Rheumatology · General Medicine
Behçet's Syndrome
Also known as Behcet syndrome · Behcet disease · Silk Road disease · Adamantiades-Behcet
Behçet's syndrome is a relapsing-remitting multisystem vasculitis (affects both arteries and veins of all sizes) classically defined by recurrent oral aphthous ulcers plus any two of: genital ulcers, eye lesions (uveitis/retinal vasculitis), skin lesions (erythema nodosum, papulopustular, pathergy), and positive pathergy test. Commonest along the Silk Road (Turkey, Mediterranean, Middle East, East Asia); rare in Northern Europeans. HLA-B51 association. Can involve vessels (venous thrombosis, arterial aneurysm, pulmonary artery aneurysm — major cause of death), eyes (panuveitis, retinal vasculitis — blindness), gastrointestinal (ileocaecal ulcers), joints (arthritis), neurological, and skin. Oral ulcers are universal and the gateway to diagnosis; genital ulcers scar. Diagnosis is clinical (ICBD criteria) with no specific test. Treat mucocutaneous disease conservatively (colchicine, apremilast); ocular, vascular, neuro and GI involvement need aggressive immunosuppression (azathioprine, ciclosporin, interferon-alpha, anti-TNF, cyclophosphamide). Thrombosis is inflammatory — immunosuppression matters more than anticoagulation.
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags
Overview & Definition
Behçet's syndrome is a relapsing-remitting, multisystem inflammatory vasculitis that affects vessels of every size, both arterial and venous — a uniqueness that explains its protean manifestations, from a trivial mouth ulcer to a fatal pulmonary artery aneurysm. It is named after the Turkish dermatologist Hulusi Behçet, who in 1937 described the classic triad of recurrent oral aphthous ulcers, genital ulcers and uveitis, though the Greek ophthalmologist Benedictos Adamantiades reported an identical case in 1930, hence the alternative name Adamantiades-Behçet disease.[2]
The clinical skill in Behçet's is twofold. First, recognising that recurrent aphthous ulcers are not always benign — when accompanied by genital ulcers, eye inflammation, skin lesions or vascular events, they are the gateway to a systemic vasculitis. Second, treating major-organ involvement aggressively: Behçet's is one of the few conditions that can cause a young person to go blind, die of massive haemoptysis from a pulmonary artery aneurysm, or suffer irreversible brainstem damage. A pivotal exam point that drives management is that thrombosis in Behçet's is inflammatory — the vessel wall is inflamed, the clotting cascade is normal, and immunosuppression, not anticoagulation, is the mainstay.[1]
Three features set Behçet's apart from the other vasculitides: (1) it involves both the venous and arterial trees (most vasculitides favour one), producing the paradoxical combination of venous thrombosis and arterial aneurysms in the same patient; (2) its striking geographic distribution along the ancient Silk Road; and (3) the pathergy phenomenon, an exaggerated skin inflammatory response to minor trauma that is both a clinical sign and a diagnostic test.[2]
Classification
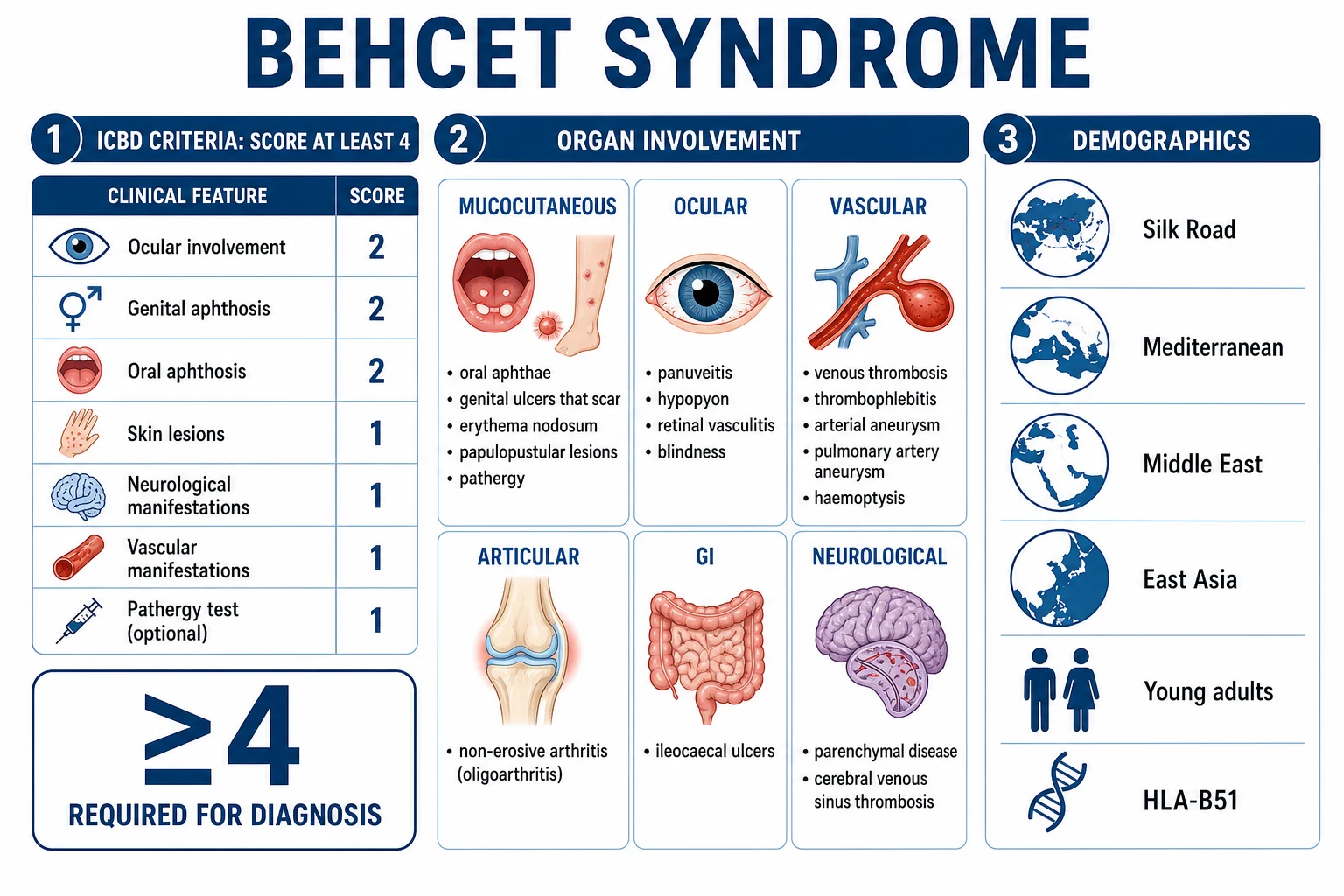
Behçet's is classified and diagnosed using the International Criteria for Behçet's Disease (ICBD), which in 2014 superseded the older 1990 International Study Group (ISG) criteria. The ICBD was developed from a multinational collaborative study of 27 countries and over 900 patients, and improved on the ISG by weighting ocular and genital disease more heavily and by simplifying the scoring system. It is entirely clinical — there is no confirmatory laboratory test — and a score of at least 4 establishes the diagnosis with markedly better sensitivity than the older ISG criteria while maintaining high specificity.[4]
Epidemiology & Risk Factors
The single highest-yield epidemiological fact is geography. Behçet's is endemic along a band that traces the ancient Silk Road — running from the Mediterranean (Turkey, Greece, Italy) through the Middle East (Iran, Iraq, Saudi Arabia) to East Asia (Japan, China, Korea). Turkey has the highest prevalence in the world, with rates up to 420 per 100,000 in some regions. Iran, Saudi Arabia and the Mediterranean littoral follow. By contrast, Behçet's is rare in Northern Europeans and North Americans (prevalence 1 to 8 per 100,000).[2][5]
This geographic gradient tracks closely with the HLA-B51 association, the strongest genetic link identified. HLA-B51 confers a relative risk of approximately 6 and is carried by over half of patients in endemic populations but is far less common in Europeans. HLA-B52 and the broader HLA-B5(5101) subtypes are also linked. The disease is more common in men in endemic regions, and men have more severe disease — particularly vascular and ocular complications. Onset is typically in young adults, between 20 and 40 years, although paediatric and late-onset forms exist.[2]
Behçet's syndrome — epidemiology at a glance
Precipitants are incompletely defined. Environmental triggers invoked include oral infection with Streptococcus sanguinis (a commensal that may trigger neutrophil activation), herpes simplex virus, and exposure to heavy metals and pesticides. A positive family history increases risk in endemic populations, consistent with polygenic inheritance centred on the HLA region but extending to non-HLA loci such as IL10, IL23R and CCR1.[2][5]
Pathophysiology
The aetiology of Behçet's is unknown, but the current model is of an aberrant inflammatory and immune response to an environmental trigger (infectious or other) in a genetically susceptible host (HLA-B51). The unifying pathological lesion is vasculitis affecting all vessel sizes and both the venous and arterial systems, with a predominantly neutrophilic and lymphocytic infiltrate of vessel walls, endothelial dysfunction and immune complex deposition.[1][2]
Three interlocking mechanisms drive the disease: [1]
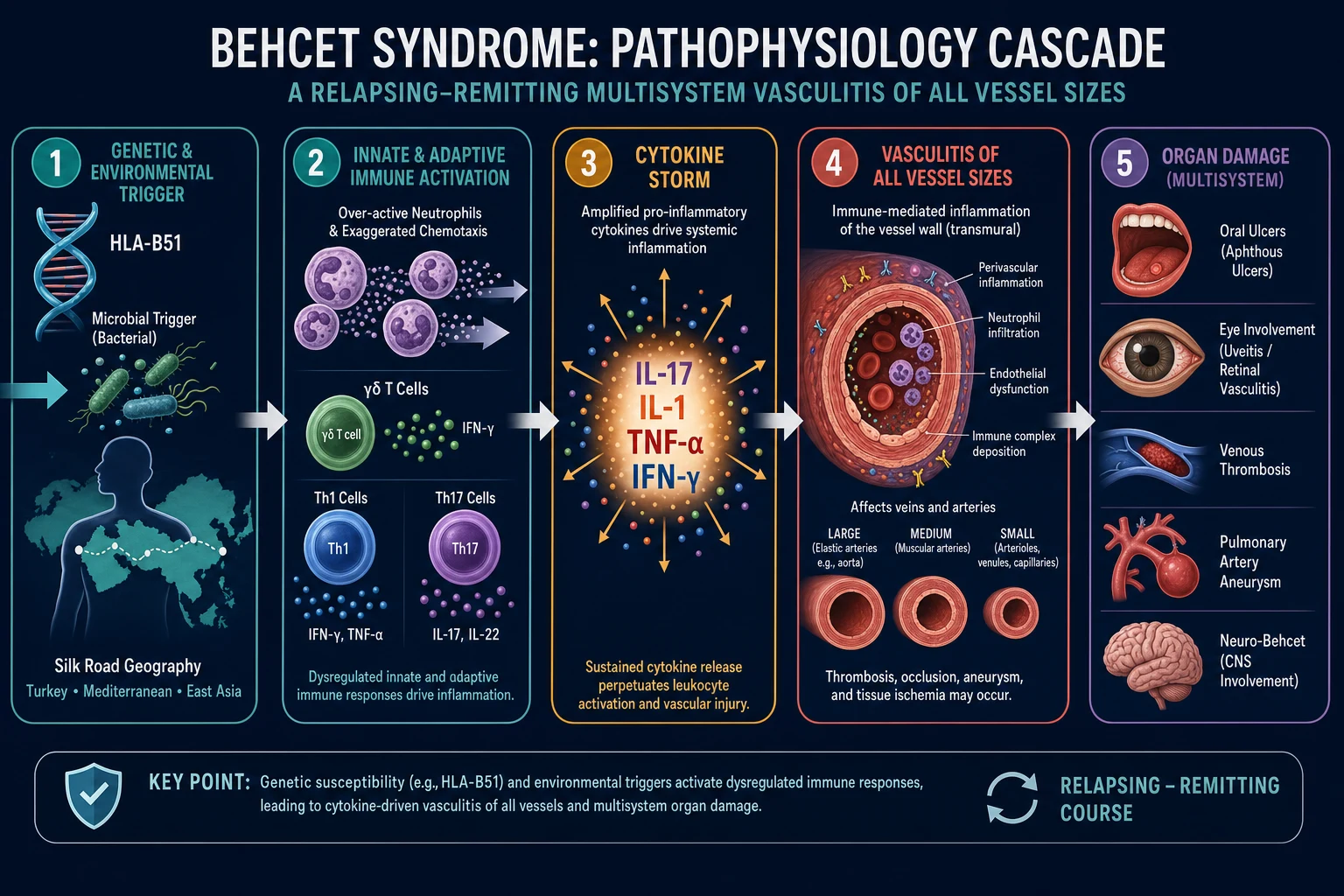
Innate immune dysregulation. Behçet's is sometimes called a neutrophilic vasculitis because neutrophils are constitutively hyperactive — they show exaggerated chemotaxis, superoxide production and phagocytosis, even when the disease is quiescent. Gamma-delta T cells are over-represented in the peripheral blood and lesions, and they produce interleukin-17, bridging innate and adaptive arms. This innate priming explains the pathergy phenomenon — the formation of a pustule at the site of a needle prick — which is the bedside signature of an exaggerated non-specific inflammatory response.[2]
Adaptive Th1 and Th17 polarisation. The adaptive response is dominated by Th1 and Th17 cells, with elevated interleukin-17, interleukin-1, interleukin-6, interleukin-8, TNF-alpha and interferon-gamma driving the relapsing inflammatory cycle. The dominance of the IL-17/IL-23 axis explains the clinical efficacy of phosphodiesterase-4 inhibition (apremilast) and the rationale for anti-TNF biologics. The cytokine profile is also why colchicine — which inhibits neutrophil chemotaxis and microtubule assembly — is effective for mucocutaneous disease.[3]
Endothelial dysfunction and a pro-thrombotic vasculitic vessel wall. Immune complex deposition and neutrophilic infiltration of vessel walls produce endothelial activation and injury. In veins this manifests as thrombophlebitis and venous thrombosis (the thrombus is inflammatory, and the clotting cascade is typically normal). In arteries it produces wall weakening and aneurysm formation, most dramatically the pulmonary artery aneurysm that causes fatal haemoptysis. This dual venous-arterial involvement is the pathophysiological signature that distinguishes Behçet's from nearly every other vasculitis.[1]
Genetic architecture and the HLA-B51 effect. Genome-wide association studies consistently identify HLA-B*51:01 as the strongest susceptibility locus, but its carriage is neither necessary nor sufficient for disease — roughly 10 to 20 per cent of patients are HLA-B51 negative, and many carriers never develop Behçet's. The mechanistic link is incompletely understood but is thought to involve aberrant peptide presentation that drives auto-reactive T-cell activation, and impaired natural killer cell function via the killer immunoglobulin-like receptor (KIR) axis. Non-HLA loci — IL10, IL23R, CCR1, STAT4 and ERAP1 — point to Th17 polarisation and antigen-processing pathways as central to pathogenesis. ERAP1 in particular shapes the MHC-I peptide repertoire, and its epistatic interaction with HLA-B51 mirrors the well-known HLA-B27 / ERAP1 interaction in ankylosing spondylitis. Together these findings paint Behçet's as a polygenic autoinflammatory disease in which an innate immune priming defect (neutrophil and gamma-delta T-cell hyper-reactivity) converges with adaptive Th1/Th17 amplification on a susceptible HLA background, expressing itself through a uniquely destructive vasculitis of both vascular circuits.[2][5]
BEHCET — the six axes of the disease
BEHCET
Vasculitis of all vessel sizes — unique in causing thrombosis AND aneurysms
Panuveitis, hypopyon, retinal vasculitis — blindness if untreated
Genetic association; Silk Road geography (Turkey highest prevalence)
For mucocutaneous disease and arthritis; apremilast for oral ulcers
Aggressive immunosuppression for eye, vascular, neuro, GI involvement
Immunosuppression over anticoagulation; coagulation tests are normal
Clinical Presentation
Behçet's runs a relapsing-remitting course with flares separated by variable intervals. Presentation is dominated by mucocutaneous lesions (universal), with variable involvement of eyes, vessels, joints, gut, skin and the nervous system. The tempo, severity and organ pattern differ markedly between patients, which is why an organ-by-organ approach to history and examination is essential.[1][2][5]
Oral aphthous ulcers — universal and mandatory. These are the gateway lesion and are present in nearly every patient. They are painful, multiple and recurrent (at least three episodes per year), and appear on the buccal mucosa, tongue, lips, gingiva and soft palate — but rarely the hard palate (a useful discriminator from herpetic lesions). They are typically round or oval with a yellow-grey floor and erythematous halo, measure a few millimetres to over a centimetre, and heal without scarring in 1 to 3 weeks. They are often the first manifestation, sometimes preceding other features by years. [1]
Genital ulcers. These are the second mucosal hallmark. They occur on the scrotum in men and the labia in women, are larger and deeper than oral aphthae, and — critically — heal with scarring, leaving a characteristic puckered or stellate scar that can be a permanent diagnostic clue even between flares. They are less recurrent than oral ulcers but more destructive. [1]
Skin lesions. Several patterns coexist: erythema nodosum (tender red subcutaneous nodules on the anterior shins, more common in women), papulopustular lesions and acneiform nodules on the trunk and face (more common in men), and a positive pathergy test — the formation of a papule or pustule 24 to 48 hours after a sterile needle prick. Skin pathergy, like the genital ulcer, reflects the innate immune hyper-reactivity that defines the disease. [1]
Ocular involvement (50 to 70 per cent, sight-threatening). Eye disease is the leading cause of disability in endemic regions. The spectrum runs from anterior uveitis with hypopyon (a layered pus level in the anterior chamber, a classic but now less common sign) to panuveitis and — most dangerously — retinal vasculitis with occlusive arterial and venular disease, retinal infiltrates, macular oedema and ischaemia. Untreated, ocular Behçet's progresses to irreversible blindness, often within five years. Any visual symptom in a Behçet's patient is an emergency requiring urgent slit-lamp examination.[6]
Vascular involvement (about 25 per cent, major morbidity and mortality). Behçet's uniquely causes both venous thrombosis and arterial aneurysms. Venous disease includes superficial thrombophlebitis, deep vein thrombosis (DVT) of the legs, thrombosis of the superior and inferior vena cava, and Budd-Chiari syndrome (hepatic vein thrombosis). Arterial disease manifests as aneurysms of the pulmonary, aortic, femoral, carotid and cerebral arteries. The pulmonary artery aneurysm is the single most feared lesion — it presents with massive haemoptysis and is the classic cause of death in Behçet's. Pulmonary arterial thrombosis and infarction also occur. Because thrombosis is vasculitic and inflammatory, the coagulation screen is normal and pulmonary embolism from a deep vein source is uncommon.[1]
Articular involvement (about 50 per cent). A non-erosive, non-deforming oligoarthritis or monoarthritis, principally affecting the knees, ankles and wrists, accompanies flares and is usually self-limiting over a few weeks. [1]
Gastrointestinal involvement. The hallmark lesion is ileocaecal ulceration, mimicking Crohn's disease. Presentation is with abdominal pain (typically right lower quadrant), diarrhoea and sometimes bleeding or perforation. Distinction from inflammatory bowel disease rests on endoscopic pattern and histology (see Differential Diagnosis). [1]
Neurological involvement (5 to 10 per cent, neuro-Behçet). Two distinct patterns exist. Parenchymal neuro-Behçet involves the brainstem and diencephalon, producing headache, pyramidal weakness (hemiparesis), ataxia, cranial nerve palsies, cognitive and behavioural change, and bladder/bowel disturbance; it carries a poor prognosis and cumulative disability. Non-parenchymal neuro-Behçet is dominated by cerebral venous sinus thrombosis, presenting with raised intracranial pressure — headache, papilloedema and sixth-nerve palsy — and generally has a better outlook. Cognitive impairment and a relapsing-remitting or secondary progressive course may follow repeated parenchymal attacks.[1]
Atypical presentations. Disease may be dominated by a single organ (e.g. isolated retinal vasculitis, a solitary pulmonary artery aneurysm, or a deep vein thrombosis) months or years before mucocutaneous features declare themselves. In endemic populations a high index of suspicion is warranted for any young man presenting with a vascular event. [1]
Renal and cardiac involvement (rare but recognised). Renal disease is uncommon and usually mild (glomerulonephritis, amyloidosis after decades of inflammation); renal artery aneurysm or thrombosis can cause hypertension or infarction. Cardiac involvement includes intracardiac thrombus (especially in right ventricle, in young men of Silk Road origin), pericarditis, myocarditis, and coronary artery aneurysm or vasculitis — all of which carry a serious prognosis and warrant aggressive immunosuppression. Pulmonary parenchymal involvement beyond the pulmonary artery aneurysm (pulmonary infarction, pleuritis) also occurs. [1]
Sex differences. A clinically critical pattern is the sex dimorphism: in endemic populations the disease is more common and more severe in men, who suffer higher rates of ocular, vascular and neurological complications, and constitute most of the mortality. Women tend to run a milder, more mucocutaneous course. This sex gradient should be factored into risk stratification and the threshold for aggressive early immunosuppression. [1]
Differential Diagnosis
The pivotal differentials are the other ulcer-plus-multisystem syndromes. Behçet's is distinguished by the combination of recurrent painful scarring genital ulcers, sight-threatening eye disease, pathergy, venous thrombosis with or without arterial aneurysm, the Silk Road / HLA-B51 background, and typically negative autoantibodies.[2][4]
Behçet's syndrome
- Recurrent PAINFUL oral aphthae (universal) + genital ulcers that SCAR
- Panuveitis, hypopyon, retinal vasculitis; pathergy positive
- Venous thrombosis AND arterial aneurysm (pulmonary artery aneurysm = haemoptysis)
- Silk Road geography, HLA-B51; ANA/ANCA negative; clinical diagnosis (ICBD, score at least 4)
- Coagulation normal; thrombosis is inflammatory
Crohn's disease (IBD)
- Oral aphthae, genital ulcers (fissures), erythema nodosum, arthritis, uveitis — overlaps heavily
- GI: transmural skip lesions, cobblestoning, perianal disease, granulomas on histology
- Ileocolonoscopy + biopsy decisive; Behçet ulcers are rounder/deeper without transmural gut change
- ANCA/ASCA may help; no pathergy, no pulmonary artery aneurysm
- Smoking worsens Crohn's; no Silk Road geography
Reactive arthritis
- Triad: urethritis, conjunctivitis/uveitis, asymmetric oligoarthritis; post-STI or GI
- Circinate balanitis and keratoderma blennorrhagicum — usually PAINLESS
- HLA-B27 association; 1 to 4 weeks after infection; sterile culture
- No venous thrombosis/aneurysm; no scarring genital ulcers; no pathergy
SLE
- Oral ulcers usually PAINLESS; malar rash, photosensitivity, arthritis
- Glomerulonephritis (Behçet has bland urine/normal renal unless vascular)
- ANA and anti-dsDNA positive; low complement; no pathergy or arterial aneurysm
- Venous thrombosis possible (antiphospholipid overlap) but different serology
Stevens-Johnson / HSV
- Stevens-Johnson: drug-triggered mucocutaneous blisters, target lesions, epidermal sloughing — acute, not relapsing
- HSV: clustered vesicles preceded by prodrome, Tzanck smear/PCR positive
- Neither causes pathergy, retinal vasculitis, pulmonary artery aneurysm or chronic relapsing course
- No Silk Road pattern; diagnosis by biopsy/PCR
A second layer of reasoning distinguishes Behçet's from the vasculitides and from mimics of neuro-Behçet. IgA vasculitis (Henoch-Schönlein purpura) and polyarteritis nodosa target different vessels and age groups and lack the mucocutaneous triad; ANCA-associated vasculitis (granulomatosis with polyangiitis, microscopic polyangiitis) produces destructive upper-airway disease, glomerulonephritis and pulmonary haemorrhage with positive ANCA — none of which are features of Behçet's. Multiple sclerosis is the chief mimic of parenchymal neuro-Behçet: both affect young adults and produce relapsing neurological deficits, but MS shows periventricular, corpus callosal and juxtacortical demyelinating plaques on MRI with oligoclonal bands in CSF, whereas neuro-Behçet has brainstem and thalamic T2 hyperintensities and typically a neutrophilic or lymphocytic CSF pleocytosis. Sweet syndrome (acute febrile neutrophilic dermatosis) shares papulonodular skin lesions but lacks mucosal ulcers and pathergy. The combination of negative autoantibodies, a normal coagulation screen, and the characteristic geographic and genetic background remains the most powerful discriminator. A useful clinical rule: if the oral ulcers are painless, think SLE; if the genital ulcers do not scar, think reactive arthritis or HSV; if there is a pulmonary artery aneurysm, think Behçet's. Recurrent aphthous stomatitis (oral ulcers alone, no systemic features) is far commoner than Behçet's and must not be over-investigated in the absence of other criteria. [1]
Clinical & Bedside Assessment
A structured history and systematic examination are required, because the diagnosis is clinical and hinges on documenting multi-organ involvement. Oral ulcers are the gateway: ask explicitly about at least three episodes per year, then screen every other organ system.[1][2]
History. Ask about recurrent oral ulcers (frequency, size, healing, scarring), genital ulcers and any residual scarring, eye symptoms (pain, redness, photophobia, blurring, floaters — all red flags for retinal vasculitis), skin lesions (leg nodules, pimples, pathergy), vascular events (leg swelling or pain suggesting DVT, haemoptysis suggesting pulmonary artery aneurysm, abdominal swelling suggesting Budd-Chiari), neurological symptoms (headache, weakness, ataxia, behavioural change), gastrointestinal symptoms (abdominal pain, diarrhoea, bleeding), and joint pain. Always record geographic and ethnic origin and a family history, both of which raise pre-test probability in endemic populations. [1]
Examination. Inspect the oral mucosa (buccal, tongue, lip, soft palate — aphthae are round, painful, with an erythematous halo) and the genitalia (scrotal or vulval ulcers and, crucially, scars from previous attacks). Examine the skin for erythema nodosum (anterior shins), papulopustules, acneiform nodules, and any pathergy test site. Perform a focused eye examination: visual acuity, slit-lamp (cells and flare, hypopyon), and fundoscopy (retinal vasculitis, infiltrates, venous occlusion). All patients — even asymptomatic — should be referred for formal ophthalmology assessment because subclinical retinal vasculitis is common and sight-threatening. [1]
Vascular examination. Palpate peripheral pulses and listen for bruits, examine for signs of DVT (calf swelling, tenderness, superficial thrombophlebitis), assess for pulmonary hypertension and right heart strain, and examine the abdomen for hepatomegaly and ascites (Budd-Chiari). Check blood pressure (renovascular disease). [1]
Neurological examination. Cranial nerves, cerebellar signs, pyramidal patterns (hemiparesis, hyperreflexia, upgoing plantar), and mental state. Fundoscopy for papilloedema (cerebral sinus thrombosis). [1]
Joints. A non-erosive oligoarthritis — knees, ankles, wrists. [1]
Pathergy test. Insert a sterile 20-gauge needle obliquely into the forearm skin to a depth of 5 mm, with or without saline. Read at 24 to 48 hours: formation of an erythematous papule or pustule at least 2 mm at the prick site is a positive test. Pathergy is highly specific in endemic (Silk Road) populations but is less sensitive in Europeans; it is now listed as optional in the ICBD.[4]
Investigations
There is no specific diagnostic test — diagnosis is clinical, using the ICBD criteria. Investigations serve three roles: to support the diagnosis, to exclude mimics, and to stage organ involvement before treatment.[1][2]
Bloods. A full blood count, CRP and ESR (raised during active disease, and useful as a longitudinal marker), urea and electrolytes, liver function tests and glucose to baseline before immunosuppression. A coagulation screen (INR, APTT, fibrinogen) is typically normal even in the presence of venous thrombosis — this is a critical teaching point, because thrombosis is vasculitic and inflammatory rather than a primary clotting disorder. There is no role for routine thrombophilia screening unless coexisting antiphospholipid syndrome is suspected. [1]
Autoantibodies. ANA, anti-dsDNA, ANCA, rheumatoid factor and complement are typically negative or normal in Behçet's. Their main value is to exclude SLE and ANCA-associated vasculitis. HLA-B51 typing is supportive but neither sensitive nor specific enough to confirm or exclude the diagnosis. [1]
Ophthalmology assessment. Slit-lamp examination and fundus fluorescein angiography are mandatory even in asymptomatic patients, because subclinical retinal vasculitis and macular oedema are common, treatable, and sight-threatening. [1]
Imaging for vascular disease. CT pulmonary angiography is the test of choice for a suspected pulmonary artery aneurysm (any haemoptysis in a Behçet's patient is a pulmonary artery aneurysm until proven otherwise). Doppler ultrasound and D-dimer assess deep vein thrombosis. CT or MR angiography defines peripheral arterial aneurysms and their suitability for embolisation. CT or MR venography documents vena cava or hepatic vein thrombosis (Budd-Chiari). [1]
Imaging for neurological disease. MRI brain is the test of choice for parenchymal neuro-Behçet, showing characteristic T2/FLAIR hyperintensities in the brainstem, thalamus and basal ganglia. MR venography detects cerebral venous sinus thrombosis. Lumbar puncture may show a neutrophilic or lymphocytic pleocytosis and elevated protein in active parenchymal disease. [1]
Endoscopy. Ileocolonoscopy with biopsy is performed when gastrointestinal symptoms suggest ileocaecal ulceration; biopsies distinguish Behçet's ulcers (round, deep, discrete, without transmural inflammation or granulomas) from Crohn's disease. [1]
Skin biopsy. Biopsy of erythema nodosum, papulopustular lesions or a pathergy site shows a neutrophilic or lymphocytic vasculitis — supportive but not diagnostic. [1]
Behçet's syndrome — key numbers
Self-test: a young man with oral and genital ulcers
A 26-year-old Turkish man has recurrent painful oral aphthous ulcers (six episodes per year), scrotal ulcers that heal with scarring, and erythema nodosum on his shins. His CRP is 58 mg/L, ANA and ANCA are negative, and the coagulation screen is normal. What is the diagnosis, the score, and the immediate next step? [1]
Diagnosis: Behçet's syndrome — the combination of oral aphthosis (2), genital aphthosis that scars (2) and skin lesions (1) gives an ICBD score of 5 (above the threshold of at least 4), with negative autoantibodies and Silk Road origin supporting the clinical diagnosis. [1]
Immediate next steps: (1) urgent ophthalmology referral with slit-lamp and fluorescein angiography to detect subclinical retinal vasculitis; (2) vascular screening — CT pulmonary angiography if any haemoptysis, Doppler ultrasound for DVT; (3) baseline bloods (FBC, U&E, LFT, glucose, TPMT) and HLA-B51 typing (supportive); (4) start colchicine 1 to 1.5 mg daily for the mucocutaneous and skin disease and arrange azathioprine if any ocular involvement is found; (5) counsel on red flags — visual change, haemoptysis, headache, leg swelling.
Management — Resuscitation
Behçet's is rarely an acute resuscitation emergency, but several scenarios are time-critical and demand immediate, coordinated action.[1]
-
Massive haemoptysis from a pulmonary artery aneurysm. Protect the airway (place the bleeding side down, consider intubation with a double-lumen tube), establish large-bore IV access, send cross-match, and correct coagulopathy. Obtain an urgent CT pulmonary angiography. Start high-dose IV corticosteroids (methylprednisolone 500 mg to 1 g daily for 3 days, then oral prednisolone 1 mg/kg/day) plus IV cyclophosphamide (pulse therapy, e.g. 15 mg/kg every 2 to 4 weeks with mesna and hydration). Involve interventional radiology for embolisation and thoracic or vascular surgery for resection if embolisation fails or is unsuitable. Anti-TNF (infliximab) is an option in refractory cases. Anticoagulation is avoided because the aneurysm will bleed.[1]
-
Acute visual loss from retinal vasculitis or panuveitis. Urgent ophthalmology assessment and high-dose corticosteroids (IV methylprednisolone pulses followed by oral prednisolone 1 mg/kg/day) with rapid institution of a steroid-sparing agent (azathioprine, or anti-TNF for severe disease). Sight is at risk within hours to days. [1]
-
Cerebral venous sinus thrombosis. Manage raised intracranial pressure (head elevation, acetazolamide, mannitol if severe). Anticoagulation is generally given for sinus thrombosis because the mechanism differs from peripheral Behçet thrombosis, but only after excluding a coexisting arterial aneurysm (CT or MR angiography). Add corticosteroids for the underlying vasculitis.[1]
-
Gastrointestinal perforation from an ileocaecal ulcer is a surgical emergency: resuscitate, broad-spectrum antibiotics, and laparoscopic or open resection. [1]
Management — Definitive & Stepwise
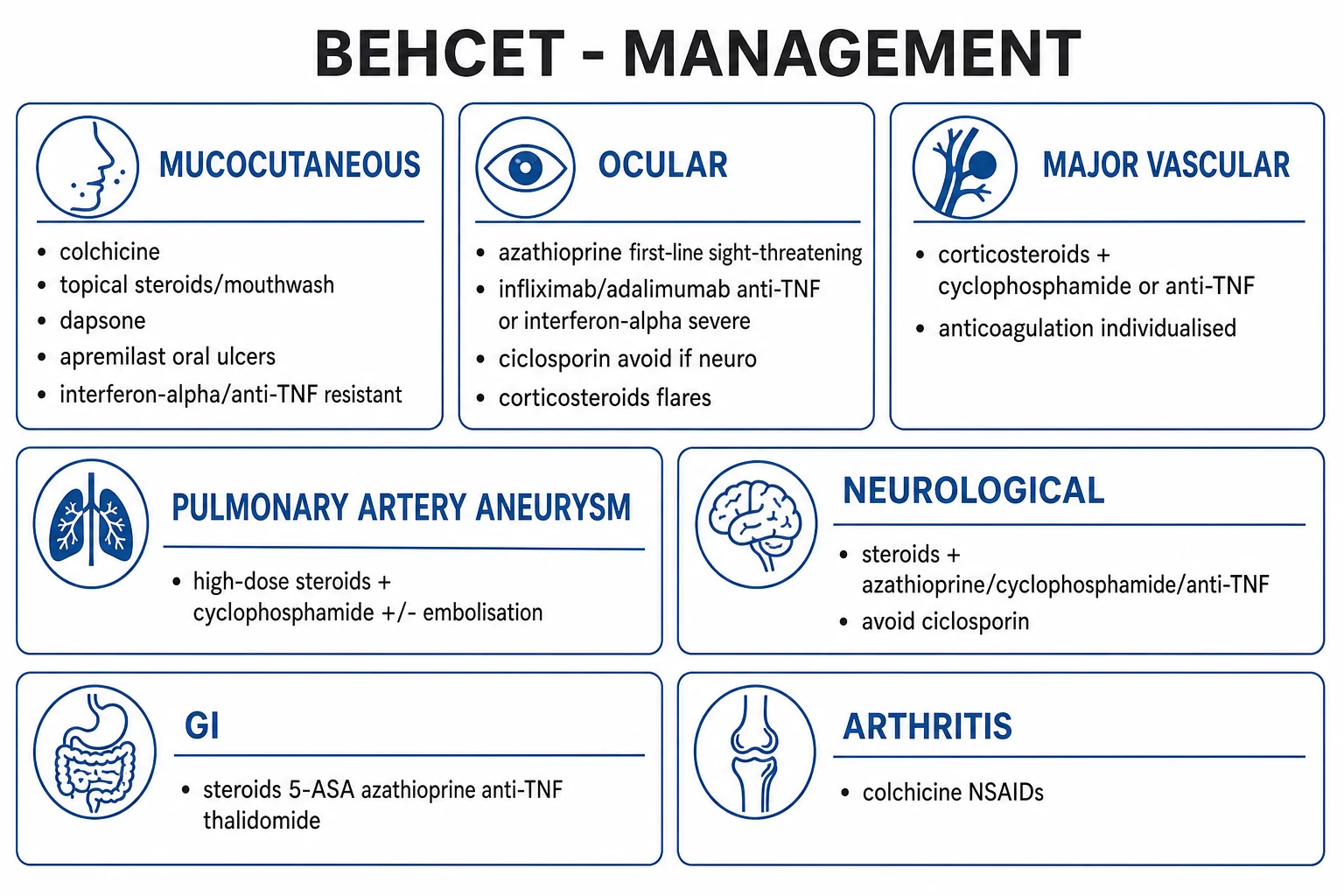
Management is organ-based and severity-stratified, following the EULAR recommendations. The central principle is that mucocutaneous disease is managed conservatively, while major-organ involvement (eye, vascular, nervous system, GI) requires aggressive immunosuppression.[1][2][3]
Mucocutaneous disease. First-line measures are topical corticosteroids (mouthwash or cream for oral and genital ulcers) and colchicine 1 to 1.5 mg daily (especially effective for erythema nodosum and arthritis, by inhibiting neutrophil chemotaxis). Dapsone is an alternative. For refractory oral ulcers, apremilast 30 mg twice daily (a phosphodiesterase-4 inhibitor) is supported by phase 3 randomised data showing significant reduction in oral ulcer burden. Interferon-alpha or anti-TNF (infliximab, adalimumab) is reserved for resistant, disabling mucocutaneous disease.[3][7]
Arthritis. NSAIDs and colchicine are first-line; intra-articular or short-course oral corticosteroids for flares; azathioprine or methotrexate for recurrent or refractory disease. [1]
Ocular disease (sight-threatening). Azathioprine is first-line steroid-sparing immunosuppression, supported by a landmark randomised trial showing that early azathioprine reduces eye disease recurrence and improves long-term visual prognosis while also controlling oral and genital ulcers and arthritis.[6] Corticosteroids (oral prednisolone 1 mg/kg/day, or IV methylprednisolone pulses for severe flares) cover the acute phase. For severe, resistant or posterior-segment disease (chorioretinitis, retinal vasculitis), anti-TNF — infliximab 5 mg/kg or adalimumab — or interferon-alpha is added or substituted. Ciclosporin is effective for anterior uveitis but is avoided when there is any neurological involvement because of its neurotoxicity. Tocilizumab (IL-6 blockade) and secukinumab (IL-17 blockade) are emerging options.
Major vascular disease (aneurysm and thrombosis). The first-line regimen is high-dose corticosteroids plus cyclophosphamide (pulse IV cyclophosphamide with mesna and hydration, oral prednisolone 1 mg/kg/day tapered over weeks). Anti-TNF (infliximab) is an alternative or adjunct for refractory aneurysms. Embolisation or surgical repair is required for bleeding, expanding or ruptured aneurysms. Anticoagulation is individualised and controversial: because thrombosis is inflammatory and because coexisting aneurysms can bleed, EULAR practice favours immunosuppression with selective anticoagulation rather than routine anticoagulation. Azathioprine is used for maintenance.[1]
Neuro-Behçet. Acute parenchymal disease is treated with high-dose corticosteroids plus cyclophosphamide; azathioprine, mycophenolate or anti-TNF is used for maintenance or refractory disease. Ciclosporin is avoided because it is neurotoxic in this context. For cerebral venous sinus thrombosis, corticosteroids plus carefully considered anticoagulation (after excluding an aneurysm) are standard. [1]
Gastrointestinal disease. First-line is corticosteroids with 5-ASA compounds (sulfasalazine, mesalazine) and azathioprine for steroid-sparing maintenance. Thalidomide (with strict contraception and monitoring for neuropathy) or anti-TNF is used for refractory ileocaecal ulcers. Surgery (segmental resection) is reserved for perforation, refractory bleeding or stricturing. [1]
Adjunctive and supportive care. Vaccination before immunosuppression (influenza, pneumococcal, COVID-19, hepatitis B; avoid live vaccines on active immunosuppression), Pneumocystis jirovecii prophylaxis (co-trimoxazole) with prolonged corticosteroids, bone protection (calcium, vitamin D, bisphosphonate) with chronic steroids, regular ophthalmology surveillance, and patient education on red flags (visual change, haemoptysis, new headache, leg swelling). Multidisciplinary care — rheumatology, ophthalmology, and as indicated vascular surgery, neurology, gastroenterology — is the standard. [1]
Specific Subtypes & Scenarios
Several recognisable phenotypes guide prognosis and therapy.[1][2][5]
-
Mucocutaneous Behçet — oral and genital ulcers with skin lesions only. The commonest and most benign phenotype, managed with colchicine, topical steroids and apremilast; excellent prognosis. [1]
-
Ocular Behçet — panuveitis, retinal vasculitis, hypopyon. Sight-threatening and the leading cause of disability in endemic regions; first-line azathioprine with corticosteroids and anti-TNF or interferon-alpha for severe disease. [1]
-
Vascular Behçet — venous thrombosis, thrombophlebitis migrans, arterial aneurysms (especially pulmonary artery), Budd-Chiari. The principal driver of mortality; treated with corticosteroids plus cyclophosphamide, anticoagulation individualised. [1]
-
Neuro-Behçet — parenchymal brainstem disease (poor prognosis, cumulative deficit) versus non-parenchymal cerebral venous sinus thrombosis (better prognosis). Ciclosporin is contraindicated. [1]
-
Gastrointestinal Behçet — ileocaecal ulceration mimicking Crohn's; treated medically, surgery reserved for complications. [1]
-
Articular Behçet — non-erosive oligoarthritis, knees and ankles; NSAIDs and colchicine. [1]
-
Paediatric Behçet — rare; family history often positive; generally milder course, with a longer interval between oral ulcers and full-blown disease. [1]
-
Hughes-Stovin syndrome — the arterial-pulmonary-aneurysm subset of Behçet's spectrum, defined by pulmonary artery aneurysms with or without systemic arterial aneurysm and venous thrombosis, but without the full mucocutaneous criteria; treated identically to vascular Behçet. [1]
Complications & Pitfalls
-
Blindness from untreated retinal vasculitis or panuveitis — the leading cause of disability in endemic regions, largely preventable with early azathioprine and anti-TNF.[6]
-
Fatal haemoptysis from pulmonary artery aneurysm rupture — the major cause of death in Behçet's. Any haemoptysis in a Behçet's patient warrants urgent CT pulmonary angiography and high-dose corticosteroids plus cyclophosphamide.[1]
-
Venous thromboembolism, Budd-Chiari syndrome (hepatic vein thrombosis), portal hypertension, vena cava obstruction and venous stasis ulcers. [1]
-
Arterial aneurysm rupture (aorta, femoral, pulmonary, cerebral) with ischaemia or infarction; embolisation or surgery may be life-saving. [1]
-
Neurological deficit, cognitive impairment and disability from cumulative parenchymal brainstem damage. [1]
-
Gastrointestinal perforation, fistulae and malnutrition from ileocaecal ulcers. [1]
-
Treatment-related harms: infections from immunosuppression; osteoporosis, cataracts, glaucoma and growth retardation from corticosteroids; haemorrhagic cystitis, malignancy and cytopenia from cyclophosphamide; nephrotoxicity and hypertension from ciclosporin; teratogenicity and neuropathy from thalidomide. [1]
The classic diagnostic pitfall is over-investigating recurrent oral ulcers as Behçet's in the absence of other criteria — simple recurrent aphthous stomatitis is far commoner and requires no immunosuppression. The classic management pitfall is treating Behçet thrombosis with anticoagulation alone — because the thrombus is inflammatory, the mainstay is immunosuppression, and anticoagulation risks bleeding from a coexisting aneurysm. A further pitfall is using ciclosporin in a patient with neurological involvement, which can precipitate or worsen neurotoxicity. [1]
Prognosis & Disposition
The course is relapsing-remitting, and disease activity often declines with age — Behçet's tends to "burn out" over decades — but vascular and neurological damage is cumulative and irreversible. Mortality is driven mainly by pulmonary artery aneurysm (haemoptysis) and severe neuro-Behçet. Young men with vascular disease have the worst prognosis, whereas isolated mucocutaneous disease carries an excellent outlook. Ocular prognosis has been transformed by early azathioprine and anti-TNF: without treatment, blindness was common within five years in endemic populations, whereas with modern immunosuppression useful vision is usually preserved. Pregnancy outcomes are generally favourable with multidisciplinary care (see below).[1][2][5]
Disposition depends on organ involvement. Patients with active eye disease, a new aneurysm, or acute neurological disease are admitted for urgent imaging and pulse immunosuppression. Stable mucocutaneous or articular disease is managed in the outpatient rheumatology clinic with at least annual ophthalmology review. Every patient receives structured education on red flags — new visual change, haemoptysis, severe headache, leg swelling — and an open route to urgent reassessment. [1]
Typical natural history of Behcet's syndrome
The disease usually declares itself with recurrent oral aphthous ulcers in a young adult (typically 20 to 40 years), often years before any other feature. This is the gateway lesion and the basis of the diagnostic question — at least three episodes per year.
Genital ulcers (which scar), skin lesions (erythema nodosum, papulopustular, acneiform), a positive pathergy test, and a non-erosive oligoarthritis of knees and ankles appear. The ICBD score typically reaches 4 or more at this stage.
Ocular disease (panuveitis, retinal vasculitis — the leading cause of disability), vascular disease (venous thrombosis, pulmonary artery aneurysm — the major cause of death), neuro-Behcet (brainstem or sinus thrombosis), and ileocaecal ulcers declare themselves, particularly in young men. This phase demands aggressive immunosuppression.
Relapsing inflammatory activity often declines with age, but vascular and neurological damage is cumulative and irreversible. Long-term immunosuppression, ophthalmology surveillance, and treatment-related harm prevention (osteoporosis, infection, malignancy) dominate management.
Special Populations
-
Young men of Silk Road origin carry the most severe phenotype: maintain a high index of suspicion for vascular and ocular complications and start aggressive early immunosuppression. This is the group in which a single pulmonary artery aneurysm can be fatal. [1]
-
Pregnancy. Multidisciplinary management is essential. Continue pregnancy-compatible immunosuppression (azathioprine, corticosteroids, colchicine, calcineurin inhibitors). Avoid thalidomide, methotrexate, cyclophosphamide, mycophenolate and leflunomide (all teratogenic). Inflammatory thrombosis is treated with immunosuppression; anticoagulation is individualised, with low-molecular-weight heparin preferred over warfarin. Disease activity often improves in pregnancy but may flare postpartum.[1]
-
Paediatric Behçet. Rare; family history is frequently positive and the course is generally milder, with a longer interval between the first oral ulcer and full clinical expression. Treatment follows adult principles, with weight-based dosing and careful attention to growth and steroid toxicity. [1]
-
Elderly. Late-onset Behçet is uncommon and tends to run a milder course, but comorbidity (diabetes, hypertension, osteoporosis) increases the risk of steroid and immunosuppressant toxicity — favour steroid-sparing strategies. [1]
-
Eye disease in any Behçet patient. Mandatory urgent ophthalmology referral is the rule, because subclinical sight-threatening retinal vasculitis is common and asymptomatic until vision is lost. [1]
-
Thrombosis in Behçet is inflammatory. The coagulation screen (INR, APTT, platelets) is normal; immunosuppression is the mainstay and anticoagulation is selective rather than universal.[1]
Evidence, Guidelines & Regional Differences
-
EULAR recommendations for Behçet's syndrome (Hatemi 2018) set the international standard for organ-based management: colchicine for mucocutaneous disease and arthritis; azathioprine (with corticosteroids) for sight-threatening ocular disease; corticosteroids plus cyclophosphamide for major vascular disease (especially pulmonary artery aneurysm); and corticosteroids plus cyclophosphamide or azathioprine for neuro-Behçet. Anti-TNF and interferon-alpha are positioned for refractory ocular, vascular, neurological and gastrointestinal disease.[1][3]
-
International Criteria for Behçet's Disease (ICBD, 2014) — a collaborative study of 27 countries that established the current clinical scoring system (score at least 4), replacing the 1990 ISG criteria and improving sensitivity while preserving specificity.[4]
- In Australia and New Zealand, where Behçet's is rare and largely seen in migrant populations from endemic regions, management follows EULAR principles with strong reliance on tertiary rheumatology-ophthalmology collaboration and access to anti-TNF through specialist initiation. [1]
-
Landmark evidence. The Hamuryudan 1997 randomised trial established that azathioprine reduces ocular disease recurrence and improves long-term visual prognosis while also controlling oral and genital ulcers and arthritis — the foundation of azathioprine as first-line steroid-sparing therapy in ocular Behçet.[6] The 2018–2022 phase 3 programme confirmed that apremilast significantly reduces the burden of oral ulcers, including in Japanese and other endemic subgroups.[7]
-
Regional delta — anticoagulation. The most contentious issue is anticoagulation for Behçet venous thrombosis. European (EULAR) practice favours immunosuppression with selective anticoagulation, on the grounds that thrombosis is inflammatory and coexisting aneurysms bleed. Some Asian centres use anticoagulation more liberally, particularly for deep vein thrombosis without aneurysm. There is no high-quality randomised evidence to settle this.[1][5]
-
Where the evidence is weak. Optimal duration of maintenance immunosuppression, the role of IL-6 (tocilizumab) and IL-17 (secukinumab) blockade, and the place of JAK inhibitors remain under investigation. Paediatric and pregnancy registries are limited. [1]
Exam Pearls
Exam application bank (NEET-PG / INICET)
One-line answer
Behçet's syndrome is a relapsing-remitting multisystem vasculitis (affects both arteries and veins of all sizes) classically defined by recurrent oral aphthous ulcers plus any two of: genital ulcers, eye lesions (uveitis/retinal vasculitis), skin lesions (erythema nodosum, papulopustular, pathergy), and positive pathergy test. Commonest along the Silk Road (Turkey, Mediterranean, Middle East, East Asia); rare in Northern Europeans. HLA-B51 association. Can involve vessels (venous thrombosis, arterial aneurysm, pulmonary artery aneurysm — major cause of death), eyes (panuveitis, retinal vasculitis — blindness), gastrointestinal (ileocaecal ulcers), joints (arthritis), neurological, and skin. Oral ulcers are universal and the gateway to diagnosis; genital ulcers scar. Diagnosis is clinical (ICBD criteria) with no specific test. Treat mucocutaneous disease conservatively (colchicine, apremi
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard. [1]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes. [1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change. [1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each. [1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory. [1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Behçet's Syndrome.
References
- [1]Ozguler Y, Leccese P, Christensen R, et al. Management of major organ involvement of Behçet's syndrome: a systematic review for update of the EULAR recommendations Rheumatology (Oxford), 2018.PMID 30107448
- [2]Hatemi G, Seyahi E, Fresko I, Yurdakul S. One year in review 2018: Behçet's syndrome Clin Exp Rheumatol, 2018.PMID 30582516
- [3]Leccese P, Ozguler Y, Christensen R, et al. Management of skin, mucosa and joint involvement of Behçet's syndrome: A systematic review for update of the EULAR recommendations for the management of Behçet's syndrome Semin Arthritis Rheum, 2019.PMID 29954598
- [4]International Team for the Revision of the International Criteria for Behcet's Disease (ITR-ICBD). The International Criteria for Behçet's Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria J Eur Acad Dermatol Venereol, 2014.PMID 23441863
- [5]Davatchi F, Chams-Davatchi C, Shams H, et al. Adult Behcet's disease in Iran: analysis of 6075 patients Int J Rheum Dis, 2016.PMID 26258691
- [6]Hamuryudan V, Ozyazgan Y, Hizli N, et al. Azathioprine in Behcet's syndrome: effects on long-term prognosis Arthritis Rheum, 1997.PMID 9125262
- [7]Takeno M, Dobashi H, Tanaka Y, et al. Apremilast in a Japanese subgroup with Behçet's syndrome: Results from a Phase 3, randomised, double-blind, placebo-controlled study Mod Rheumatol, 2022.PMID 34894266