Rheumatology · General Medicine
Dermatomyositis & Polymyositis (Idiopathic Inflammatory Myopathies)
Also known as Dermatomyositis · Polymyositis · Idiopathic inflammatory myopathy · IIM · Heliotrope rash · Gottron papules · Antisynthetase syndrome · Inclusion body myositis
Idiopathic inflammatory myopathies (IIM) are acquired autoimmune muscle diseases causing symmetric, proximal muscle weakness (difficulty combing hair, rising from a chair, climbing stairs) with raised creatine kinase. Dermatomyositis (DM) features characteristic skin manifestations (heliotrope rash — violaceous periorbital; Gottron papules/sign — over knuckles; shawl/V-sign, nailfold changes); polymyositis (PM) is muscle-only. Other IIM: inclusion body myositis (IBM) (older, distal + quadriceps/finger flexor weakness, poor steroid response), immune-mediated necrotising myopathy (IMNM) (anti-SRP/HMGCR), antisynthetase syndrome (anti-Jo-1, ILD, fever, mechanic's hands, arthritis), juvenile DM. Strong association of adult DM with occult malignancy and of antisynthetase/anti-MDA5 disease with interstitial lung disease (ILD). Diagnosis combines proximal weakness + high CK + myopathic EMG + muscle biopsy (DM perifascicular; PM endomysial) plus myositis-specific antibodies and MRI. Treat with high-dose steroids then MTX/azathioprine/MMF/IVIG/rituximab; IBM is largely refractory.
On this page & tools
Your progress
Saved locally on this device.
Exam tags
Red flags
Overview & Definition
The idiopathic inflammatory myopathies (IIM) are a heterogeneous group of acquired, immune-mediated skeletal-muscle disorders unified by three clinical anchors: subacute onset (weeks to months, not days and not lifelong), symmetric, proximal muscle weakness, and muscle inflammation with raised creatine kinase (CK).[1][2] Five subtypes are now distinguished because they behave very differently in cause, treatment response, and prognosis:
- Dermatomyositis (DM) — muscle plus characteristic skin disease; malignancy- and ILD-associated; driven by a type-I-interferon, complement-mediated microangiopathy.[2]
- Polymyositis (PM) — muscle only, no rash; driven by CD8+ T-cell cytotoxicity; rarer than historical accounts suggested (many old "PM" cases were undiagnosed IBM).[2]
- Inclusion body myositis (IBM) — older patients, asymmetric quadriceps and finger-flexor weakness, steroid-refractory, with a degenerative component (rimmed vacuoles).[5]
- Immune-mediated necrotising myopathy (IMNM) — antibody-defined (anti-SRP, anti-HMGCR), very high CK, necrosis with minimal inflammation, often statin-exposed.[8][9]
- Antisynthetase syndrome — antibody-defined (anti-Jo-1 and others), ILD-predominant with fever, mechanic's hands, arthritis.[1]
Juvenile DM (onset before age 18) is conventionally considered a sixth group with prominent vasculopathy and calcinosis.[1]
The pivotal clinical skill is recognising the phenotype — proximal weakness plus the DM skin signs — and then risk-stratifying: malignancy screen in DM, ILD screen in antisynthetase/MDA5, and avoiding futile immunosuppression in IBM. Two contrasts anchor the diagnosis. First, IIM causes weakness, not pain: the cardinal distinction from polymyalgia rheumatica (PMR), which is pain and stiffness with normal strength and normal CK. Second, IIM is myopathic, not neuropathic: proximal, symmetric, myopathic EMG, raised muscle enzymes — distinguishing it from myasthenia, motor neuron disease, and the muscular dystrophies.[2]
Classification
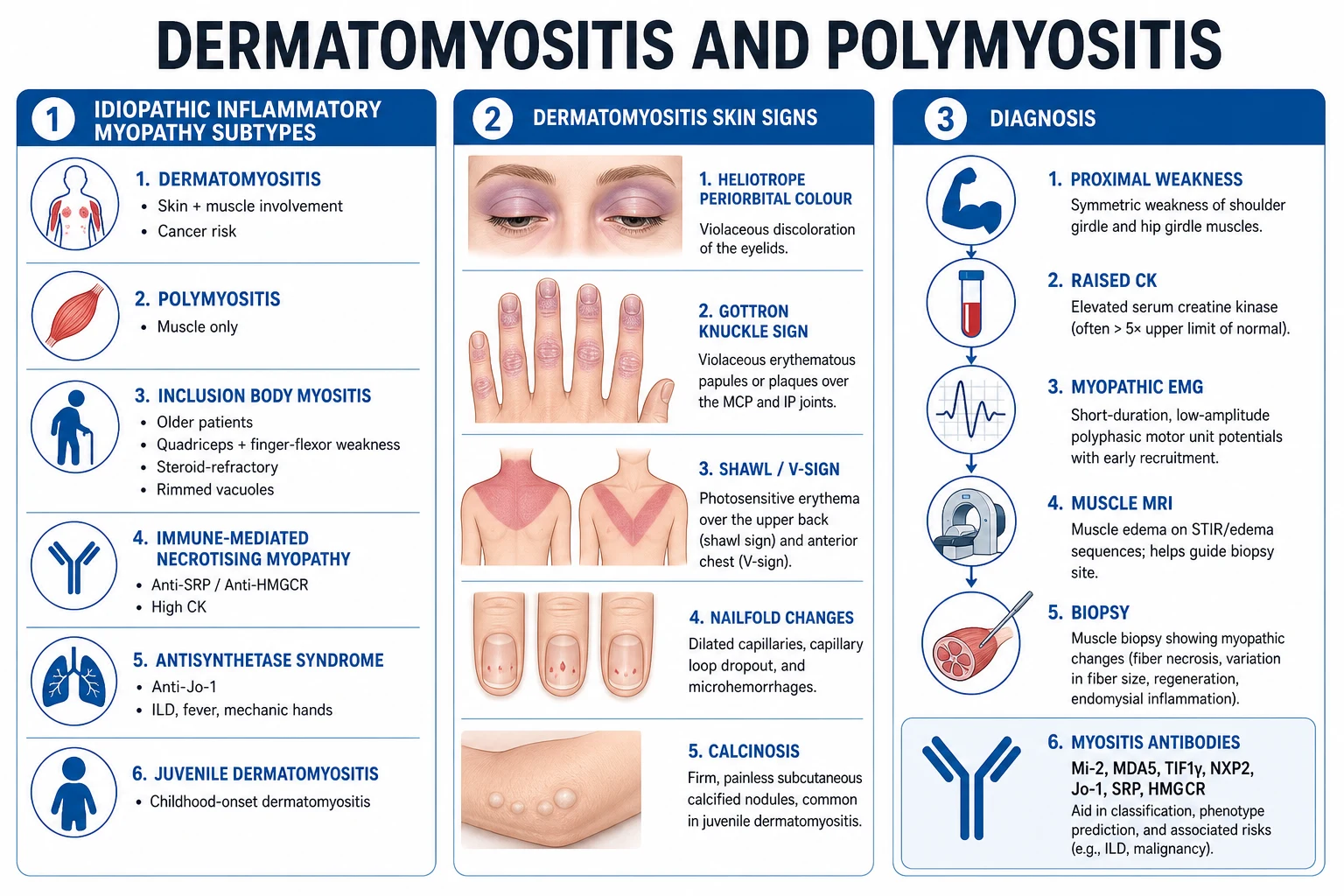
Classification has evolved in three waves. The 1975 Bohan and Peter criteria are the historic, clinically intuitive scheme that examiners still ask for.[3][4] The 2004 ENMC (119th workshop) workshop refined the categories by introducing antibody-defined and biopsy-defined subgroups and codified IBM criteria.[6] The current standard is the 2017 EULAR/ACR classification criteria of Lundberg and colleagues, a data-driven, probability-scored instrument that incorporates clinical, enzymatic, antibody, and biopsy variables.[1]
Bohan and Peter (1975) — the five classic criteria
BP criterion 1 — Weakness
- Symmetric proximal muscle weakness developing subacutely
- Shoulder girdle: difficulty lifting the arms, combing the hair
- Hip girdle: difficulty rising from a chair, climbing stairs, stepping onto a bus
- Neck flexors weak; distal muscles usually spared early
BP 2 — Enzymes
- Elevation of serum muscle enzymes
- Creatine kinase (CK), aldolase, lactate dehydrogenase (LDH), and transaminases (AST/ALT) are all of muscle origin
- CK is the most sensitive and specific for active disease
BP 3 — EMG
- Myopathic electromyographic triad
- Short, small, low-amplitude polyphasic motor unit potentials
- Fibrillations, positive sharp waves, and insertion irritability
- Bizarre high-frequency repetitive discharges
BP 4 — Biopsy
- Muscle biopsy showing degeneration, regeneration, necrosis, phagocytosis
- Interstitial mononuclear inflammatory infiltrate (perivascular/perimysial in DM)
- Perifascicular atrophy — the histological hallmark of DM
BP 5 — Rash (DM only)
- Heliotrope rash (violaceous discolouration of the eyelids)
- Gottron papules over MCP and interphalangeal joints
- Scaling erythema over elbows, knees, malleoli, face, neck, upper trunk
The Bohan and Peter diagnostic tiers assign confidence on the number of criteria met. For polymyositis, definite requires four of the first four; probable three of four; possible two of four. For dermatomyositis the rash (criterion 5) is added — definite DM is the rash plus any three of the first four, probable DM the rash plus any two, possible DM the rash plus any one.[3][4] The scheme's principal limitation is that it over-diagnoses PM by absorbing IBM and IMNM cases, which is why the modern antibody- and biopsy-driven 2017 criteria supersede it for research and subclassification.
2017 EULAR/ACR classification — the current standard
The 2017 EULAR/ACR criteria combine sixteen weighted variables across age at onset, clinical features (symmetric proximal weakness, distal weakness, neck weakness, dysphagia, Gottron papules/sign, heliotrope rash, shawl/V-sign, mechanic's hands, Raynaud), laboratory features (raised CK, ANA, anti-Jo-1) and muscle biopsy (perifascicular, endomysial, perivascular/perimysial inflammation, rimmed vacuoles, necrosis/regeneration).[1] A patient is classified as having IIM when the probability score is at least 55 per cent, using a tree that first separates juvenile-onset from adult-onset disease. After the diagnosis is established, a subclassifying algorithm — incorporating the biopsy pattern, anti-Jo-1, and clinical phenotype — assigns one of six mutually exclusive subgroups: polymyositis, dermatomyositis (clinically amyopathic DM included), inclusion body myositis, juvenile DM, cancer-associated myositis, or overlap myositis. The criteria deliberately exclude patients with an alternative cause for the myopathy (neuromuscular disease, drug-induced myopathy, endocrine myopathy, rhabdomyolysis).[1]
IBM — the dedicated criteria
IBM is classified using the Griggs 1995 research criteria and the ENMC 119th workshop framework, which require a constellation that is quite different from PM/DM:[5][6] onset over age 30 (usually over 50) with male predominance; knee extensor (quadriceps) weakness equal to or greater than hip flexor weakness; finger flexor weakness (especially the deep flexors of the forearm) greater than shoulder abductor weakness; biopsy with endomysial inflammation and rimmed vacuoles; and slow progression with a poor response to glucocorticoids. Recognising this pattern is critical because IBM is mislabelled as refractory PM more often than any other myopathy.
Epidemiology & Risk Factors
The inflammatory myopathies are uncommon but not rare, with an overall annual incidence of approximately 5 to 11 cases per million and a prevalence of around 14 to 18 per 100,000.[2] Several patterns decide the differential:
- Bimodal incidence of DM: a juvenile peak (juvenile DM, ages 5 to 15) and an adult peak (ages 40 to 60). DM is more common than PM. IBM is the commonest inflammatory myopathy over age 50 and is systematically under-recognised, often mislabelled as refractory PM before a biopsy demonstrates rimmed vacuoles.[5]
- Sex: DM and PM are female-predominant (about 2 to 1); IBM is male-predominant.
- Cancer-associated myositis: roughly 15 to 25 per cent of adults with DM have an associated malignancy — substantially more than in PM. Risk is highest in the first year around diagnosis and is most strongly enriched in patients with anti-TIF1-gamma and anti-NXP2 antibodies; intensified screening and rescreening for up to three years are now recommended by the 2023 International Myositis Society guideline.[13]
- Antisynthetase syndrome: anti-Jo-1 is the commonest antisynthetase antibody (about a fifth of IIM); other antisynthetases (PL-7, PL-12, EJ, OJ) are rarer but carry a higher ILD burden.
- Anti-MDA5 disease: characteristically amyopathic or mildly myopathic DM with rapidly progressive ILD and cutaneous ulcers — high early mortality, especially aggressive in East Asian cohorts (Japan, China).[7]
- IMNM: anti-HMGCR disease often follows statin exposure and may persist and progress despite statin withdrawal.[8]
- Environmental triggers: viral infections (coxsackievirus, parvovirus, retroviruses), ultraviolet light (a documented latitude gradient for DM), drugs (statins for anti-HMGCR IMNM), and malignancy.[2]
Dermatomyositis and polymyositis — key numbers
Pathophysiology
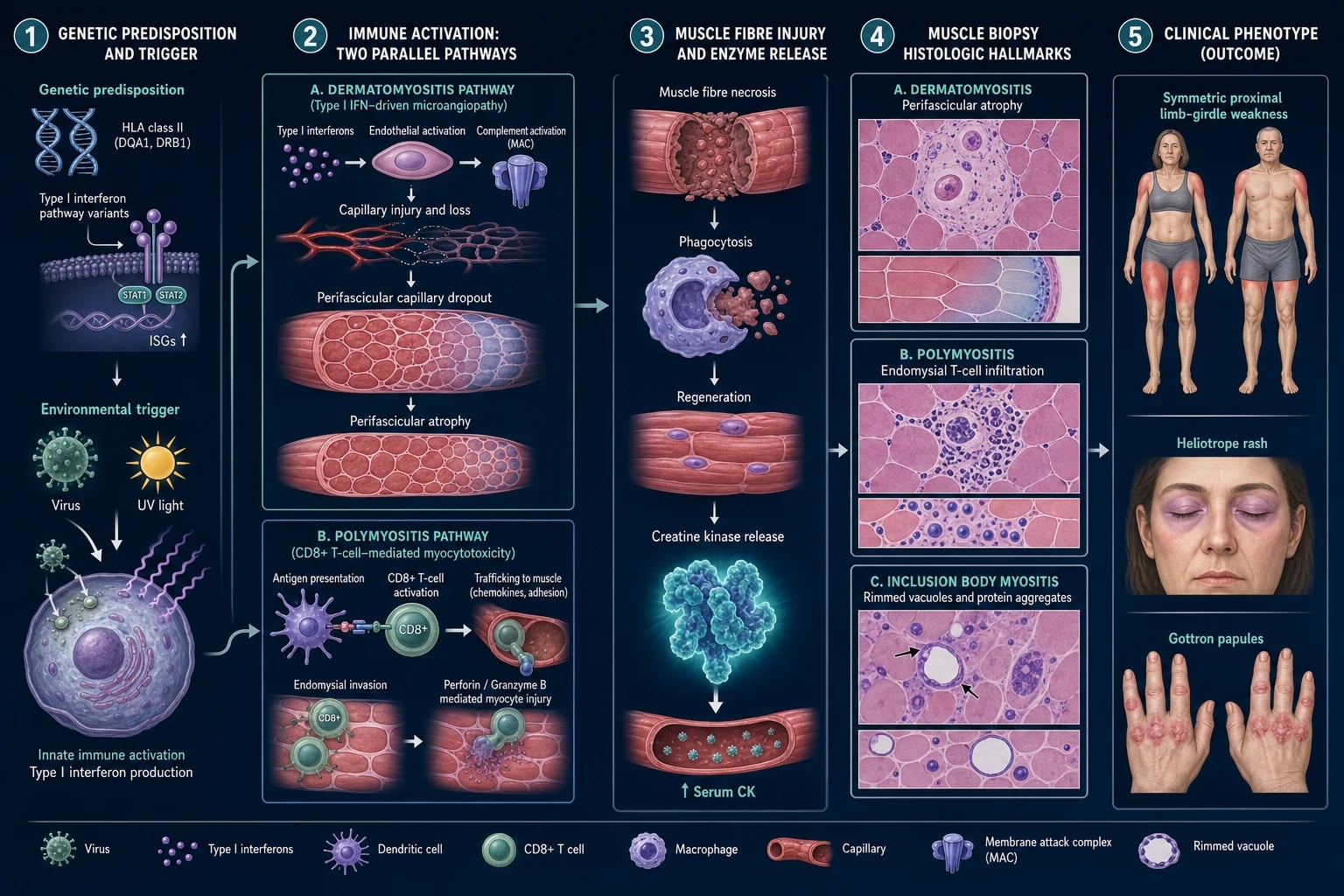
Two fundamentally different immune mechanisms underlie the myopathy, which explains the divergent biopsy findings and treatment responses.[1][2]
In dermatomyositis, a type-I-interferon-driven, complement-mediated microangiopathy is central. Immune-complex deposition activates complement, generating the membrane attack complex (MAC, C5b-9) on endomysial capillaries; this causes capillary destruction, ischaemia, and the hallmark perifascicular atrophy — the perifascicular region is the watershed zone most vulnerable to ischaemia because it lies furthest from the perforating arteriole. Plasmacytoid dendritic cells drive interferon-alpha/beta over-expression, which is also why the skin and microvasculature are involved and why JAK inhibitors are mechanistically rational. MAC deposition on endomysial capillaries is the earliest biopsy change and precedes inflammation.[2]
In polymyositis and IBM, the mechanism is cell-mediated cytotoxicity: CD8+ cytotoxic T cells and macrophages surround and invade MHC-class-I-upregulated, otherwise healthy muscle fibres through the endomysium, releasing perforin and granzyme and inducing necrosis. MHC class I is aberrantly upregulated on the surface of muscle fibres in PM and IBM (but not in normal muscle), enabling T-cell recognition.[2]
IBM adds a degenerative layer on top of the T-cell attack — rimmed vacuoles containing protein aggregates (TDP-43, p62, beta-amyloid, tau) and mitochondrial abnormalities with ragged-red or cytochrome-oxidase-negative fibres. This mixed autoimmune-degenerative biology is the principal reason IBM is steroid-refractory: immunosuppression cannot reverse a degenerative process.[5]
Immune-mediated necrotising myopathy (anti-SRP, anti-HMGCR) causes marked muscle-fibre necrosis with minimal inflammatory infiltrate and MAC deposition on non-necrotic fibres; the antibody targets are the signal recognition particle (ribonucleoprotein complex essential for protein translocation) and HMG-CoA reductase (the rate-limiting enzyme of cholesterol synthesis and the pharmacological target of statins — explaining the statin exposure link).[8][9]
DIMPLE — distinguishing the inflammatory myopathies
DIMPLE
Skin plus muscle; type-I-interferon/complement microangiopathy; perifascicular atrophy; cancer and ILD risk
Older men; quadriceps plus finger-flexor weakness; rimmed vacuoles; STEROID-REFRACTORY
Symmetric shoulder and hip girdle weakness; difficulty combing hair, rising from a chair, climbing stairs
Muscle only; CD8+ endomysial cytotoxic T cells; no skin signs; rarer than once thought
Antisynthetase (anti-Jo-1) and anti-MDA5 lead to interstitial lung disease; screen with HRCT and PFTs
Creatine kinase markedly raised (also AST/ALT/aldolase/LDH of muscle origin) — do not mislabel as liver disease
Clinical Presentation
Weakness develops subacutely over weeks to months, the tempo that distinguishes IIM from acute rhabdomyolysis (days) and the chronic muscular dystrophies (years). The symptom complex divides into muscle, skin, systemic/overlap, and subtype-specific clusters.[1][2]
Muscle — symmetric, proximal, weakness not pain
The classic pattern is symmetric proximal weakness of the shoulder and hip girdles. Patients describe difficulty lifting the arms overhead (combing hair, reaching a high shelf, hanging washing), rising from a low chair or the floor, climbing stairs, and stepping onto a bus. Neck-flexor weakness produces a head drop and fatigue holding the head up. Dysphagia ( solids more than liquids) and dysphonia (nasal voice) reflect pharyngeal and proximal oesophageal striated-muscle involvement and are prognostically important — they predict aspiration. Respiratory muscle weakness (rare but dangerous) presents with morning headache, orthopnoea, and a falling vital capacity. Pain is usually mild or absent; weakness is the cardinal feature, and this is the single most useful point of distinction from polymyalgia rheumatica. [1]
Dermatomyositis skin — pathognomonic clues
The DM rash is photosensitive, often precedes the myopathy by weeks or months, and several features are pathognomonic:[1][2]
- Heliotrope rash — a symmetric, violaceous (lilac) erythema with oedema of the periorbital skin, especially the upper eyelid; the most specific DM sign.
- Gottron papules — violaceous, scaly, flat-topped papules over the metacarpophalangeal and interphalangeal joints of the fingers; pathognomonic.
- Gottron sign — the same violaceous macular eruption over extensor surfaces of elbows, knees, and malleoli.
- Shawl sign (posterior shoulders and neck) and V-sign (anterior chest in the V of the neck) — photosensitive distributions.
- Holster sign — lateral thighs.
- Mechanic's hands — hyperkeratotic, fissured, dirty-appearing lateral fingers and palms; characteristic of antisynthetase syndrome but also seen in overlap.
- Nailfold changes — dilated and tortuous capillary loops, capillary dropout, dystrophic/ragged cuticles (Samitz sign), and periungual erythema — a microvascular stigmata shared with systemic sclerosis.
- Poikiloderma (salt-and-pepper atrophy with telangiectasia), flagellate erythema (linear streaks on the trunk), calcinosis cutis (subcutaneous calcium deposits, especially in juvenile DM), and cutaneous ulceration (a hallmark of anti-MDA5 disease with palmar papules). [1]
Antisynthetase syndrome
The classic tetrad is myositis, interstitial lung disease, non-erosive inflammatory arthritis, and fever, often accompanied by mechanic's hands and Raynaud phenomenon. ILD dominates prognosis: a dry cough and exertional dyspnoea may be the presenting complaint, and the myositis may be mild or amyopathic. Anti-Jo-1 is the prototypic antibody; PL-7, PL-12, EJ, and OJ are rarer and carry a higher ILD burden.[1]
Amyopathic and hypomyopathic DM
Approximately one in five DM patients have the characteristic rash for at least six months with no or minimal muscle weakness and a near-normal CK — amyopathic DM. Hypomyopathic DM has subclinical myopathy on investigation. Crucially, malignancy and ILD risk persist in amyopathic DM, so screening is unchanged.[2]
Inclusion body myositis — the pattern that is missed
IBM presents differently and must be actively sought in any older patient labelled "refractory PM". The pattern is asymmetric, mixed distal and proximal weakness with early quadriceps involvement (knee buckling, frequent falls, difficulty rising from a chair out of proportion to hip weakness) and deep finger-flexor involvement of the forearms (difficulty gripping, buttoning). Dysphagia is common and may dominate. Onset is over age 50 with male predominance, progression is slow (years), and the response to corticosteroids is poor. The CK is often only mildly to moderately raised, which contributes to delayed diagnosis.[5]
Differential Diagnosis
The pivotal skill is separating true inflammatory myopathy from its mimics. The combination of proximal weakness plus high CK plus myopathic EMG plus muscle biopsy narrows the field, after which the DM rash and antibody profile define the subtype.[2]
Dermatomyositis / polymyositis
- Symmetric PROXIMAL weakness (subacute) with markedly raised CK; weakness not pain
- DM: heliotrope rash, Gottron papules/sign, shawl/V-sign, nailfold telangiectasia
- Myopathic EMG; biopsy — perifascicular atrophy (DM) or endomysial CD8 (PM); myositis-specific antibodies
- Cancer and ILD associations (anti-TIF1-gamma/NXP2, anti-MDA5/Jo-1); steroid-responsive (NOT IBM)
Polymyalgia rheumatica
- Pain and STIFFNESS (not weakness) of shoulder and hip girdles; strength is NORMAL
- CK is NORMAL; ESR and CRP markedly raised (often above 40 mm/h); age over 50 (often over 70)
- Dramatic, rapid response to low-dose corticosteroids (under 24 to 72 hours)
- Associated with giant cell arteritis; no rash, no muscle biopsy inflammation
Drug-induced and endocrine myopathy
- Statin myopathy (or anti-HMGCR necrotising myopathy), glucocorticoid myopathy, colchicine, hydroxychloroquine, alcohol
- Endocrine: hypothyroidism (raised CK, normal strength), hyperthyroidism, Cushing, vitamin D deficiency
- Often proximal weakness with normal or mildly raised CK; reversible on withdrawal or correction
- No DM rash; biopsy non-specific (or type-2 fibre atrophy for steroids)
Neuromuscular mimics
- Myasthenia gravis — FATIGABLE, fluctuating weakness; ocular/bulbar prominent; CK NORMAL; AChR/MuSK antibodies
- Motor neuron disease (ALS) — mixed UMN plus LMN signs, asymmetric, progressive; CK mildly raised
- Muscular dystrophies — chronic, family history, pseudohypertrophy; genetic testing diagnostic
- Guillain-Barre — acute, ascending, areflexic, neuropathic; CSF albuminocytologic dissociation
Other entities to consider are infectious myositis (viral — influenza, coxsackie, HIV; pyomyositis; trichinosis), overlap connective tissue disease with myositis (systemic sclerosis, SLE, mixed connective tissue disease, Sjogren syndrome), and rhabdomyolysis with acute kidney injury. [1]
Clinical & Bedside Assessment
Take a structured history and examine systematically. The pattern of weakness (proximal versus distal, symmetric versus asymmetric), the presence and type of rash, and systemic screening for ILD and malignancy decide the diagnosis.[1][2]
- History: onset and tempo; difficulty with specific tasks (combing hair, rising from a chair, climbing stairs); dysphagia or dysphonia; skin changes and photosensitivity; dry cough and exertional dyspnoea (ILD); fever, arthritis, Raynaud (antisynthetase); weight loss or constitutional symptoms (malignancy); drug exposure (statins); family history; sun exposure.
- Muscle examination: test shoulder abduction, elbow flexion, hip flexion, knee extension, neck flexion; grade with the Medical Research Council 0 to 5 scale. In suspected IBM, specifically test distal finger flexors (deep flexors of the forearm) and quadriceps — these are easily missed and are the IBM signature.
- Skin examination: heliotrope rash, Gottron papules/sign, shawl/V-sign, mechanic's hands, nailfold (dilated capillary loops, dropout, ragged cuticles), poikiloderma, calcinosis, ulceration and palmar papules (anti-MDA5). Nailfold capillaroscopy using a dermatoscope or ophthalmoscope plus immersion oil reveals dilated, tortuous capillary loops and dropout.
- Systemic: lung auscultation (basal Velcro crackles of ILD), joints (non-erosive arthritis), Raynaud, dysphagia assessment (bedside swallow screen), cardiac examination (myocarditis, conduction disease), and a full malignancy review by system. [1]
Investigations
Diagnosis combines muscle enzymes, autoantibodies, EMG, MRI, and muscle biopsy, plus targeted screening for ILD and malignancy.[1][2]
- Muscle enzymes. CK is the key marker — markedly raised in IMNM (often many times normal, 10 to 50 times the upper limit of normal), moderately raised in DM and PM, and may be normal or near-normal in IBM and amyopathic DM. AST, ALT, LDH, and aldolase are also of muscle origin — a raised AST/ALT in the setting of proximal weakness is muscle-derived, not liver disease, and is one of the classic examination pitfalls. Aldolase is particularly useful when CK is normal (for example, in some IBM and amyopathic DM cases).
- Autoantibodies. ANA is often positive in DM. Myositis-specific antibodies (MSA) define phenotype and prognosis and should be requested in every suspected IIM. Myositis-associated antibodies (anti-Ro52, anti-La, anti-U1-RNP, anti-Ku, anti-PM-Scl) suggest overlap syndromes.
- EMG. Myopathic motor unit potentials (small, short, polyphasic) with spontaneous activity (fibrillations, positive sharp waves, bizarre high-frequency repetitive discharges). EMG excludes a neurogenic process and helps select a muscle for biopsy (biopsy the contralateral paired muscle to avoid end-stage artefact).
- MRI (STIR sequences). Demonstrates oedema in inflamed muscles and guides the biopsy site. MRI is increasingly the first-line imaging modality in suspected IIM and may obviate biopsy in classic DM with a pathognomonic rash and antibody.
- Muscle biopsy (diagnostic gold standard). DM shows perifascicular atrophy with perifascicular MAC deposition and perivascular/perimysial inflammation. PM shows endomysial CD8+ T-cell infiltration with MHC-class-I upregulation. IBM shows endomysial inflammation with rimmed vacuoles and protein aggregates (TDP-43, p62). IMNM shows necrosis with minimal inflammation and MAC on non-necrotic fibres.[2]
- ILD screen (mandatory in every patient). High-resolution CT chest (ground-glass, reticulation, honeycombing — typically non-specific interstitial pneumonia pattern) and pulmonary function tests (reduced FVC and DLCO, restrictive pattern). A falling FVC or DLCO on serial testing signals progression.
- Malignancy screen (mandatory in adult DM). Chest, abdomen, and pelvis CT plus age-appropriate tests (mammography, colonoscopy, cervical screening, prostate-specific antigen, pelvic ultrasound). Screening is intensified and extended for up to three years in patients with anti-TIF1-gamma or anti-NXP2 antibodies.[13]
- Other. TSH (exclude thyroid myopathy), HIV and hepatitis serology, echocardiogram and ECG (myocarditis, conduction disease), and skin biopsy (interface dermatitis with basement membrane thickening in DM).
Management — Resuscitation

Most IIM is subacute rather than an emergency, but several scenarios are time-critical and demand same-day escalation:[2]
- Rapidly progressive ILD (anti-MDA5) — hypoxaemia can evolve over days; urgent HRCT and aggressive combination immunosuppression (high-dose corticosteroids plus a calcineurin inhibitor or cyclophosphamide, with rituximab or a JAK inhibitor). High early mortality; early combination therapy is associated with improved survival.
- Severe dysphagia with aspiration risk or respiratory muscle weakness — assess swallow (speech and language therapy), arrange nasogastric feeding, and monitor forced vital capacity (a falling FVC approaching 1.5 L or 50 per cent predicted, or a negative inspiratory force worse than minus 60 cm water, signals the need for non-invasive ventilation as in neuromuscular respiratory failure).
- Myocarditis or arrhythmia — cardiology input, troponin, echocardiogram, and a rhythm monitor.
- Acute, very high CK with dark urine — rhabdomyolysis and acute kidney injury (more typical of IMNM or statin toxicity); intravenous fluids, urine alkalinisation, and renal protection. [1]
Management — Definitive & Stepwise
Treatment is phenotype-stratified. The central principle: induce remission with high-dose corticosteroids, then transition early to a steroid-sparing agent because steroid monotherapy is inadequate and toxic; treat the phenotype (ILD in antisynthetase/MDA5, cancer in cancer-associated DM, skin in DM) and avoid futile immunosuppression in IBM.[1][2]
Induction — corticosteroid backbone
- Oral prednisolone 0.5 to 1 mg/kg/day (typically 60 to 80 mg once daily) for 4 to 8 weeks, then a slow taper (approximately 10 per cent every 1 to 2 weeks) over 6 to 12 months guided by CK trend and muscle strength.[2]
- Intravenous methylprednisolone (500 to 1000 mg daily for 3 to 5 days) for severe disease — dysphagia, ILD, respiratory involvement, or markedly raised CK — followed by oral prednisolone.
- Co-prescribe gastric protection (proton pump inhibitor), bone protection (calcium 1000 to 1200 mg and vitamin D 800 to 1000 IU daily, with a bisphosphonate if osteoporotic), and Pneumocystis jirovecii prophylaxis (co-trimoxazole 480 mg once daily or 960 mg three times weekly) when the prednisolone dose exceeds 20 mg daily for more than four weeks.
Steroid-sparing agents — start early
Steroid monotherapy is inadequate for most IIM. Start a steroid-sparing agent early in parallel with the taper:[2]
- Methotrexate 10 to 25 mg once weekly (oral or subcutaneous, with folic acid 5 mg the day after); first-line steroid-sparing for DM and PM without ILD; monitor liver function and consider a chest CT before use (rare methotrexate pneumonitis).
- Azathioprine 2 to 3 mg/kg/day (typically 150 to 200 mg daily); check thiopurine methyltransferase (TPMT) activity before starting to avoid fatal myelosuppression; first-line in pregnancy.
- Mycophenolate mofetil 2 to 3 g/day in two divided doses (or mycophenolate sodium 1440 to 2160 mg daily); favoured when ILD coexists.
- Tacrolimus (trough 4 to 12 ng/mL) and ciclosporin (2.5 to 5 mg/kg/day) — calcineurin inhibitors, favoured for ILD and overlap disease. [1]
Biological and advanced therapy
- Intravenous immunoglobulin (IVIG) 2 g/kg/month (often split across two to five days; for example, 0.4 to 1 g/kg daily for two to five days), titrated to response. Particularly effective in DM refractory to first-line therapy and in severe dysphagia, with a rapid onset of action (within one to two weeks). The 1993 Dalakas randomised controlled trial established IVIG efficacy in steroid-resistant DM.[11]
- Rituximab (either two 1 g intravenous infusions two weeks apart, or 375 mg per square metre weekly for four weeks, repeated at six months if needed). For refractory DM and PM; the RITUXIMAB in Myositis (RIM) trial was negative on its primary combined endpoint but post-hoc analysis and a subsequent predictors analysis favoured dermatomyositis.[10]
- Cyclophosphamide (intravenous pulse 500 to 1000 mg per square metre monthly or 1 to 2 mg/kg/day oral) — reserved for severe or progressive ILD and overlap disease with organ threat.
- JAK inhibitors (tofacitinib 5 to 11 mg daily; baricitinib 2 to 4 mg daily) — increasingly used for refractory DM and anti-MDA5 ILD given the type-I-interferon axis, and an active area of investigation.
Skin
Rigorous photoprotection (broad-spectrum SPF 50-plus sunscreen, protective clothing, sun avoidance), hydroxychloroquine 200 to 400 mg daily, topical corticosteroids or calcineurin inhibitors, and IVIG for refractory rash. Hydroxychloroquine is less effective than in cutaneous lupus and rarely causes a drug-induced myopathy that can mimic active DM (check CK trend). [1]
IBM — do not over-treat
IBM is largely refractory to immunosuppression — avoid the harm of escalating steroids and immunosuppressants. The RESILIENT trial of bimagrumab (an activin-receptor type IIA antagonist targeting muscle growth) in IBM was negative for its primary endpoint.[12] Management is supportive: physiotherapy with a focus on quadriceps strengthening and fall prevention, swallowing therapy (and where needed cricopharyngeal myotomy or dilatation for severe dysphagia), and assistive devices (rolling walkers, knee braces, ankle-foot orthoses). A trial of low-dose steroids or IVIG for dysphagia is reasonable, with a clear stop date.
Malignancy and cancer-associated myositis
Search for and treat the underlying malignancy — resection may improve the myositis. Follow the 2023 International Myositis Society cancer-screening guideline: age-appropriate screening plus CT chest/abdomen/pelvis, intensified and extended for up to three years in patients with anti-TIF1-gamma or anti-NXP2 antibodies.[13]
Specific Subtypes & Scenarios
- Juvenile DM — the commonest IIM of childhood. Calcinosis cutis, vasculopathy (including intestinal vasculitis), and lipodystrophy are prominent and often overshadow the myopathy. Treatment is high-dose corticosteroids (often intravenous methylprednisolone pulses) plus methotrexate, with aggressive early therapy to prevent calcinosis and contractures. IVIG is used for refractory disease and to spare steroids.[1]
- Amyopathic DM — the classic rash for at least six months with no or minimal weakness and near-normal CK. Malignancy and ILD risk are unchanged, so a full malignancy screen and HRCT/PFTs are mandatory. Anti-MDA5 is over-represented and demands close respiratory surveillance.
- Antisynthetase syndrome (anti-Jo-1, PL-7, PL-12, EJ, OJ) — interstitial lung disease dominates prognosis. Treat with aggressive immunosuppression (often a steroid plus mycophenolate, tacrolimus, or cyclophosphamide, with rituximab for refractory ILD) and PFT and HRCT surveillance every three to six months.[1]
- Immune-mediated necrotising myopathy (anti-SRP, anti-HMGCR) — very high CK, necrosis with minimal inflammation. Statin withdrawal alone is insufficient for anti-HMGCR disease; aggressive immunosuppression (high-dose steroids plus MTX, azathioprine, or rituximab, often IVIG) is required. Anti-SRP disease tends to be severe, refractory, and relapsing.[8][9]
- Inclusion body myositis — older men; steroid-refractory. Supportive care, physiotherapy, fall-prevention, swallowing therapy, and assistive devices; avoid immunosuppression harm.[5][12]
- Overlap myositis — with systemic sclerosis, SLE, mixed connective tissue disease, or Sjogren; treat the dominant connective tissue disease. Anti-PM-Scl, anti-Ku, and anti-U1-RNP antibodies suggest overlap.
- Cancer-associated myositis — strongest with anti-TIF1-gamma and anti-NXP2 in adults; find and treat the malignancy, rescreen for up to three years.[13]
Complications & Pitfalls
- Interstitial lung disease — the leading cause of IIM morbidity and mortality (especially anti-MDA5 and antisynthetase). Screen every patient with HRCT and PFTs at diagnosis and serially.
- Occult malignancy — strongest with anti-TIF1-gamma/anti-NXP2 in adult DM; rescreen for up to three years.[13]
- Aspiration pneumonia from pharyngeal and oesophageal weakness — assess swallow early and involve speech and language therapy.
- Respiratory muscle weakness — monitor FVC; risk of ventilatory failure requiring non-invasive ventilation.
- Calcinosis cutis — especially juvenile DM; difficult to treat (topical sodium thiosulfate, bisphosphonates, surgical excision of symptomatic deposits).
- Myocarditis and cardiac conduction disease — a less-appreciated but real cause of mortality; baseline ECG and echocardiogram.
- Steroid toxicity — osteoporosis, diabetes, hypertension, infection, cataracts, avascular necrosis. Always co-prescribe bone and gastric protection and PJP prophylaxis.
- Misdiagnosis pitfalls: treating IBM as refractory PM with escalating immunosuppression (futile and harmful); mislabelling a raised AST/ALT as liver disease when it is muscle-derived; confusing PMR (pain, normal CK) with IIM (weakness, high CK); and under-recognition that amyopathic DM carries the same malignancy and ILD risk as classic DM.
Prognosis & Disposition
DM and PM generally respond to corticosteroids and steroid-sparing agents with good functional recovery; modern three- and five-year survival exceeds 80 per cent. Outcomes are driven by three factors: ILD, malignancy, and treatment complications.[1][2]
- Anti-MDA5 ILD carries the highest early mortality — death often within three to six months of presentation — and demands early aggressive combination immunosuppression.
- IBM is slowly progressive and refractory; patients accumulate disability (falls, dysphagia, quadriceps weakness requiring a wheelchair) over five to fifteen years. Life expectancy is relatively preserved.
- Juvenile DM has a good prognosis with modern aggressive therapy, though calcinosis, vasculopathy, and lipodystrophy may persist.
- Cancer-associated myositis prognosis tracks the underlying malignancy — resection may improve the myositis.
- Multidisciplinary follow-up — rheumatology, neurology, respiratory, dermatology, oncology (where relevant), and speech and language therapy — with serial CK, PFTs, and malignancy surveillance is essential. [1]
Special Populations
- Adult dermatomyositis and malignancy — strongest association; full age-appropriate malignancy screen, intensified if anti-TIF1-gamma or anti-NXP2, rescreen for up to three years.[13]
- Anti-MDA5 disease — rapidly progressive ILD with amyopathic DM; early aggressive combination immunosuppression and close respiratory monitoring, with a low threshold for intensive care.
- Juvenile DM — calcinosis, vasculopathy, and lipodystrophy predominate; aggressive early therapy to prevent calcinosis and contractures; weight-based dosing.
- Inclusion body myositis — older men; steroid-refractory; supportive care, physiotherapy, swallowing therapy; avoid immunosuppression harm.[5]
- Pregnancy — plan conception during remission. Prefer azathioprine or calcineurin inhibitors (comparatively safe in pregnancy and lactation). Avoid methotrexate, mycophenolate, and cyclophosphamide (teratogenic). Hydroxychloroquine and low-dose prednisolone are acceptable. IVIG is safe in pregnancy for flares.
- Statin-exposed patient with very high CK and weakness — consider anti-HMGCR immune-mediated necrotising myopathy; statin withdrawal alone is insufficient and immunosuppression is required.[8]
- Elderly and immunocompromised — at higher risk of steroid toxicity, infection, and drug interactions; favour IVIG and lower cumulative steroid exposure.
Evidence, Guidelines & Regional Differences
- NICE and the British Society for Rheumatology endorse high-dose oral prednisolone with early methotrexate as first-line steroid-sparing, azathioprine as first-line in pregnancy, IVIG for refractory DM and severe dysphagia, and rituximab for refractory disease. NHS commissioning tightly governs biologic access. [1]
- The ACR/Vasculitis Clinical Research Consortium and the International Myositis Society favour the 2017 EULAR/ACR criteria, the 2023 International Myositis Society cancer-screening guideline, and a growing role for JAK inhibitors in refractory DM and anti-MDA5 ILD (interferon-axis rationale). The RITUXIMAB in Myositis (RIM) trial, although negative on its primary endpoint, established the safety of rituximab and a post-hoc predictor analysis favoured DM.[10]
- Japan and East Asia — anti-MDA5 ILD is particularly aggressive, and Japanese centres have pioneered combination immunosuppression (cyclosporine or tacrolimus plus intravenous cyclophosphamide with high-dose steroids) for MDA5-ILD, with whole-body MRI for juvenile DM.[7]
- Europe (EULAR) — the 2017 EULAR/ACR criteria are standard; calcineurin inhibitors are favoured for ILD, and pan-European registries drive antibody-phenotype definition.
- India and resource-limited settings — diagnosis rests on CK plus muscle biopsy plus EMG; access to myositis-specific antibody panels, IVIG, and rituximab is often limited, and management is built around affordable steroid-sparing agents (methotrexate, azathioprine) with opportunistic infection prophylaxis.
Landmark evidence
- 2017 EULAR/ACR classification criteria (Lundberg 2017) — the current data-driven, probability-scored criteria for IIM and its subgroups, superseding the Bohan and Peter scheme; incorporates biopsy and anti-Jo-1.[1]
- Pathophysiology framework (Dalakas 2015, NEJM) — defines the two distinct mechanisms: complement-mediated microangiopathy (DM) versus T-cell-mediated cytotoxicity (PM/IBM), underpinning biopsy interpretation and the steroid-refractory nature of IBM.[2]
- Griggs 1995 / ENMC 2004 IBM criteria — codified the IBM phenotype (quadriceps plus finger-flexor weakness, rimmed vacuoles, steroid-refractory).[5][6]
- Mammen 2011 (anti-HMGCR) — defined the statin-associated autoimmune necrotising myopathy.[8]
- Sato 2005 (CADM-140/anti-MDA5) — described the amyopathic DM antibody that predicts rapidly progressive ILD.[7]
- Dalakas 1993 (IVIG in DM, NEJM) — the landmark randomised controlled trial establishing IVIG for steroid-resistant DM.[11]
- RESILIENT 2019 (bimagrumab, Lancet Neurology) — negative primary endpoint in IBM; reaffirmed the absence of disease-modifying therapy.[12]
- 2023 International Myositis Society cancer-screening guideline — risk-stratified malignancy screening, intensified for anti-TIF1-gamma and anti-NXP2, rescreen for up to three years.[13]
Red Flags
Exam application bank (NEET-PG / INICET)
One-line answer
Idiopathic inflammatory myopathies (IIM) are acquired autoimmune muscle diseases causing symmetric, proximal muscle weakness (difficulty combing hair, rising from a chair, climbing stairs) with raised creatine kinase. Dermatomyositis (DM) features characteristic skin manifestations (heliotrope rash — violaceous periorbital; Gottron papules/sign — over knuckles; shawl/V-sign, nailfold changes); polymyositis (PM) is muscle-only. Other IIM: inclusion body myositis (IBM) (older, distal + quadriceps/finger flexor weakness, poor steroid response), immune-mediated necrotising myopathy (IMNM) (anti-SRP/HMGCR), antisynthetase syndrome (anti-Jo-1, ILD, fever, mechanic's hands, arthritis), juvenile DM. Strong association of adult DM with occult malignancy and of antisynthetase/anti-MDA5 disease with interstitial lung disease (ILD). Diagnosis combines proximal weakness + high CK + myopathic EMG + mu
Worked stems (answer without another resource)
Stem 1 — Classic presentation. Map symptoms to mechanism; name the first investigation and first treatment step with dose/route if drug therapy is standard. [1]
Stem 2 — Unstable / complicated. List red flags that force immediate resuscitation, theatre, ICU, antidote, or reperfusion — and what you do in the first 15 minutes. [1]
Stem 3 — Atypical group. Elderly, pregnancy, child, or immunocompromised: how presentation and thresholds change. [1]
Stem 4 — Differential trap. Name the three closest mimics and one discriminator for each. [1]
Stem 5 — Disposition. Who goes home with safety-netting, who is admitted, who needs HDU/ICU/theatre, and what follow-up is mandatory. [1]
Rapid viva checklist
- Definition + classification
- Pathophysiology chain
- Bedside signs / criteria
- Score with exact components (if any)
- Emergency bundle
- Definitive therapy with doses
- Complications of disease and of treatment
- Special populations
- Guideline/trial name if classic
- Three exam traps
Coverage self-check
If you cannot answer any stem above from this page alone, re-read the matching section — the page is intended to be self-sufficient for final-prof and NEET-PG/INICET questions on Dermatomyositis & Polymyositis (Idiopathic Inflammatory Myopathies).
Exam Pearls
References
- [1]Lundberg IE, Tjärnlund A, Bottai M, et al. 2017 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Adult and Juvenile Idiopathic Inflammatory Myopathies and Their Major Subgroups Arthritis Rheumatol, 2017.PMID 29106061
- [2]Dalakas MC. Inflammatory muscle diseases N Engl J Med, 2015.PMID 25923553
- [3]Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts) N Engl J Med, 1975.PMID 1090839
- [4]Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts) N Engl J Med, 1975.PMID 1089199
- [5]Griggs RC, Askanas V, DiMauro S, et al. Inclusion body myositis and myopathies Ann Neurol, 1995.PMID 7486861
- [6]Hoogendijk JE, Amato AA, Lecky BR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10-12 October 2003, Naarden, The Netherlands Neuromuscul Disord, 2004.PMID 15099594
- [7]Sato S, Hirakata M, Kuwana M, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis Arthritis Rheum, 2005.PMID 15880816
- [8]Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy Arthritis Rheum, 2011.PMID 21360500
- [9]Lescure FX, Mescam-Mancini L, Allenbach Y, et al. Myositis-specific autoantibodies, a cornerstone in immune-mediated necrotizing myopathy Autoimmun Rev, 2019.PMID 30639649
- [10]Aggarwal R, Bandos A, Reed AM, et al. Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis Arthritis Rheumatol, 2014.PMID 24574235
- [11]Dalakas MC, Illa I, Dambrosia JM, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis N Engl J Med, 1993.PMID 8247075
- [12]Amato AA, Saperstein DS, Bodzin AE, et al. Safety and efficacy of intravenous bimagrumab in inclusion body myositis (RESILIENT): a randomised, double-blind, placebo-controlled phase 2b trial Lancet Neurol, 2019.PMID 31397289
- [13]Fiorentino D, Albayda J, Cushman C, et al. International Guideline for Idiopathic Inflammatory Myopathy-Associated Cancer Screening: an International Myositis Assessment and Clinical Studies Group (IMACS) initiative Nat Rev Rheumatol, 2023.PMID 37945774