulmonary-renal syndrome with both antibodies; urgent daily plasma exchange to remove anti-GBM, plus induction immunosuppression; high recurrence risk. [1]
Differential Diagnosis
The pivotal decisions are: (1) recognise the pulmonary-renal syndrome and distinguish AAV from other small-vessel vasculitides, (2) exclude infection and malignancy (which can mimic AAV and which immunosuppression would worsen), and (3) consider drug-induced AAV (which remits on withdrawal).[1]
AAV (GPA/MPA/EGPA)
- Necrotising small-vessel vasculitis plus ANCA (PR3 or MPO)
- Pulmonary-renal syndrome; ENT (GPA); asthma and eosinophilia (EGPA)
- Pauci-immune necrotising crescentic GN on renal biopsy
- Treat: steroids plus rituximab or cyclophosphamide; PLEX if severe
Polyarteritis nodosa (PAN)
- Medium muscular-artery vasculitis; transmural fibrinoid necrosis and microaneurysms
- Mononeuritis multiplex, abdominal angina, renovascular hypertension, testicular pain, livedo
- Bland urinalysis (no GN), ANCA negative, lungs spared
- HBV-associated: antivirals plus plasma exchange; idiopathic: steroids plus cyclophosphamide
IgA vasculitis (Henoch-Schonlein)
- Children; post-upper-respiratory-infection
- Palpable purpura (lower limbs), abdominal pain, arthritis, IgA nephritis
- Mesangial IgA deposition on biopsy; normal complement
- Supportive; steroids for severe gut or renal disease
Anti-GBM (Goodpasture)
- Pulmonary-renal syndrome with linear IgG on GBM
- Anti-GBM antibody positive (ELISA); rapidly progressive GN
- Daily plasma exchange until antibody undetectable plus cyclophosphamide plus steroids
- Can coexist with AAV (double-positive) — add ANCA induction
Cryoglobulinaemic vasculitis
- Type II or III mixed cryoglobulins; hepatitis C association
- Palpable purpura, arthralgia, weakness, neuropathy, GN
- Low C4; immune-complex deposition on biopsy
- Treat HCV with direct-acting antivirals; rituximab for severe
Drug-induced AAV
- Propylthiouracil, hydralazine, levamisole-cocaine, anti-TNF
- High-titre p-ANCA plus PR3; cutaneous purpura (retiform in levamisole)
- Often renal-limited; remits on drug withdrawal
- Immunosuppress if organ-threatening; supportive care
Other considerations in selected contexts: systemic lupus erythematosus (ANA, low complement, immune-complex GN), IgG4-related disease (mass-like lesions, elevated serum IgG4, storiform fibrosis), sarcoidosis (non-caseating granulomas, bilateral hilar lymphadenopathy), lymphoma (sinonasal NK/T-cell lymphoma) mimicking GPA ENT disease (biopsy is essential in destructive sinus disease), tuberculosis and atypical infection (visible on biopsy and culture), cocaine-induced midline destructive lesion (without vasculitis on biopsy, no ANCA response to immunosuppression). [1]
Clinical & Bedside Assessment
A focused multi-system examination is mandatory at first contact and at every flare assessment. [1]
ENT: inspect the nasal bridge (saddle deformity), nasal cavity (crusting, ulceration, septal perforation), oral cavity (painless ulcers on hard palate or gingiva), oropharynx; otoscopy (serous otitis media, conductive hearing loss); examine for proptosis (orbital pseudotumour), red eye (scleritis, episcleritis), and salivary glands. [1]
Respiratory: assess for stridor (subglottic stenosis — a surgical airway emergency), added sounds (crackles, bronchial breathing in alveolar haemorrhage or pneumonia), respiratory failure (hypoxaemia, accessory muscle use). Falling haemoglobin plus rising KCO supports diffuse alveolar haemorrhage even when haemoptysis is absent. [1]
Renal and fluid status: blood pressure (hypertension in renal AAV), jugular venous pressure, fluid balance, oedema; bedside urine dipstick for blood and protein, then microscopy for dysmorphic red cells and red cell casts (the cardinal sign of glomerular disease). [1]
Neurological: examine for mononeuritis multiplex (asymmetric — wrist drop, foot drop, sensory loss in nerve territories), sensorimotor neuropathy, cranial nerves (especially sensorineural hearing loss in GPA), and signs of cerebral vasculitis (rare). [1]
Skin and abdomen: palpable purpura (lower limbs, non-blanching), nodules, ulcers, digital infarcts, livedo; abdominal tenderness (mesenteric vasculitis); testicular tenderness (more characteristic of PAN). [1]
Standardised activity and prognosis scores
Two named scores structure AAV assessment: [1]
BVAS — Birmingham Vasculitis Activity Score (multi-domain)
BVAS
standardised multi-domain activity tool for AAV (and other vasculitides)
general, cutaneous, mucous membranes/ENT, chest, cardiovascular, abdominal, renal, nervous system
weights new/worsening features; persisted damage goes to VDI not BVAS
BVAS over 15 = severe active disease requiring induction; falls with remission
The Five Factor Score (FFS, 2009 revision — Guillevin) is the prognostic instrument for necrotising vasculitides (AAV and PAN): [1]
The Vasculitis Damage Index (VDI) records cumulative damage (non-reversible scarring from disease or treatment) and is distinct from activity. A useful mnemonic for the assessment trio: BVAS = activity (drives induction intensity); FFS = prognosis (drives escalation); VDI = damage (tracks the chronic burden). [1]
Investigations
A cost-sensitive staged workup confirms AAV, characterises organ involvement, excludes mimics, and informs treatment intensity. [1]
First-line bloods: FBC (anaemia of chronic disease; eosinophilia in EGPA; thrombocytosis as an acute-phase response); ESR and CRP (elevated, but non-specific); U&E and creatinine (renal involvement); LFT (low albumin in active inflammation; hepatitic pattern in drug-induced or viral overlap); serum IgG and IgE (IgE elevated in EGPA); creatine kinase (myositis overlap). [1]
Urinalysis and urine microscopy: the single most important bedside test in suspected AAV. Look for microscopic haematuria, dysmorphic red cells and red cell casts (the cardinal sign of glomerulonephritis — AAV, anti-GBM, SLE, IgA nephritis). Send urine protein-to-creatinine ratio (sub-nephrotic proteinuria in AAV). [1]
ANCA testing — the two-step strategy: [1]
- Indirect immunofluorescence (IIF) on ethanol-fixed neutrophils — pattern reading: c-ANCA (diffuse cytoplasmic, granular) usually corresponds to anti-PR3; p-ANCA (perinuclear) usually corresponds to anti-MPO. Atypical patterns are common in drug-induced and inflammatory bowel disease.
- Confirmatory antigen-specific ELISA for anti-PR3 and anti-MPO — the diagnostic standard, and the basis of the modern PR3-AAV vs MPO-AAV split. [1]
ANCA yield by subtype: PR3-ANCA positive in approximately 75 to 90 percent of GPA, MPO-ANCA positive in approximately 40 to 80 percent of MPA, and MPO-ANCA positive in approximately 40 percent of EGPA. ANCA may be negative in limited GPA (ENT only), in some drug-induced cases, and early in disease — biopsy is still diagnostic.[1]
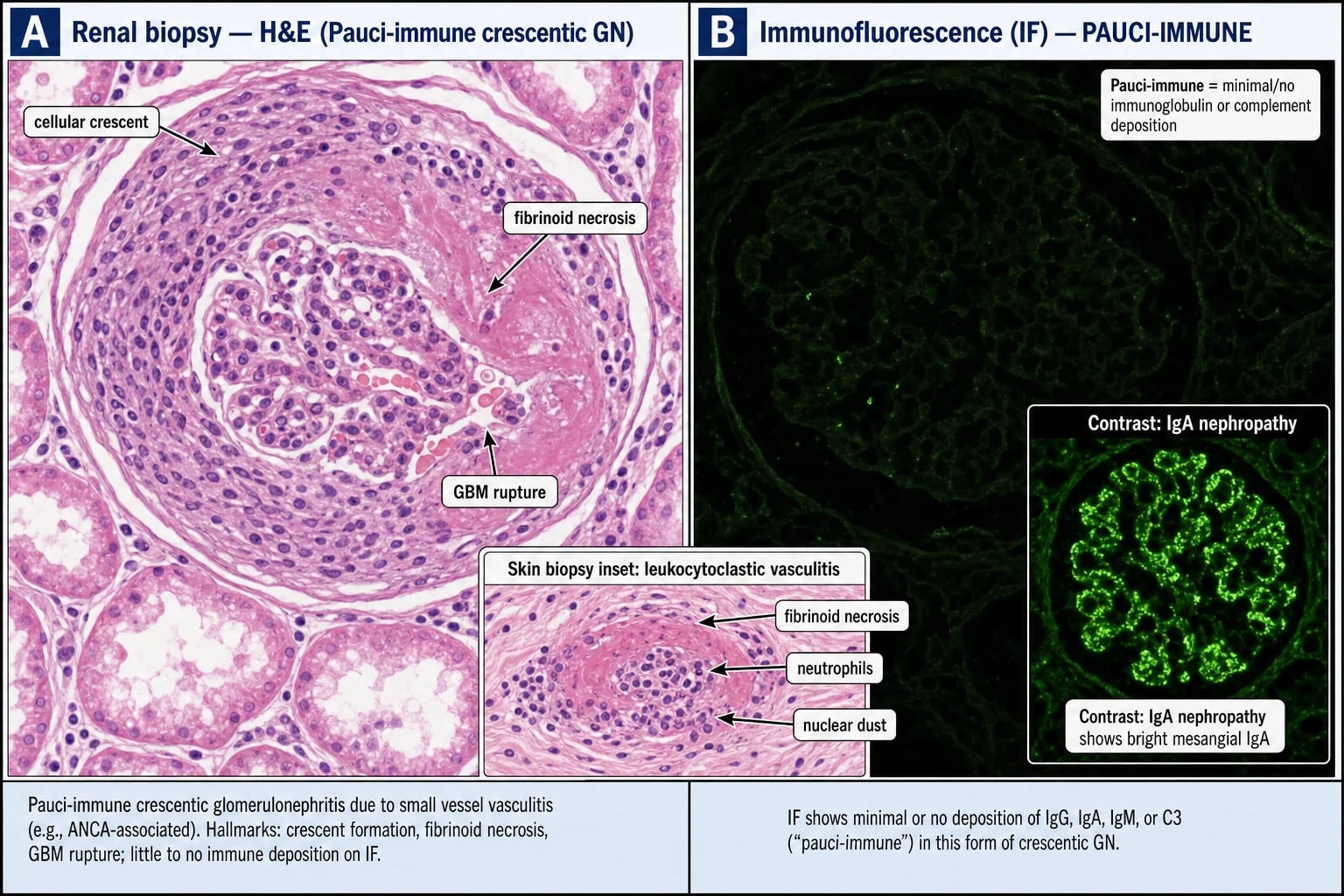
Tissue biopsy — the diagnostic gold standard: [1]
- Renal biopsy (the highest-yield single specimen in pulmonary-renal presentation): pauci-immune necrotising crescentic glomerulonephritis — segmental fibrinoid necrosis of the capillary tuft with cellular crescents in Bowman space, little or no IgG/C3 on immunofluorescence (in contrast to the linear IgG of anti-GBM or the full-house deposition of SLE). The chronicity index (tubulointerstitial fibrosis, glomerular sclerosis) strongly predicts renal recovery.
- Lung biopsy (thoracoscopic or transbronchial): necrotising granulomatous inflammation with geographic necrosis and a palisading histiocyte rim — diagnostic of GPA, but rarely required when ENT and renal biopsy already establish the diagnosis.
- Sinus or nasal mucosal biopsy: often non-specific (acute and chronic inflammation, occasional giant cells) but excludes sinonasal lymphoma, squamous carcinoma, tuberculosis, and cocaine-related destruction. Repeated biopsies may be necessary.
- Skin biopsy of palpable purpura: leukocytoclastic vasculitis with nuclear debris (karyorrhexis); eosinophils suggest EGPA. [1]
Imaging: [1]
- High-resolution CT chest: pulmonary nodules (often cavitating), consolidations, ground-glass change (DAH), pleural effusion; tracheobronchial thickening in GPA.
- CT sinuses: bony destruction (septal perforation), sinus opacification, mucosal thickening — characteristic of GPA.
- Angiography (renal, mesenteric, hepatic): NOT for AAV; reserved for polyarteritis nodosa (microaneurysms and beading of medium arteries).
- Bronchoscopy in DAH: progressively bloodier lavage from serial aliquots, negative microbiology; not diagnostic but supports alveolar haemorrhage and excludes infection. [1]
Adjuncts to exclude mimics: anti-GBM antibody (Goodpasture), ANA, anti-dsDNA and complement C3/C4 (SLE), hepatitis B and C serology (PAN and cryoglobulinaemia), HIV, cryoglobulins and serum electrophoresis (cryoglobulinaemia, myeloma), serum IgG4 (IgG4-related disease), blood cultures, echocardiogram and atypical infection screens (infective endocarditis), QuantiFERON-TB / IGRA (pre-immunosuppression). [1]
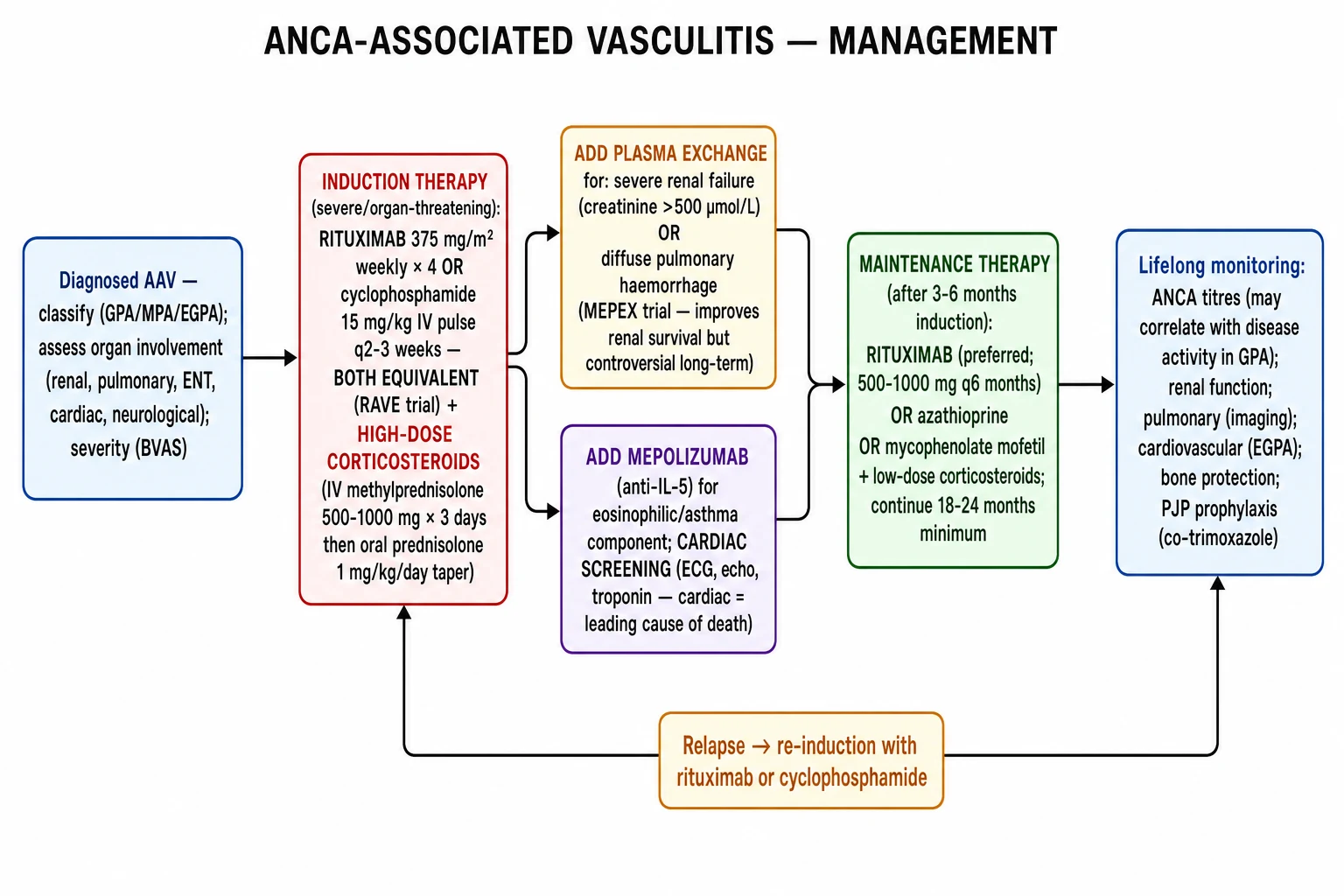
Management — Resuscitation
Recognise the pulmonary-renal syndrome, DAH, and rapidly progressive GN as emergencies. Untreated severe AAV is fatal within months. The aims of resuscitation are to stabilise airway, breathing and circulation, treat hypoxaemia and acute kidney injury, secure the diagnosis, and start induction within hours to days. [1]
Immediate measures: [1]
- ABC plus oxygenation for hypoxaemia (DAH), with lung-protective ventilation if intubated.
- Establish IV access, cross-match (ongoing alveolar haemorrhage may require transfusion), and treat hypovolaemia cautiously — over-resuscitation worsens pulmonary haemorrhage and hypertension in renal AAV.
- Avoid nephrotoxins (NSAIDs, aminoglycosides, contrast where possible); manage fluids and electrolytes for acute kidney injury; nephrology early for potential dialysis.
- Admit for suspected severe AAV (creatinine rising rapidly, haemoptysis/DAH, hypoxaemia, severe neuropathy, carditis, mesenteric ischaemia); multidisciplinary input — nephrology, rheumatology, respiratory and ICU.
- Treat coexisting anti-GBM overlap (double-positive) with daily plasma exchange until anti-GBM is undetectable, plus induction immunosuppression. [1]
Investigations BEFORE induction (because induction will be profoundly immunosuppressive): [1]
- ANCA (PR3/MPO), anti-GBM, ANA and complement.
- Urinalysis and urine protein-to-creatinine ratio; renal biopsy within days.
- Hepatitis B and C, HIV; QuantiFERON / IGRA; HBV DNA / HBsAg / HBcore for pre-emptive antiviral therapy before rituximab or cyclophosphamide.
- Infection screen — cultures, chest imaging, urinalysis; consider echocardiogram if bacteraemia or endocarditis suspected.
- Baseline FBC, U&E, LFT, glucose, lipids, bone profile (steroid and cyclophosphamide toxicity monitoring). [1]
Immediate induction (within hours of presentation of organ-threatening disease): IV methylprednisolone 500 mg to 1 g daily for 3 days, then oral prednisolone 1 mg/kg (max 60 to 80 mg daily), tapering over months.[1] Add rituximab 375 mg/m² weekly for 4 doses OR cyclophosphamide 15 mg/kg IV every 2 to 4 weeks as soon as the diagnosis is secure — do not delay beyond the diagnostic window.
Plasma exchange (PLEX) is considered for: SCr over 500 micromol/L (approximately 5.7 mg/dL); dialysis-dependent renal AAV; severe diffuse alveolar haemorrhage with hypoxaemia; or anti-GBM overlap. The PEXIVAS trial (2020) showed no overall benefit on death or ESKD, so use is now selective rather than routine; many units still follow the MEPEX evidence and KDIGO guidance for the most severe renal presentations.[5][6]
Management — Definitive & Stepwise
Definitive therapy is structured as induction (achieving remission over 3 to 6 months), maintenance (sustaining remission for 24 to 36 months or longer), relapse management, and supportive care. [1]
Induction (severe, organ-threatening AAV)
- Glucocorticoids: IV methylprednisolone 500 mg to 1 g daily for 3 days, then oral prednisolone 1 mg/kg (max 60 to 80 mg daily), tapered to approximately 10 to 15 mg by 3 months and to a maintenance dose (5 to 7.5 mg) by 6 months, then weaned slowly if possible. PEXIVAS supports a reduced-dose glucocorticoid regimen to limit infection and steroid toxicity.[5]
- Rituximab (anti-CD20): 375 mg per square metre IV weekly for 4 doses (RAVE regimen) OR 1 g IV at day 0 and day 14. RAVE (Stone, NEJM 2010) established rituximab as non-inferior to cyclophosphamide for severe AAV and superior in relapsing disease. Preferred in PR3-AAV, relapsing disease, women of childbearing potential, and where cyclophosphamide toxicity is a concern.[4]
- Cyclophosphamide: IV pulse 15 mg/kg every 2 to 4 weeks (CYCLOPS regimen) OR oral 2 mg/kg daily for 3 to 6 months. CYCLOPS (de Groot, Ann Intern Med 2009) showed pulse IV non-inferior to daily oral with lower cumulative dose, but slightly higher relapse at long-term follow-up. Dose-reduce in elderly and renal impairment.[7]
- Plasma exchange (PLEX): 7 to 14 sessions of 60 mL/kg exchange over 2 to 4 weeks, with human albumin and (if bleeding or recent biopsy) fresh-frozen plasma. PEXIVAS (Walsh, NEJM 2020) showed no overall benefit on death or ESKD; use selectively for SCr over 500, dialysis-dependent renal AAV, severe DAH with hypoxaemia, or anti-GBM overlap.[5]
- Avacopan (C5a receptor blocker): 30 mg orally twice daily as a glucocorticoid-sparing induction agent (ADVOCATE trial — Jayne, NEJM 2021). Now a licensed option for selected patients intolerant of high-dose steroids.[1]
Maintenance (after remission at 3 to 6 months)
- Rituximab (preferred, especially in PR3-AAV and relapsing disease): 1 g IV every 4 months OR 500 mg IV every 6 months for at least 24 to 36 months; longer in PR3-AAV.
- Azathioprine 2 mg/kg/day — long-standing first-line maintenance.
- Mycophenolate mofetil 2 g/day — useful in patients intolerant of azathioprine, but IMPROVE (Hiemstra, JAMA 2010) showed azathioprine superior for relapse-free maintenance.[8]
- Methotrexate 20 to 25 mg/week (with folate) — for non-severe / limited AAV only.
- Low-dose prednisolone continued for at least 12 to 18 months, then tapered under BVAS and ANCA monitoring.
EGPA-specific therapy
- Mepolizumab (anti-IL-5) 300 mg subcutaneously every 4 weeks for the eosinophilic phenotype with asthma (MIRRA trial — Wechsler, NEJM 2017): improved remission and steroid-sparing in relapsing or refractory EGPA.[9]
- Cyclophosphamide plus glucocorticoids for severe organ-threatening EGPA (cardiac, severe neuropathy, mesenteric, renal).
- Avoid long-term high-dose steroids — cardiac and bone toxicity dominate.
Relapse management
- Re-induce with rituximab (preferred) or cyclophosphamide plus glucocorticoids.
- Consider fixed-schedule rituximab maintenance to prevent relapse in PR3-AAV.
- Cotrimoxazole (trimethoprim-sulfamethoxazole) prophylaxis reduces GPA relapse by clearing Staphylococcus aureus nasal carriage. [1]
Non-severe or limited AAV
- Methotrexate or mycophenolate plus rituximab increasingly used.
- ENT-dominant GPA: topical nasal steroids, saline irrigation; intralesional steroid for subglottic stenosis; cotrimoxazole 960 mg twice daily for staphylococcal carriage. [1]
Supportive care bundle
- Pneumocystis jirovecii prophylaxis: cotrimoxazole 480 mg daily (or 960 mg three times weekly) on cyclophosphamide, rituximab, or sustained high-dose steroids (over 20 mg prednisolone for over 4 weeks).
- Bone protection: calcium and vitamin D, plus a bisphosphonate or denosumab on long-term steroids; bone densitometry.
- Gastric protection: PPI for high-risk patients on steroids (caution hypomagnesaemia with avacopan).
- Vaccination: influenza, pneumococcal, COVID-19, hepatitis B, shingles (zoster vaccine — recombinant preferred if immunosuppressed) before immunosuppression if possible; avoid live vaccines during immunosuppression.
- Screening: latent TB (IGRA), hepatitis B (HBsAg, HBcore — pre-emptive antiviral such as entecavir or tenofovir), hepatitis C, HIV.
- Cardiovascular risk: aggressive risk factor modification (smoking cessation, statin, blood pressure control) — chronic inflammation accelerates atherosclerosis.
- VTE prophylaxis during hospitalisation (active AAV is prothrombotic). [1]
Specific Subtypes & Scenarios
GPA (Wegener granulomatosis)
PR3-ANCA in 75 to 90 percent. ENT plus pulmonary plus renal with granulomatous inflammation on biopsy. Saddle nose, subglottic stenosis, orbital pseudotumour, refractory sinusitis. Relapsing course (the highest relapse rate of any AAV subtype). Rituximab preferred for both induction and maintenance. Cotrimoxazole reduces staphylococcal carriage and GPA relapse. [1]
MPA
MPO-ANCA in 40 to 80 percent. Necrotising small-vessel vasculitis with renal and pulmonary capillaritis but NO granulomatous ENT disease. Idiopathic RPGN and diffuse alveolar haemorrhage are the dominant emergencies. Neuropathy and palpable purpura common. Less relapsing than GPA. Treat as PR3/MPO-directed: rituximab or cyclophosphamide induction, rituximab or azathioprine maintenance. [1]
EGPA (Churg-Strauss)
Three phases — allergic, eosinophilic, vasculitic. MPO-ANCA in approximately 40 percent (ANCA-positive phenotype more vasculitic and renal; ANCA-negative more eosinophilic and cardiac). Cardiomyopathy is the leading cause of death — request echocardiography and troponin at diagnosis. Mepolizumab 300 mg subcutaneously every 4 weeks for the eosinophilic phenotype with asthma (MIRRA). Cyclophosphamide for severe organ-threatening EGPA. Steroid-sparing is paramount because of long disease course.[2][9]
Renal-limited vasculitis (RLV)
Pauci-immune necrotising crescentic GN with ANCA positivity, no extrarenal disease. Treated as MPA-equivalent. Outcomes depend on creatinine at presentation and biopsy chronicity index (tubulointerstitial fibrosis, glomerular sclerosis). Consider plasma exchange for the most severe presentations. [1]
Drug-induced AAV
Propylthiouracil, hydralazine, levamisole-adulterated cocaine, anti-TNF agents. High-titre p-ANCA plus PR3 (sometimes both p-ANCA and c-ANCA simultaneously in levamisole-cocaine cases), often with ANA and anti-histone antibodies. Cutaneous purpura (especially retiform, ear, and necrotic in levamisole-cocaine), arthralgia, often renal-limited disease. Stop the offending drug and immunosuppress if organ-threatening; remission usually follows drug withdrawal, but kidney injury may persist. [1]
Anti-GBM overlap (double-positive)
Pulmonary-renal syndrome with both ANCA and anti-GBM antibodies. Urgent daily plasma exchange until anti-GBM is undetectable, plus rituximab or cyclophosphamide induction. High recurrence risk of the ANCA-arm disease (anti-GBM disease does not usually relapse, but the coexisting AAV does), so ongoing maintenance is essential. [1]
Complications & Pitfalls
Disease-related complications
- End-stage kidney disease (ESKD): dialysis or transplant; outcomes depend on creatinine at presentation and chronicity index on biopsy.
- Saddle-nose deformity and conductive deafness: chronic structural damage from granulomatous ENT inflammation.
- Subglottic tracheal stenosis: surgical dilatation, intralesional steroid, sometimes tracheostomy.
- Peripheral neuropathy (mononeuritis multiplex): often incomplete recovery.
- EGPA cardiomyopathy: heart failure, arrhythmia, eosinophilic endomyocardial fibrosis — leading cause of death in EGPA.
- Mesenteric vasculitis: abdominal pain, bleeding, perforation.
- Pulmonary fibrosis: after recurrent DAH.
- Venous thromboembolism: high VTE risk in active AAV — prophylax in hospitalised patients. [1]
Treatment-related complications
- Infection: Pneumocystis jirovecii pneumonia, CMV, fungal, bacterial sepsis — the leading cause of early death. Cotrimoxazole prophylaxis is mandatory on cyclophosphamide or sustained high-dose steroids.
- Cyclophosphamide: cytopenias, haemorrhagic cystitis, bladder cancer, infertility (sperm and oocyte banking for younger patients).
- Glucocorticoids: diabetes, osteoporosis, avascular necrosis, hypertension, cataract, psychosis, infection, peptic ulcer.
- Rituximab: late-onset neutropenia, hypogammaglobulinaemia (monitor IgG before each course), HBV reactivation, infusion reactions, rare PML.
- Plasma exchange: catheter-related infection and thrombosis, bleeding (especially if fresh-frozen plasma not given post-biopsy), hypocalcaemia (citrate). [1]
Classic pitfalls
- Missing limited GPA (seronegative, ENT only): biopsy despite negative ANCA.
- Treating ANCA titre alone: ANCA titre is not a reliable flare marker in isolation — relapse can occur without titre rise, and titre can rise without relapse.
- Failing to biopsy new proteinuria: missing a treatable flare or an alternative diagnosis.
- Confusing DAH with infective pneumonitis: bronchoscopy and culture, and rising KCO, distinguish.
- Giving cyclophosphamide to elderly without dose reduction: unacceptable toxicity.
- Missing double-positive (ANCA plus anti-GBM) disease: requires both rituximab/cyclophosphamide and daily plasma exchange.
- Ignoring Pneumocystis prophylaxis: PJP is a preventable killer on induction.
- Confusing GPA ENT disease with sinonasal lymphoma, sarcoidosis, TB, or cocaine-induced destruction: tissue biopsy is essential in destructive sinus disease. [1]
Prognosis & Disposition
Survival has improved dramatically with modern therapy: 5-year survival now approximately 75 to 90 percent in severe AAV, compared with fatality within months untreated. The leading causes of death are infection (early), active vasculitis (especially DAH and EGPA cardiomyopathy), cardiovascular disease (late), and ESKD.[1]
Five Factor Score (FFS 2009) stratifies prognosis across AAV and PAN: age over 65 years, cardiac involvement, renal involvement (creatinine over 1.7 mg/dL or 150 micromol/L), gastrointestinal involvement, absence of ENT involvement — each plus one. Five-year mortality is approximately 12 percent for FFS 0, 26 percent for FFS 1, 46 percent for FFS 2 or more. The FFS guides escalation (consider plasma exchange, more intensive induction) and risk counselling. [1]
Relapse prediction: PR3-AAV relapses more than MPO-AAV (relapse rate up to 50 percent at 5 years versus 20 to 30 percent). ENT involvement, persistent PR3 positivity, and Staphylococcus aureus nasal carriage predict relapse. Rituximab maintenance reduces relapse compared with azathioprine, especially in PR3-AAV. [1]
Renal outcomes: predictors of ESKD include dialysis-dependence at presentation, creatinine over 5.7 mg/dL (500 micromol/L), crescents in over 50 percent of glomeruli, severe tubulointerstitial fibrosis on biopsy, and older age. Renal recovery is unusual if dialysis is required beyond 12 weeks, but recovery beyond that window still occurs — do not prematurely label ESKD. [1]
Maintenance duration: at least 24 to 36 months (longer in PR3-AAV), with slow taper under BVAS and ANCA monitoring. Premature discontinuation risks relapse; prolonged immunosuppression risks infection and toxicity. [1]
Renal transplant in ESKD after disease quiescence (typically 6 to 12 months of remission) — recurrence post-transplant is uncommon but possible, especially in PR3-AAV. [1]
Special Populations
Pregnancy
Plan conception during remission (at least 6 months). Rituximab is acceptable in early pregnancy (avoid third trimester — neonatal B-cell depletion, often reversible). Avoid cyclophosphamide, mycophenolate, methotrexate, leflunomide (teratogenic); switch to azathioprine maintenance. Steroids as required (prefer non-fluorinated — prednisolone — in first trimester). Low-dose aspirin if antiphospholipid overlap. Active AAV in pregnancy carries high maternal and foetal risk — multidisciplinary care with maternal-foetal medicine. [1]
Children (juvenile AAV)
Rare; more often GPA phenotype. Weight-based dosing of rituximab (375 mg per square metre) and cyclophosphamide. Growth, fertility, and bone concerns with chronic steroids — minimise exposure, consider bisphosphonate in selected cases. [1]
Elderly
More MPO-AAV and renal-predominant disease. Worse tolerance of cyclophosphamide and high-dose steroids — favour rituximab and reduced-dose glucocorticoids (PEXIVAS-supported). Age over 65 is an FFS point and predicts higher mortality; lower threshold for plasma exchange and intensive support. [1]
Renal impairment and dialysis
Dose-adjust cyclophosphamide (creatinine-based nomograms); rituximab dosing unaffected by renal function. Avacopan useful for steroid-sparing. Transplant in ESKD after disease quiescence. [1]
Immunosuppression and infection screen
Screen and pre-emptively treat: latent TB (IGRA; treat with standard regimen before induction), hepatitis B (antiviral prophylaxis with entecavir or tenofovir for HBsAg-positive or HBcore-positive patients throughout therapy and for 12 months after rituximab), hepatitis C (direct-acting antivirals before or alongside induction where possible). Provide Pneumocystis prophylaxis (cotrimoxazole 480 mg daily). Vaccinate (avoid live vaccines during immunosuppression). [1]
Anticoagulated patients
Balance VTE risk of active AAV (high) against bleeding risk from induction and PLEX. LMWH preferred in acute VTE. Avoid NSAIDs in renal disease. Use PPI for gastric protection on steroids plus anticoagulation. [1]
[1] [1]Evidence, Guidelines & Regional Differences
Landmark trials and classifications that shaped modern AAV therapy
Key trials summarised: [1]
- RAVE (Stone, NEJM 2010 — PMID 20647199): multicentre randomised double-blind non-inferiority trial of rituximab (375 mg per square metre weekly for 4 doses) versus cyclophosphamide in severe AAV. Rituximab was non-inferior (64 percent vs 53 percent achieving 6-month remission off steroids) and superior in relapsing disease. Established rituximab as standard induction.[4]
- PEXIVAS (Walsh, NEJM 2020 — PMID 32053298): 2-by-2 factorial trial in 704 patients with severe AAV (creatinine over 300 micromol/L or DAH). Plasma exchange did not reduce death or ESKD at 12 months (28.4 percent vs 31.0 percent). Reduced-dose glucocorticoid was non-inferior to standard dose with fewer serious infections.[5]
- MEPEX (Jayne, JASN 2007 — PMID 17582159): plasma exchange versus IV methylprednisolone as adjunctive therapy in severe renal AAV (creatinine over 500 micromol/L). Plasma exchange increased dialysis independence at 3 months (69 percent vs 49 percent); long-term advantage attenuated. Underpins selective use in the most severe renal presentations.[6]
- CYCLOPS (de Groot, Ann Intern Med 2009 — PMID 20505178): pulse IV cyclophosphamide non-inferior to daily oral for remission induction with lower cumulative dose; long-term follow-up showed slightly higher relapse with pulse therapy.[7]
- IMPROVE (Hiemstra, JAMA 2010 — PMID 21060104): azathioprine superior to mycophenolate mofetil for relapse-free maintenance after cyclophosphamide induction.[8]
- MIRRA (Wechsler, NEJM 2017 — PMID 28514601): mepolizumab 300 mg subcutaneously every 4 weeks improved remission (28 percent vs 3 percent on placebo) and steroid-sparing in relapsing or refractory EGPA.[9]
Exam Pearls
PEARLS — the seven pearls that decide an AAV answer
PEARLS
haemoptysis plus RPGN; ANCA, anti-GBM, urinalysis, renal biopsy; immediate induction
saddle nose, subglottic stenosis, nodules, pauci-immune GN; relapsing; rituximab preferred
PR3 corresponds to c-ANCA; MPO corresponds to p-ANCA; PR3-AAV vs MPO-AAV now the primary split
segmental fibrinoid necrosis, cellular crescents, little/no IgG/C3 — distinguishes from anti-GBM (linear) and lupus (full-house)
MPO-ANCA; idiopathic RPGN, DAH, neuropathy, purpura; less relapsing than GPA
PLEX for SCr over 500, dialysis-dependent, severe DAH, or anti-GBM overlap; rituximab or azathioprine maintenance
EGPA — the three phases of Churg-Strauss
EGPA
adult-onset asthma, allergic rhinitis, nasal polyposis, eosinophilia over 10 percent — prodromal phase
tissue eosinophilia, fleeting pulmonary infiltrates, eosinophilic GI infiltrate — eosinophilic phase
vasculitic phase — mononeuritis multiplex (commonest), cardiomyopathy (leading cause of death), mesenteric vasculitis, GN
ANCA-positive phenotype more vasculitic and renal; ANCA-negative more eosinophilic and cardiac; mepolizumab for eosinophilic phenotype
Exam application bank (NEET-PG / INICET)
One-line answer
ANCA-associated vasculitis (AAV) comprises three necrotising small-vessel vasculitides associated with anti-neutrophil cytoplasmic antibodies (ANCA): granulomatosis with polyangiitis (GPA, formerly Wegener) — PR3-ANCA (c-ANCA), ENT (epistaxis, saddle nose, deafness) plus respiratory (sinusitis, lung nodules/cavitation) plus renal (pauci-immune glomerulonephritis); microscopic polyangiitis (MPA) — MPO-ANCA (p-ANCA), renal plus pulmonary without granulomatous ENT; eosinophilic granulomatosis with polyangiitis (EGPA, Churg-Strauss) — asthma, eosinophilia, sinus/nasal polyposis then vasculitic phase (neuropathy, cardiomyopathy, renal). The feared pulmonary-renal syndrome (alveolar haemorrhage plus rapidly progressive glomerulonephritis) is a medical emergency. Diagnosis combines ANCA (immunofluorescence plus PR3/MPO ELISA), urinalysis, renal biopsy (pauci-immune necrotising crescentic GN), a
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard. [1]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes. [1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change. [1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each. [1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory. [1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on ANCA-Associated Vasculitis (GPA, MPA & EGPA).
References
- [1]Chevet B, Cornec D, Casal Moura M, et al. Diagnosing and treating ANCA-associated vasculitis: an updated review for clinical practice Rheumatology (Oxford), 2023.PMID 36315063
- [2]White J, Dubey S, Salil A, et al. Eosinophilic granulomatosis with polyangiitis: A review Autoimmun Rev, 2023.PMID 36283646
- [3]Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides Arthritis Rheum, 2013.PMID 23045170
- [4]Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis N Engl J Med, 2010.PMID 20647199
- [5]Walsh M, Merkel PA, Peh CA, et al. Plasma Exchange and Glucocorticoids in Severe ANCA-Associated Vasculitis N Engl J Med, 2020.PMID 32053298
- [6]Jayne DRW, Gaskin G, Rasmussen N, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis J Am Soc Nephrol, 2007.PMID 17582159
- [7]de Groot K, Harper L, Jayne DRW, et al. Infectious mononucleosis N Engl J Med, 2010.PMID 20505178
- [8]Hiemstra TF, Walsh M, Mahr A, et al. Mycophenolate mofetil vs azathioprine for remission maintenance in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized controlled trial JAMA, 2010.PMID 21060104
- [9]Wechsler ME, Akuthota P, Jayne D, et al. Mepolizumab or Placebo for Eosinophilic Granulomatosis with Polyangiitis N Engl J Med, 2017.PMID 28514601